

Abstract
Over the past decade, fluorescence‐based single‐molecule studies significantly contributed to characterizing the mechanism of RNA polymerase at different steps in transcription, especially in transcription initiation. Transcription by bacterial DNA‐dependent RNA polymerase is a multistep process that uses genomic DNA to synthesize complementary RNA molecules. Transcription initiation is a highly regulated step in E. coli, but it has been challenging to study its mechanism because of its stochasticity and complexity. In this review, we describe how single‐molecule approaches have contributed to our understanding of transcription and have uncovered mechanistic details that were not observed in conventional assays because of ensemble averaging.
Keywords: single molecule, transcription, RNA polymerase, FRET, alternating laser excitation
Introduction
Transcription of the bacterial genome is implemented by a multisubunit RNA polymerase (RNAP or E) enzyme1 that shares many basic structural features and molecular mechanisms with eukaryotic RNA polymerases.1 Most striking among them is the conserved RNAP architecture resembling a “crab‐claw” [Fig. 1(A)]. RNA synthesis by RNAP is achieved via the nucleotide addition cycle, in which each iteration incorporates an incoming ribonucleoside triphosphate (NTP). Incoming NTPs are selected based upon sequence complementarity to the DNA template. The nucleotide addition mechanism for RNAP employs two Mg2+ ions for catalysis. The tightly bound Mg2+ is coordinated by a conserved triad of aspartic acid residues in a highly conserved NADFDGD motif within the RNAP β′ subunit, while a second Mg2+ associates with the incoming NTP.2, 3 The 370 kDa E. coli RNAP core enzyme is composed of five subunits (αI, αII, β′, β, ω)4 that can be grouped into three functional classes: assembly platform subunits that nucleate RNAP assembly (αI, αII), catalytic subunits (β′, β), and an auxiliary function subunit (ω).1, 3 The cleft and channels formed between the β and β′ subunits (pincers of the crab‐claw) includes: (i) the duplex DNA‐binding clamp where downstream DNA is bound, (ii) the primary channel that holds the RNA‐DNA hybrid, and (iii) the RNA exit channel (reviewed in Ref. 2) [Fig. 1(A)]. The active site for RNA polymerization is located at the base of the cleft between the pincers. Several mobile elements of the β′ subunit, including the bridge helix and trigger loop, divide the primary channel and form a secondary channel through which diffusing NTPs can access the active site [Fig. 1(B)]. The two identical α subunits (αI and αII) form a functionally asymmetric dimer through different interactions with the β and β′ subunits. The core RNAP is catalytically competent for transcription elongation; however, sequence‐specific recognition of promoter DNA requires association with a sigma (σ) factor to form a holoenzyme, holoRNAP (Eσ) (Eσ structure reviewed in Refs. 5, 6). Bacteria typically have one major σ factor (σ70 in E. coli) that drives expression of “housekeeping” genes and varying numbers of alternative σ factors (6 in E. coli, but up to 60 in Streptomyces coelicolor) that direct expression of genes in response to specific environmental conditions or stress.1, 4, 7, 8 The σ70 subunit consists of four domains (R1–4), connected by flexible linkers, that interact with separate DNA sequence elements in the promoter.4
Figure 1.
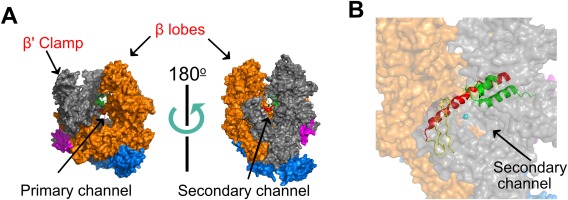
High‐resolution structure of Thermus aquaticus core RNAP (PDB 1HQM). Figure generated using PyMol. (A) The five RNAP subunits are represented with different colors: The two α subunits are blue, β′ is grey, β is orange, and ω is magenta. Two orientations (related by ∼180° rotation) are shown, with the LEFT showing the trailing edge of RNAP facing upstream DNA and the RIGHT showing the leading edge facing downstream DNA. The cleft between the β′ clamp and β lobes (the pincers) forms the primary channel, while the secondary channel is arranged on the opposite face of RNAP. (B) The catalytic magnesium (cyan sphere), bridge helix (red), trigger loop (green), and F‐loop (yellow) of the active site can be seen from the downstream facing side of RNAP.
Transcription can be divided into three major stages: initiation, elongation, and termination [Fig. 2(A)]. Each stage is a multistep process that offers multiple targets for regulation. Initiation is the most regulated step in transcription; it begins with Eσ binding to promoter DNA to form the Eσ‐closed promoter complex (RPC), in which the DNA remains double stranded. The RPC undergoes a series of conformational changes (reviewed in Ref. 4 resulting in the unwinding of ∼13 bp DNA near the transcription start site (TSS).9, 10 This forms an Eσ‐open promoter complex (RPO) either spontaneously (σ70) or with assistance of ATP hydrolysis (σ54).11 The unwound region of DNA (open bubble) in the RPO has the template strand placed at the wall of the primary channel with the DNA base corresponding to TSS (+1) positioned at the active site, and an initiating NTP (iNTP) and a second NTP can bind to the “i” and “i + 1” sites of the enzyme, respectively. Phosphodiester bond formation between the initial two NTPs leads to a transition from RPO to the initial transcribing complex (RPITC) that extends the RNA in the 5′ to 3′ direction. After reaching a length of 4–5 bases,9, 10, 12, 13 the RNA 5′‐end begins to sterically and electrostatically clash with the σ factor at the “σ finger” or σR3.2 domain (for σ70). This σR3.2 domain blocks access to the RNA exit channel, resulting in either the ejection of the RNA through the secondary channel (abortive initiation) or structural re‐organization of the σ factor (regions σR3.2 and σ70‐R4) to open the RNA exit channel.14 Structural re‐organization marks the entry of RPITC into elongation through the process of promoter escape. The highly processive ternary RNAP–DNA–RNA elongation complex (RPE) eventually terminates transcription and releases the RNA product either after reaching an intrinsic termination signal encoded in the DNA template, or by the action of termination factors such as Rho.
Figure 2.
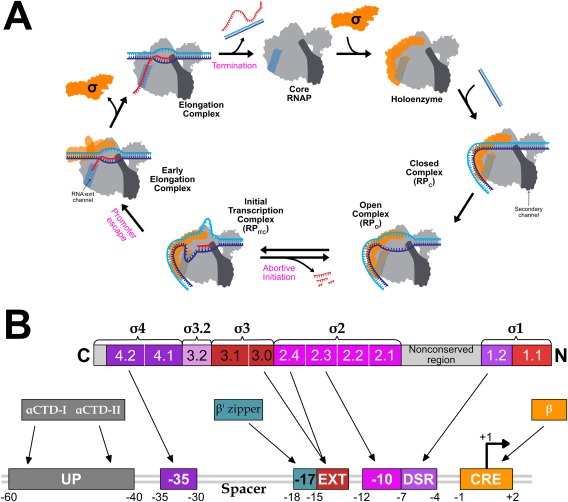
Schematic of transcription cycle and σ70 interactions with promoter DNA. (A) Association of σ (green) with core RNAP forms the RNAP holoenzyme (grey). The secondary channel (darker grey highlight) and the RNA exit channel (medium grey highlight) are represented on the right side and left side of the RNAP cartoon, respectively. σR3.2 and σR4 is shown protruding into the primary channel and occupying the RNA exit channel, respectively. HoloRNAP associates with promoter DNA (template strand in blue, and nontemplate strand in cyan) to form RPC, which isomerizes to RPO. Initial transcription by RPITC produces short abortive RNA products. Nascent RNA can displace σR4 from the RNA exit channel by clashing with σR3.2 and can enter the RNA exit channel to form the elongation complex. Eventually, RPE undergoes transcription termination resulting in RNAP dissociation from DNA. (B) Evolutionarily conserved structural domains and conserved regions of σ are shown as numbered and color‐coded boxes. Promoter DNA is shown underneath, with arrows indicating interactions between promoter DNA sequence elements and regions of σ or RNAP.
Much of what we understand about transcription derives from ensemble‐averaged experiments; however, single‐molecule (sm) experiments performed over the past decade have also contributed significantly. The application of sm techniques was driven by their utility in providing mechanistic information that is typically concealed in the averaged data from ensemble measurements. In particular, information about rare sub‐species, transient interactions between biomolecules, distinct subpopulations, or states for static or dynamic heterogeneous populations has been revealed through sm approaches.15, 16, 17, 18 A variety of sm methods have been applied in the study of bacterial RNAP.19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31
In fluorescence microscopy and spectroscopy, FRET (Förster Resonance Energy Transfer) is a valuable tool for distance‐dependent measurements. FRET is the nonradiative transfer of energy from a donor fluorophore excited by a higher energy photon to an adjacent acceptor fluorophore. FRET is calculated by the ratio of acceptor photons to the sum of donor and acceptor photons. FRET occurs if the following conditions are met: (i) the respective orientation of the donor and acceptor dipoles is not orthogonal, (ii) the donor and acceptor fluorophores are within a 2–10 nm range for resonance energy transfer, and (iii) there is an overlap between the donor emission and the acceptor excitation spectra.32 A powerful advancement for smFRET (single‐molecule FRET) was the development of alternating laser excitation (ALEX), in which the excitation source is continuously alternated between donor and acceptor excitation wavelengths by acoustic optical modulation.32, 33, 34 Application of smFRET‐ALEX enables separation of subpopulations based on the stoichiometry of donor/acceptor fluorophore labeling.
Here, we review and highlight contributions mainly from diffusion‐based smFRET‐ALEX and some other sm techniques that have further elucidated key steps in RNAP transcription initiation. These include transcription bubble opening and dynamics related to TSS selection, the mechanism of DNA scrunching, changes in RNAP clamp conformation during the transcription cycle, the characterization of a paused‐backtracked state in initiation, and retention of σ70 in early elongation.
RNAP RPO Formation and TSS Selection
Promoter search
Transcription of a particular set of E. coli genes requires the proper σ factor to position RNAP at specific promoter sequences. Determining the mechanism by which Eσ scans the genome for target promoters is a challenge. Although the precise mechanism remains obscure, sm techniques have contributed to the elucidation of molecular mechanisms that govern this process. Facilitated diffusion or random 3‐dimensional (3D) diffusion mechanisms have been proposed.35, 36 Potential facilitated diffusion mechanisms include: (i) a series of local Eσ–DNA binding and unbinding events, called 1‐dimensional (1D) 'hopping, (ii) the directional movement of Eσ along DNA without dissociation, called 1D sliding, and (iii) the movement of Eσ from one DNA site to another that is juxtaposed through DNA looping, called intersegmental transfer (discussed in Refs. 20, 26). Facilitated diffusion of RNAP has been demonstrated using a variety of sm techniques including AFM and sm fluorescence microscopy.26, 37, 38, 39, 40 However, the random 3D diffusion of Eσ is assumed to be the predominant mechanism.41, 42
The strength of the Eσ–promoter interaction is primarily determined by the sequence properties of the promoter. Different promoter elements interact with different regions of σ or core RNAP. The six nucleotide‐long (hexameric) −10 and −35 elements with “TATAAT” and “TTGACA” consensus sequences,43 respectively, and the length of the spacer between them, are the major determinants of promoter strength43 [Fig. 2(B)]. Additionally, the UP element located upstream of the −35 element to which the α subunits of RNAP bind, is required for efficient transcription from some promoters.43, 44 A summary of contacts between Eσ70 and different promoter elements is shown in Figure 2(B) and are extensively reviewed elsewhere.43, 45
Characterizing the RNAP clamp domain conformation during RPO formation using smFRET‐ALEX
Ensemble and sm experiments have uncovered many kinetic and structural details of the RPC transition to RPO. The mechanism for transcription initiation at all σ70 promoters is proposed to entail a three‐step promoter opening process.46 Initially, the isomerization of RPC to RPO was thought to be a single step process;47 however, recent biophysical and biochemical data points to the existence of relatively unstable intermediate complexes (reviewed in Ref. 4). During the isomerization of RPc into the first intermediate complex (RPI1), the DNA upstream of the −35 element wraps around Eσ, while the DNA downstream of the −10 element bends towards and into the RNAP cleft.4 DNA unwinding in the RNAP cleft then leads to the formation of a 13‐base DNA bubble, converting RPI1 into a second intermediate complex, RPI2. Finally, additional conformational changes in the RNAP clamp that binds downstream DNA forms stable RPO from RPI2.4, 46 Crystal structures of RNAP–DNA complexes revealed critical conformational changes in RNAP that occur during transcription initiation.48, 49 Among these, the most prominent structural transformations involved the RNAP clamp, a large mobile domain within the β′ subunit that is connected to the “β′ switch‐2” hinge region at the base of the β′ pincer.4
Conformational changes in the β′ switch‐2 region enable the β′ clamp to swing relative to the β pincer, which leads to either opening or closing of the pincers. Depending on crystallization conditions, the β′ clamp adopted different conformations, varying from an open to a closed state because of clamp swinging by at least 20°.49 To confirm that the clamp conformations were not artifacts of crystallization, and to probe their functional relevance, the clamp conformations at different steps of transcription were examined using diffusion‐based smFRET‐ALEX.49 Changes in spatial organization of the clamp domain were monitored by measuring shifts in FRET efficiency—a function of the distance between two FRET dyes positioned at the tips of the RNAP pincers [Fig. 3(A)]. For example, swinging of the β′ clamp away from the β pincer would result in a relative decrease in FRET efficiency, as the distance between the two pincers would increase. The smFRET‐ALEX measurements by Chakraborty et al.49 identified three different clamp conformations, each with a characteristic mean FRET, based on Gaussian fitting of FRET efficiency histograms [Fig. 3(B)]. The three clamp conformations corresponded to an (i) open, (ii) closed (inward rotation by ∼14°), and (iii) collapsed clamp, with the collapsed clamp reflecting a conformation more closed than any crystal structure had shown (inward rotation by ∼22°).
Figure 3.
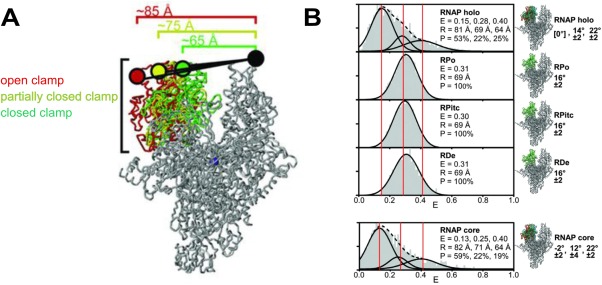
RNAP clamp conformation during different steps of transcription. Reprinted from Chakraborty et al.49 with permission. (A) The labeling positions of the FRET dye pair at the tip of the RNAP pincers (Cy3B donor on β′ clamp tip, Alexa647N acceptor on β lobe tip) distinguishes three clamp conformations (distances shown) during different steps of transcription: the closed clamp‐higher FRET configuration (green), the partially closed clamp‐medium FRET configuration (yellow), and the open clamp‐lower FRET configuration (red). (B) FRET histograms from single molecule FRET experiments. Three vertical lines mark the mean efficiency of FRET transfer “E” for each of the three clamp conformations. The mean distance calculated from the mean efficiency is shown as is the percentage of each subpopulation.
Given the short lifetime of Eσ70 RPC and its rapid isomerization to RPO at room temperature, the ATP hydrolysis‐dependent Eσ54 mechanism for bubble opening was used to monitor changes in clamp conformation for RPC, RPi1, and RPi2. After initial promoter binding, E‐σ54 remains in RPC until an AAA+ ATPase (e.g., NtrCl) mediates the transition to RPO. The RPi1 and RPi2 intermediates were trapped using ATP analogs that mimic the ground (ADP‐BeFx) and transition state (ADP‐AlFx) intermediates during ATP hydrolysis. The RNAP clamp exhibited an open conformation for all initial steps until formation of RPO, in which a closed clamp conformation was observed. However, the flexibility in clamp conformations during the early steps of transcription initiation was implied by the minor subpopulations in the FRET histogram corresponding to the closed clamp.49 An open clamp conformation for Eσ up until RPi2 is consistent with the hypothesis that opening of the RNAP pincers permits DNA loading into the RNAP cleft and for subsequent conformational changes in DNA until formation of RPO. The closed clamp conformation in RPO suggests an interaction between the positively charged inner surface of the RNAP cleft and the negatively charged backbone of the single stranded DNA (ssDNA) in the bubble.
A smFRET‐ALEX assay for measuring open bubble formation and dynamics
RPO formation is a common rate‐limiting step in transcription initiation. RPO stability is controlled by various factors, including the global transcription regulators guanosine (penta‐ or tetra‐) phosphate ((p)ppGpp) and DksA.50, 51 To measure promoter opening kinetics, several ensemble and sm approaches have been used, including chemical perturbation footprinting assays (discussed in Refs. 52, 53), DNA topology assays,52 magnetic tweezers,54 and sm fluorescence techniques (discussed in Ref. 29). To sense conformational change in the transcription bubble during RPO formation, a modified diffusion‐based smFRET‐ALEX assay53 with a FRET dye pair positioned in the transcription bubble was used to conduct end‐point and real‐time monitoring experiments. In this FRET assay, the donor/acceptor dye pair was placed on opposite DNA strands within a region that would form an open bubble in initiation.53 Importantly, the dyes were in such close proximity (2 bp apart) that one dye quenched the fluorescence of the other, whereas quenching was abolished upon bubble opening.53 We modified this assay by generating a library of DNA probes carrying donor/acceptor dyes at various positions within the transcription bubble and monitored the FRET changes in different regions of the bubble. This library of FRET dye DNA probes can be a powerful tool for elucidating the conformational dynamics of transcription bubble opening during Eσ70 RPO formation.
Several analysis methods have expanded upon the information obtained from FRET histograms to identify the dynamic behavior of single molecules in the millisecond and sub‐millisecond time‐scales. Burst variance analysis (BVA) is one such method.55 Because of photon counting statistics (shot‐noise), a smFRET measurement of any molecule with a fixed distance between donor and acceptor results in a distribution of FRET efficiencies instead of a single value. The width of the distribution increases as the number of photons decreases. The expected width (standard deviation, σE) of a distribution only broadened by shot‐noise can be computed from the binomial law. A distribution broader than the shot‐noise limited width indicates the superimposition of multiple heterogeneities in a FRET distribution. This heterogeneity could either be because of the presence of multiple species with distinct FRET efficiencies (static heterogeneity) or a single species dynamically interconverting between different states (dynamic heterogeneity). BVA determines if these heterogeneities are static or dynamic. BVA achieves this by equally dividing each burst into smaller sub‐bursts for which a FRET efficiency is computed. The standard deviation of sub‐bursts from the FRET efficiency of the burst is then computed (si). Shot‐noise because of the lower number of photons per sub‐burst predicts a distribution of si around σE. For static heterogeneity, the distribution of si is consistent with σE. However, if the broadening is caused by dynamic heterogeneity, then the distribution of si is deviated from σE. Application of BVA by Robb et al.56 demonstrated that FRET efficiency for the RPO transcription bubble fluctuated more than the expected shot noise limit. This indicated that the millisecond time‐scale dynamics of the transcription bubble could be associated with dynamics that are important for TSS selection [Fig. 4(A)]. Both eukaryotic and bacterial RNAPs can initiate transcription from different TSSs on the same promoter. Promoter sequence characteristics play an important role in TSS selection, which in turn could affect abortive RNA synthesis, RNA stability, and translation efficiency. For bacterial RNAPs, transcription is primarily initiated by purine NTPs at sites located 4–12 bp downstream of the −10 element. The process of alternative TSS selection is thought to proceed through a scrunching and anti‐scrunching mechanism56, 57, 58 [Fig. 4(B)]. For start sites located downstream of the canonical TSS, scrunching of the transcription bubble unwinds additional downstream DNA, incorporating it into the transcription bubble while bringing it towards the active site. However, positioning of registers upstream to the canonical TSS in the active site requires rewinding of DNA at the downstream edge of the bubble.56 Recently, the correlation between TSS position and scrunching was demonstrated using a high‐throughput approach termed MASTER (massively systematic transcript end readout), which relied on next generation sequencing to report on the TSS and transcript yields generated from a library of DNA templates.59 The increase of downstream TSSs for negatively supercoiled DNA templates, which promote DNA unwinding and bubble expansion, suggested a scrunching mechanism for downstream TSS selection.59 Further evidence implicating DNA scrunching dynamics for TSS selection came from combining leading edge and trailing edge RNAP crosslinking to the DNA template with MASTER.58
Figure 4.
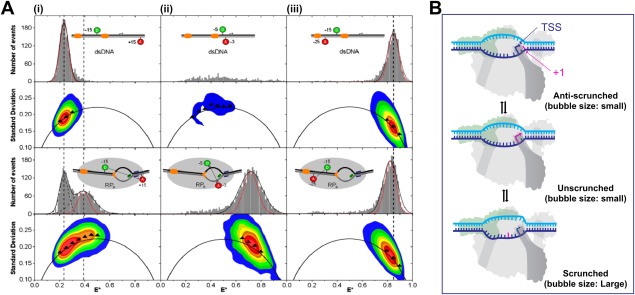
Transcription‐bubble dynamics in RPo and TSS. Reprinted from Robb NC et al.56 with permission. (A) FRET histograms from diffusion‐based smFRET‐ALEX measurements were derived from the mean values of (at least) triplicate experiments. Sizable FRET distributions were fitted with a Gaussian function (black curve) to determine the center and width of the distribution, while the calculated shot‐noise‐limited width is shown as a red Gaussian fit. For BVA plots the black arc represents σE, colored contour plots represent frequency distributions (red contour, most abundant region; blue, less abundant) and triangles represent the standard deviation of a particular part of the two‐dimensional histogram from the experimental data. Fluorophore labeling registers are relative to TSS. (i) DNA template with nontemplate strand donor (NTD) at register −15 and template strand acceptor (TA) at register +15 demonstrated a low‐FRET population with no dynamics (black triangles fall on σE line). However, RPO population with a mid‐FRET population showed dynamic heterogeneity (black triangles for mid‐FRET population deviate from σE) (ii) DNA template with −5 NTD and −3 TA alone gave few bursts in the FRET histogram and in the BVA plot. After RPO formation, the quenching was abolished and a high‐FRET population is seen. Dynamics in the transcription bubble was observed (black triangles deviate from σE) (iii) DNA template with −15 NTD and −25 TA showed no change in FRET (high‐FRET for both) for DNA alone or in RPO. No dynamics was seen as the dyes fall outside of RNAP–DNA interaction regions (iii) (B) Schematic showing potential alternative transcription start sites, achieved through different bubble dynamics, including changes in bubble size (scrunching, unscrunching, and antiscrunching).
Transcription initiation proceeds through a scrunching mechanism and can generate a paused, backtracked state
The mechanism of initial transcription and detection of abortive initiation
RNAP can form very stable RPO complexes because of the high binding affinity between Eσ and promoter elements. However, strong Eσ–promoter interactions may impede promoter escape. A characteristic of such promoters (LacUV5, λPR, T5N25) is the increased production of short abortive RNA products (2–10 nucleotides for lacUV5 promoter60). Initial transcription by RPITC ultimately results in promoter escape or abortive initiation, in which the abortive products are released through the secondary channel. Determinants of abortive initiation include: (i) the stability of the RNA‐DNA hybrid (dictated by the sequence and the length of the nascent transcript), (ii) repulsive interactions between the nascent transcript 5′ end and the σR3.2 finger that blocks the RNA exit channel,9, 13, 61 and (iii) the interactions between Eσ and promoter DNA.62, 63, 64 Abortive initiation returns the complex to RPO, which can continue to undergo multiple initial transcription/abortive initiation cycles until promoter escape occurs.
RNAP abortive initiation has been observed by equipping a total internal reflection (TIRF) optical microscopy setup with smFRET‐ALEX and imaging surface‐immobilized Eσ70 complexes labeled with a FRET dye pair.65 FRET changes between the leading edge of Eσ70 and downstream DNA were evaluated for RPO and different RPITC states; these states were achieved through NTP starvation (i.e. by excluding one or more NTPs from the reaction) to reach a specific RPITC state (e.g., ITC 2, 4, or 7). The mean FRET efficiency increased gradually in going from RPO (or earlier ITC states) to later RPITC states, which suggested that longer nascent RNAs increasingly reduce the distance between the downstream DNA and the leading edge of RNAP.65
Insight into the mechanism of initial transcription by RPITC was offered in early DNA footprinting studies,60, 66, 67, 68 in which three possible models were proposed [Fig. 5(A)]: (1) transient excursions, (2) RNAP inchworming, and (3) scrunching. The transient excursion model proposed that RNAP translocates downstream during initial transcription before returning back to the TSS following abortive initiation. The inchworming mechanism hypothesized structural flexibility in RNAP that would permit the leading edge of RNAP to translocate downstream and return, while the trailing edge of RNAP remained stationary. The scrunching mechanism hypothesized that a stationary RNAP reeled downstream DNA into the RNAP cleft during initiation, resulting in a larger “scrunched” bubble that recoils and returns to the original bubble size during abortive transcript release.69 These mechanisms were studied by smFRET‐ALEX experiments in solution, which supported a scrunching mechanism.69
Figure 5.
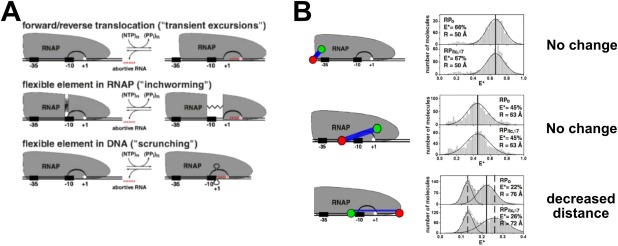
Initial transcription by RNAP proceeds through a scrunching mechanism. Reprinted from Kapanidis et al.69 with permission. (A) The three mechanistic models for initial transcription. Top, transient excursions; middle, inchworming; bottom, scrunching. (B) Schematic of the relative dye positions used in the experiments, and the corresponding FRET histograms comparing RPO to RPITC (FRET histogram: RPo is shown in the top panel, while RPITC is shown immediately beneath). TOP ROW: Since no change in FRET efficiency in going from RPO to RPITC for the upstream DNA‐RNAP trailing edge dye configuration, then the transient excursion model is ruled out. MIDDLE ROW: Since no change in FRET efficiency in going from RPO to RPITC for the spacer‐RNAP leading edge configuration, then the inchworming model was is ruled out (a change is expected if there was a flexible element in RNAP with the leading edge stretching into downstream DNA). BOTTOM ROW: The decreased FRET efficiency in going from RPO to RPITC for the spacer‐downstream DNA configuration supports the scrunching model for initial transcription mechanism.
The smFRET‐ALEX experiments tested different configurations of FRET pairs on RNAP and DNA: (1) the leading edge of RNAP and downstream DNA, (2) the trailing edge (σ70‐R4) of the Eσ70 and upstream DNA, (3) the trailing and leading edges of Eσ70 relative to the spacer element (position −20 relative to TSS), and (4) upstream and downstream DNA [Fig. 5(B)]. The transient excursion model was refuted because during initial transcription, no changes in FRET were observed in the upstream DNA‐RNAP trailing edge configuration for RPITC≤7 compared to RPO [Fig. 5(B), top row]; by contrast, a shift toward lower FRET was expected according to the model. The inchworming model was ruled out based on the absence of FRET changes between the spacer region and the leading edge of the Eσ70 (for RPITC≤7 compared to RPO) [Fig. 5(B), middle row]. The inchworming model, however, predicted decreased FRET between the leading edge of RNAP and the promoter spacer region. The observed FRET changes in the downstream DNA‐leading edge and the upstream‐downstream DNA configurations supported the scrunching model [Fig. 5(B), bottom row]. The scrunching mechanism has also been demonstrated by Revyakin et al. using a different sm method, DNA nanomanipulation with magnetic tweezers.70 Subsequent studies showed viral and eukaryotic RNA polymerases also perform initial transcription through DNA scrunching.71, 72 Thus, DNA scrunching may be a universal mechanism for initial transcription throughout all domains of life.
Despite significant progress in our understanding of transcription initiation mechanisms, many details remain obscure. One major open question is how extra ssDNA in the scrunched states are accommodated by RNAP. The structural data on transcription initiation complexes suggest that the internal space within the RNAP cleft is the main limiting factor9 for accommodation of ssDNA. However, analysis of the size distribution of abortive products from strong promoters shows the presence of abortive products of significant length (up to 18–25 nt).56, 58, 59, 61, 66 A recent RNAP–DNA cross‐linking and footprinting study demonstrated that the initiation complex may accommodate the additional scrunched bases of the nontemplate strand by extruding them out of the cleft and into the solvent.73 Moreover, Ploetz and Lerner et al. reached the same conclusion using sm experiments that simultaneously involved FRET and protein‐induced fluorescence enhancement (PIFE) measurements.29 These experiments utilized the Cy3 dye, which increases its fluorescence in response to a rise in local viscosity or steric restriction. Internally labeled bases on the nontemplate strand at position +1 or +3 (relative to the TSS) acted as FRET donors to an acceptor positioned within the spacer element of the template strand. According to Winkelman et al.,73 the nontemplate base at position +1 should be exposed to solvent during scrunching, hence the steric restriction should be reduced. Increased FRET for the +1‐Cy3 reported on open bubble formation and the intensity of Cy3 fluorescence also increased (i.e. the PIFE effect) upon RPO formation, indicating that the +1‐Cy3 was in close proximity to RNAP. Upon further extension of the nascent RNA, the PIFE effect increased for +3‐Cy3 (i.e. became part of the bubble owing to scrunching); however, the PIFE effect decreased for +1‐Cy3 due its extrusion into solution.
Initial transcription by RPITC can produce a paused and backtracked intermediate
Extension of the RNA chain by RPITC involves a series of concerted motions in the holoenzyme and DNA.2 Nucleotide addition occurs in the active site of RNAP (see Fig. 1), which is composed of several β′ and β subunit domains, including: the α‐helical bridge helix (BH) that bifurcates the cleft into the primary and secondary channels, the mobile trigger loop (TL) that controls access to the active site from the secondary channel, the F‐Loop, and the catalytic loop (β′), which coordinates a Mg2+ ion2, 45 [see Fig. 1(B)]. The BH and TL are the primary actors during nucleotide addition and play a central role in RNAP translocation. The nucleotide addition cycle can be generally described as a sequence of four repeating events: (1) RNAP translocation along DNA, which moves the newly incorporated NTP from the insertion site (i + 1) to the “i” site, (2) incoming NTP binding in the “i + 1” site, (3) phosphodiester bond formation between RNA 3′‐end in “i” site and the NTP in “i + 1” site, and (4) pyrophosphate release.2, 74 Completion of a nucleotide addition cycle results in an RNAP conformation termed the pretranslocated state, in which the 3′‐end of the newly incorporated base occupies the insertion site. RNAP translocation relative to DNA shifts the RNA 3′‐end from the “i + 1” site to the upstream “i” site to form the post‐translocated state (forward translocation). Alternative translocation states, defined by the relative position of the RNA 3′‐end to the active site, are believed to alternate through rapid thermal (Brownian) fluctuations that transpire on the microsecond timescale. The driving force for forward translocation is suggested to be the stabilization energy provided by correct nucleotide binding.2, 74 The backward translocation of the pretranslocated state results in formation of a “backtracked” state in which the RNA 3′‐end enters the secondary channel.
For certain promoters (e.g., lacUV5,75 phage T5 N25,76 malT77, 78), the rate‐limiting step in transcription initiation is promoter escape. Despite efficient RNAP binding and formation of stable open complexes, these promoters exhibit an increase in the amount and size distribution of abortive products, with a reciprocal decrease in promoter escape efficiency. Previous studies suggested the existence of a paused, backtracked state in initial transcription.14, 66, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89
Lately, we confirmed the existence of a paused‐backtracked intermediate in RNAP transcription initiation. A paused‐backtracked intermediate was characterized using a single‐round quenched kinetics assay and DNA nanomanipulation with magnetic tweezers.90 The single‐round quenched kinetics assay utilized the diffusion‐based smFRET‐ALEX technique to detect transcripts over time [Fig. 6(A)], until terminated (quenched) at different time points. Because smFRET‐ALEX is a sm technique, single‐round quenched kinetics assays allowed accurate quantification of transcripts generated by RNAP–DNA complexes. To confirm the existence of the paused state in RNAP initiation, the promoter escape kinetics, starting from different RPITC states, were monitored by single‐round quenched kinetics assays. The assays revealed a delay (∼3.5 times slower) when starting from RPITC≤7 compared to RPITC=2 [Fig. 6(B)]. This delay was nearly abolished by inclusion of GreA, indicating a paused‐backtracked intermediate during initiation [Fig. 6(C)]. This finding was corroborated with DNA nanomanipulation assays with magnetic tweezers, which can detect changes in DNA topology (e.g., changes in the size of transcription bubble) in real time. Importantly, DNA nanomanipulation assays demonstrated RNAP transcription complexes could be paused‐backtracked during initial transcription in the presence of all four NTPs at biologically relevant concentrations. A modified transcription initiation model was suggested that included the paused‐backtracked Eσ [Fig. 6(D)]. This paused‐backtracked intermediate in RNAP initiation was also reported by Duchi et al. using ALEX‐TIRF techniques.91 In their report, Duchi et al. showed that RPITC could form a stable paused‐backtracked intermediate and σR3.2 played a key role in controlling the properties of this intermediate.
Figure 6.
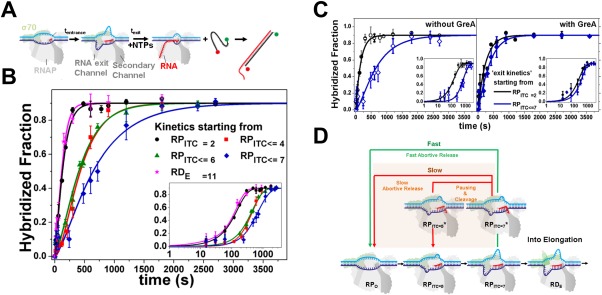
Paused backtracked intermediate in RNAP initiation. Pausing in initiation by RPITC is stabilized by backtracking. Reprinted from Lerner and Chung et al.90 with permission. (A) Schematic of transcription kinetics starting from particular NTP starved ITC states (incubation with a partial set of NTPs for tentrance) using single‐round quenched kinetics assays. Upon adding all NTPs, transcription kinetics start and stop at different incubation times (texit). Transcripts were quantified via hybridization to a ssDNA FRET probe. (B) Quenched kinetics results identify an initiation‐related stalled state. Shown are run‐off kinetics from various NTP‐starved states. Kinetics starting from late initiation states (e.g., RPITC ≤ 7, blue) are slower than from an earlier initiation state (e.g., RPITC = 2, black). (C) GreA suppresses the kinetic delay in transcription initiation. Left; Run‐off transcription kinetics are slower when starting from RPITC ≤ 7 (blue) than from RPITC = 2 (black). Right; with 1 µM GreA, the delay in transcription initiation is reduced. All data are represented as points and solid lines represent best‐fit as described in90. (D) A modified transcription initiation model. RNAP transcription initiation branches to promoter clearance and transitions into elongation (black arrows) or into release of abortive transcripts (green and red arrows). After initial backtracking steps (e.g., from RPITC = 7 to RPITC = 7*), the complex can continue with either fast abortive transcript release (classic model, green arrow) or transition into a paused‐backtracked state. Exit from the paused–backtracked state can occur either by successive slow backtracking steps (red arrow) or through intrinsic cleavage of RNA bases in the secondary channel, which prepares RNAP in, e.g., the RPITC = 5 state. Upon cleavage, the complex can release the abortive transcript or re‐establish RNA polymerization (e.g., from the RPITC = 5 state).
RNAP promoter escape and σ70 retention into elongation
For the RPITC complex, the nascent RNA eventually reaches a required length (∼9–15 bases) for promoter escape. The process of promoter escape requires an extensive set of conformational changes to allow Eσ to disengage from the promoter and form the processive RPE complex. The most notable structural transitions underlying promoter escape include the following. (1) Displacement of σR3.2, which otherwise impedes the nascent RNA 5′‐end; removal of σR3.2 together with σR4 allows nascent RNA to enter the RNA exit channel. (2) Weakening of RNAP interactions with σ, mainly through destabilization of the σR4–β interaction.5, 92 (3) Breaking σ70 interactions with the promoter (σR2.3–R2.4 with the −10 element, σR4.2 with −35 element). (4) And finally, reannealing of the upstream edge of the transcription bubble to form a 12‐base pair transcription bubble, which is characteristic of elongating RNAP.
It was previously hypothesized that σ release from RNAP during promoter escape was obligatory, and a “σ cycle” was proposed in which σ associates with RNAP for initiation and is released when RNAP enters elongation (cited in Ref. 93). However, the harsh separation techniques (e.g., gel electrophoresis and chromatography) used to test for σ presence in elongation could also promote σ dissociation. Several studies conducted in the last 10 years have provided sufficient evidence for σ retention during transcription elongation. Kapanidis et al. demonstrated σ retention in RPE up to position +50 from the TSS using diffusion‐based smFRET‐ALEX.93 Retention of σ70 in early elongation complexes was also observed during investigation of the mechanism of σ70‐dependent promoter‐proximal pausing.94, 95
Concluding Remarks and Future Perspective
RNAP has been and continues to be among the most extensively studied biomolecular machines, which is a testament to its complexity and its fundamental importance in biology. Over the past decade, sm assays have resolved heterogeneities in molecular distributions and have revealed fundamental mechanisms through which RNAP functions. In this review, we highlighted some of the contributions of sm fluorescence, mainly diffusion‐based smFRET, and other sm approaches that have uncovered RNAP transcription mechanisms.
The future questions and challenges facing the sm transcription field are numerous; many questions surround the rapid transformations that occur from the promoter search mechanism to the formation of RPO. Interestingly, the process of TSS selection, which was linked to bubble dynamics in the millisecond timescale,56 was postulated to also occur through a scrunching mechanism. Furthermore, it was proposed that DNA scrunching and anti‐scrunching may underlie alternate TSS selection in the absence of NTPs.57, 58, 96 This implies that the millisecond bubble dynamics are primarily caused by DNA scrunching dynamics in RPO. If correct, additional BVA experiments will be important to define different conformational modes and their potential contribution to the bubble dynamics.
The intermediate‐resolution structure of a paused, backtracked bacterial RNAP elongation complex has been reported,97 but the structure of a paused‐backtracked initiation complex90, 91 remains unavailable. It is assumed that the “ratcheted” form of RNAP is a structural feature common to all paused and backtracked RNAPs. A “ratcheted” form of RNAP is distinct from the processive RPE in the conformations of several prominent structural elements of RNAP97 (e.g., a kinked/bent BH, a partially open TL, an open clamp, and expansion of DNA:RNA hybrid binding site). Whereas heterogeneity among a population of paused, backtracked RNAP complexes will complicate any structural analysis, recent advances in cryoEM98, 99 may allow detailed structural information to be uncovered.
Despite the increasing capability of other techniques—such as single particle cryo‐EM, immobilized smFRET imaging, and magnetic or optical tweezers—to resolve structural intermediates hidden in ensemble data, diffusion‐based smFRET maintains several advantages. For example, diffusion‐based smFRET is better at characterizing structural dynamics and the kinetics in going between different intermediates than cryo‐EM. It is also higher a throughput method with greater temporal resolution (a few milliseconds) in comparison to immobilized FRET imaging (tens of milliseconds) and magnetic or optical tweezers (∼ 1 second).
Acknowledgments
We thank Dr. Antonino Ingargiola for fruitful discussions and Maya Lerner for preparation of illustrations.
The authors declare no conflict of interest
References
- 1. Werner F, Grohmann D (2011) Evolution of multisubunit RNA polymerases in the three domains of life. Nat Rev Microbiol 9:85–98. [DOI] [PubMed] [Google Scholar]
- 2. Nudler E (2009) RNA polymerase active center: the molecular engine of transcription. Annu Rev Biochem 78:335–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Werner F (2007) Structure and function of archaeal RNA polymerases. Mol Microbiol 65:1395–1404. [DOI] [PubMed] [Google Scholar]
- 4. Ruff EF, Record MT, Artsimovitch I (2015) Initial events in bacterial transcription initiation. Biomolecules 5:1035–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Murakami KS, Darst SA (2003) Bacterial RNA polymerases: the wholo story. Curr Opin Struct Biol 13:31–39. [DOI] [PubMed] [Google Scholar]
- 6. Opalka N, Brown J, Lane WJ, Twist K‐AF, Landick R, Asturias FJ, Darst SA (2010) Complete structural model of Escherichia coli RNA polymerase from a hybrid approach. PLoS Biol 8:e1000483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maeda H (2000) Competition among seven Escherichia coli sigma subunits: relative binding affinities to the core RNA polymerase. Nucleic Acids Res 28:3497–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feklistov A, Sharon BD, Darst SA, Gross CA (2014) Bacterial sigma factors: a historical, structural, and genomic perspective. Annu Rev Microbiol 68:357–376. [DOI] [PubMed] [Google Scholar]
- 9. Zuo Y, Steitz TA (2015) Crystal structures of the E. coli transcription initiation complexes with a complete bubble. Mol Cell 58:534–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bae B, Feklistov A, Lass‐Napiorkowska A, Landick R, Darst SA (2015) Structure of a bacterial RNA polymerase holoenzyme open promoter complex. Elife 4:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buck M, Gallegos M, Studholme DJ, Guo Y, Gralla JD, Gallegos A, Gralla JAYD (2000) The bacterial enhancer‐dependent sigma(54) (sigma(N)) transcription factor. J Bacteriol 54:4129–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Y, Feng Y, Chatterjee S, Tuske S, Ho MX, Arnold E, Ebright RH (2012) Structural basis of transcription initiation. Science 338:1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Basu RS, Warner BA, Molodtsov V, Pupov D, Esyunina D, Fernández‐Tornero C, Kulbachinskiy A, Murakami KS (2014) Structural basis of transcription initiation by bacterial RNA polymerase holoenzyme. J Biol Chem 289:24549–24559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hsu LM (2008) Promoter escape by Escherichia coli RNA polymerase. EcoSal Plus 3:1–16. [DOI] [PubMed] [Google Scholar]
- 15. Weiss S (1999) Fluorescence spectroscopy of single biomolecules. Science 283:1676–1683. [DOI] [PubMed] [Google Scholar]
- 16. Dietrich A, Buschmann V, Müller C, Sauer M (2002) Fluorescence resonance energy transfer (FRET) and competing processes in donor‐acceptor substituted DNA strands: a comparative study of ensemble and single‐molecule data. Rev Mol Biotechnol 82:211–231. [DOI] [PubMed] [Google Scholar]
- 17. Bai L, Santangelo TJ, Wang MD (2006) Single‐molecule analysis of RNA polymerase transcription. Annu Rev Biophys Biomol Struct 35:343–360. [DOI] [PubMed] [Google Scholar]
- 18. Tomov TE, Tsukanov R, Masoud R, Liber M, Plavner N, Nir E (2012) Disentangling subpopulations in single‐molecule FRET and ALEX experiments with photon distribution analysis. Biophys J 102:1163–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heyduk T, Niedziela‐Majka A (2002) Fluorescence resonance energy transfer analysis of Escherichia coli RNA polymerase and polymerase‐DNA complexes. Biopolymers 61:201–213. [DOI] [PubMed] [Google Scholar]
- 20. Herbert KM, Greenleaf WJ, Block SM (2008) Single‐molecule studies of RNA polymerase: motoring along. Annu Rev Biochem 77:149–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Santoso Y, Hwang LC, Le Reste L, Kapanidis AN (2008) Red light, green light: probing single molecules using alternating‐laser excitation. Biochem Soc Trans 36:738–744. [DOI] [PubMed] [Google Scholar]
- 22. Strick TR (2008) Optical investigations of the RNA polymerase molecular motor. J Biophotonics 1:269–279. [DOI] [PubMed] [Google Scholar]
- 23. Billingsley DJ, Bonass WA, Crampton N, Kirkham J, Thomson NH (2012) Single‐molecule studies of DNA transcription using atomic force microscopy. Phys Biol 9:21001. [DOI] [PubMed] [Google Scholar]
- 24. Michaelis J, Treutlein B (2013) Single molecule studies of RNA polymerases. Chem Rev 113:8377–8399. [DOI] [PubMed] [Google Scholar]
- 25. Collins BE, Ye LF, Duzdevich D, Greene EC (2014) DNA curtains: novel tools for imaging protein‐nucleic acid interactions at the single‐molecule level, 1st ed. Elsevier Inc.. [DOI] [PubMed] [Google Scholar]
- 26. Dangkulwanich M, Ishibashi T, Bintu L, Bustamante C (2014) Molecular mechanisms of transcription through single‐molecule experiments. Chem Rev 114:3203–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Friedman LJ, Gelles J (2015) Multi‐wavelength single‐molecule fluorescence analysis of transcription mechanisms. Methods 86:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Suzuki Y, Endo M, Sugiyama H (2015) Studying RNAP–promoter interactions using atomic force microscopy. Methods 86:4–9. [DOI] [PubMed] [Google Scholar]
- 29. Ploetz E, Lerner E, Husada F, Roelfes M, Chung S, Hohlbein J, Weiss S, Cordes T (2016) Fluorescence resonance energy transfer and protein‐induced fluorescence enhancement as synergetic multi‐scale molecular rulers. Sci Rep 6:33257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fujita K, Iwaki M, Yanagida T, (2016) Transcriptional bursting is intrinsically caused by interplay between RNA polymerases on DNA. Nat Commun 7:13788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang G, Hauver J, Thomas Z, Darst SA, Pertsinidis A (2016) Single‐molecule real‐time 3D imaging of the transcription cycle by modulation interferometry. Cell 167:1839–1852.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hohlbein J, Craggs TD, Cordes T (2014) Alternating‐laser excitation: single‐molecule FRET and beyond. Chem Soc Rev 43:1156–1171. [DOI] [PubMed] [Google Scholar]
- 33. Kapanidis AN, Laurence TA, Nam KL, Margeat E, Kong X, Weiss S (2005) Alternating‐laser excitation of single molecules. Acc Chem Res 38:523–533. [DOI] [PubMed] [Google Scholar]
- 34. Lee NK, Kapanidis AN, Wang Y, Michalet X, Mukhopadhyay J, Ebright RH, Weiss S (2005) Accurate FRET measurements within single diffusing biomolecules using alternating‐laser excitation. Biophys J 88:2939–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Feklistov A (2013) RNA polymerase: in search of promoters. Ann NY Acad Sci 1293:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Svetlov V, Nudler E (2013) Looking for a promoter in 3D. Nat Struct Mol Biol 20:141–142. [DOI] [PubMed] [Google Scholar]
- 37. Kabata H, Kurosawa O, Arai I, Washizu M, Margarson S, Glass R, Shimamoto N (1993) Visualization of single molecules of RNA polymerase sliding along DNA. Science 262:1561–1563. [DOI] [PubMed] [Google Scholar]
- 38. Bustamante C, Guthold M, Zhu X, Yang G (1999) Facilitated target location on DNA by individual Escherichia coli RNA polymerase molecules observed with the scanning force microscope operating in liquid. J Biol Chem [VOL]:16665–16668. [DOI] [PubMed] [Google Scholar]
- 39. Guthold M, Zhu X, Rivetti C, Yang G, Thomson NH, Kasas S, Hansma HG, Smith B, Hansma PK, Bustamante C (1999) Direct observation of one‐dimensional diffusion and transcription by Escherichia coli RNA polymerase. Biophys J 77:2284–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Harada Y, Funatsu T, Murakami K, Nonoyama Y, Ishihama A, Yanagida T (1999) Single‐molecule imaging of RNA polymerase‐DNA interactions in real time. Biophys J 76:709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Friedman LJ, Mumm JP, Gelles J (2013) RNA polymerase approaches its promoter without long‐range sliding along DNA. Proc Natl Acad Sci USA 110:9740–9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang F, Redding S, Finkelstein IJ, Gorman J, Reichman DR, Greene EC (2013) The promoter‐search mechanism of Escherichia coli RNA polymerase is dominated by three‐dimensional diffusion. Nat Struct Mol Biol 20:174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Haugen SP, Ross W, Gourse RL (2008) Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat Rev Microbiol 6:507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gourse RL, Ross W, Gaal T (2000) UPs and downs in bacterial transcription initiation: the role of the alpha subunit of RNA polymerase in promoter recognition. Mol Microbiol 37:687–695. [DOI] [PubMed] [Google Scholar]
- 45. Lee J, Borukhov S (2016) Bacterial RNA polymerase–DNA interaction—the driving force of gene expression and the target for drug action. Front Mol Biosci 3:[PAGE #S]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen J, Darst SA, Thirumalai D (2010) Promoter melting triggered by bacterial RNA polymerase occurs in three steps. Proc Natl Acad Sci USA 107:12523–12528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Djordjevic M, Bundschuh R (2008) Formation of the open complex by bacterial RNA polymerase—a quantitative model. Biophys J 94:4233–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Murakami K (2015) Structural biology of bacterial RNA polymerase. Biomolecules 5:848–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chakraborty A, Wang D, Ebright YW, Korlann Y, Kortkhonjia E, Kim T, Chowdhury S, Wigneshweraraj S, Irschik H, Jansen R, Nixon BT, Knight J, Weiss S, Ebright RH. (2012) Opening and closing of the bacterial RNA polymerase clamp. Science 337:591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Potrykus K, Cashel M (2008) (p)ppGpp: still magical?. Annu Rev Microbiol 62:35–51. [DOI] [PubMed] [Google Scholar]
- 51. Corrigan RM, Bellows LE, Wood A, Gründling A (2016) ppGpp negatively impacts ribosome assembly affecting growth and antimicrobial tolerance in Gram‐positive bacteria. Proc Natl Acad Sci USA 113:201522179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tchernaenko V, Halvorson HR, Kashlev M, Lutter LC (2008) DNA bubble formation in transcription initiation. Biochemistry 47:1871–1884. [DOI] [PubMed] [Google Scholar]
- 53. Cordes T, Santoso Y, Tomescu AI, Gryte K, Hwang LC, Camará B, Wigneshweraraj S, Kapanidis AN (2010) Sensing DNA opening in transcription using quenchable Förster resonance energy transfer. Biochemistry 49:9171–9180. [DOI] [PubMed] [Google Scholar]
- 54. Revyakin A, Ebright RH, Strick TR (2004) Promoter unwinding and promoter clearance by RNA polymerase: detection by single‐molecule DNA nanomanipulation. Proc Natl Acad Sci USA 101:4776–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Torella JP, Holden SJ, Santoso Y, Hohlbein J, Kapanidis AN (2011) Identifying molecular dynamics in single‐molecule fret experiments with burst variance analysis. Biophys J 100:1568–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Robb NC, Cordes T, Hwang LC, Gryte K, Duchi D, Craggs TD, Santoso Y, Weiss S, Ebright RH, Kapanidis AN (2013) The transcription bubble of the RNA polymerase‐promoter open complex exhibits conformational heterogeneity and millisecond‐scale dynamics: implications for transcription start‐site selection. J Mol Biol 425:875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Winkelman JT, Chandrangsu P, Ross W, Gourse RL (2016) Open complex scrunching before nucleotide addition accounts for the unusual transcription start site of E. coli ribosomal RNA promoters. Proc Natl Acad Sci USA 113:E1787–E1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Winkelman JT, Vvedenskaya IO, Zhang Y, Zhang Y, Bird JG, Taylor DM, Gourse RL, Ebright RH, Nickels BE (2016) Multiplexed protein‐DNA cross‐linking: scrunching in transcription start site selection. Science 351:1090–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vvedenskaya IO, Zhang Y, Goldman SR, Valenti A, Visone V, Taylor DM, Ebright RH, Nickels BE (2015) Massively Systematic Transcript End Readout, “MASTER”: transcription start site selection, transcriptional slippage, and transcript yields. Mol Cell 60:953–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Carpousis AJ, Gralla JD (1985) Interaction of RNA polymerase with lacUV5 promoter DNA during mRNA initiation and elongation. J Mol Biol 183:165–177. [DOI] [PubMed] [Google Scholar]
- 61. Pupov D, Kuzin I, Bass I, Kulbachinskiy A (2014) Distinct functions of the RNA polymerase σ subunit region 3.2 in RNA priming and promoter escape. Nucleic Acids Res 42:4494–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hsu LM, Vo NV, Kane CM, Chamberlin MJ (2003) In vitro studies of transcript initiation by Escherichia coli RNA polymerase. 1. RNA chain initiation, abortive initiation, and promoter escape at three bacteriophage promoters. Biochemistry 42:3777–3786. [DOI] [PubMed] [Google Scholar]
- 63. Vo NV, Hsu LM, Kane CM, Chamberlin MJ (2003) In vitro studies of transcript initiation by Escherichia coli RNA polymerase. 3. Influences of individual DNA elements within the promoter recognition region on abortive initiation and promoter escape. Biochemistry 42:3798–3811. [DOI] [PubMed] [Google Scholar]
- 64. Hsu LM (2009) Monitoring abortive initiation. Methods 47:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Margeat E, Kapanidis AN, Tinnefeld P, Wang Y, Mukhopadhyay J, Ebright RH, Weiss S (2006) Direct observation of abortive initiation and promoter escape within single immobilized transcription complexes. Biophys J 90:1419–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hsu LM (2002) Promoter clearance and escape in prokaryotes. Biochim Biophys Acta 1577:191–207. [DOI] [PubMed] [Google Scholar]
- 67. Straney DC, Crothers DM (1987) A stressed intermediate in the formation of stably initiated RNA chains at the Escherichia coli lac UV5 promoter. J Mol Biol 193:267–278. [DOI] [PubMed] [Google Scholar]
- 68. Krummel B, Chamberlin MJ (1989) RNA chain initiation by Escherichia coli RNA polymerase. Structural transitions of the enzyme in early ternary complexes. Biochemistry 28:1829–7842. [DOI] [PubMed] [Google Scholar]
- 69. Kapanidis AN, Margeat E, Ho SO, Kortkhonjia E, Weiss S, Ebright RH (2006) Initial transcription by RNA polymerase proceeds through a DNA‐scrunching mechanism. Science 314:1144–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Revyakin A, Liu C, Ebright RH, Strick TR (2006) Abortive initiation and productive initiation by RNA polymerase involve DNA scrunching. Science 314:1139–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tang GQ, Roy R, Ha T, Patel SS (2008) Transcription initiation in a single‐subunit RNA polymerase proceeds through DNA scrunching and rotation of the N‐terminal subdomains. Mol Cell 30:567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fazal FM, Meng CA, Murakami K, Kornberg RD, Block SM (2015) Real‐time observation of the initiation of RNA polymerase II transcription. Nature 525:274–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Winkelman JT, Winkelman BT, Boyce J, Maloney MF, Chen AY, Ross W, Gourse RL (2015) Crosslink mapping at amino acid‐base resolution reveals the path of scrunched DNA in initial transcribing complexes. Mol Cell 59:768–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zuo Y, Steitz TA (2017) A structure‐based kinetic model of transcription. Transcription 1264: [PAGE #S]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Stefano JE, Gralla J (1979) Lac UV5 transcription in vitro. Rate limitation subsequent to formation of an RNA polymerase‐DNA complex. Biochemistry 18:1063–1067. [DOI] [PubMed] [Google Scholar]
- 76. Kammerer W, Deuschle U, Gentz R, Bujard H (1986) Functional dissection of Escherichia coli promoters: information in the transcribed region is involved in late steps of the overall process. Embo J 5:2995–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Menendez M, Kolb A, Buc H (1987) A new target for CRP action at the malT promoter. Embo J 6:4227–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Eichenberger P, Déthiollaz S, Buc H, Geiselmann J (1997) Structural kinetics of transcription activation at the malT promoter of Escherichia coli by UV laser footprinting. Proc Natl Acad Sci USA 94:9022–9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ellinger T, Behnke D, Bujard H, Gralla J (1994) Stalling of Escherichia coli RNA polymerase in the +6 to +12 region in vivo is associated with tight binding to consensus promoter elements. J Mol Biol 239:455–465. [DOI] [PubMed] [Google Scholar]
- 80. Brodolin K, Zenkin N, Mustaev A, Mamaeva D, Heumann H (2004) The sigma 70 subunit of RNA polymerase induces lacUV5 promoter‐proximal pausing of transcription. Nat Struct Mol Biol 11:551–557. [DOI] [PubMed] [Google Scholar]
- 81. Samanta S, Martin CT (2013) Insights into the mechanism of initial transcription in Escherichia coli RNA polymerase. J Biol Chem 288:31993–32003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shaevitz JW, Abbondanzieri EA, Landick R, Block SM (2003) Backtracking by single RNA polymerase molecules observed at near‐base‐pair resolution. Nature 426:684–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Stepanova E, Lee J, Ozerova M, Semenova E, Datsenko K, Wanner BL, Severinov K, Borukhov S (2007) Analysis of promoter targets for Escherichia coli transcription elongation factor GreA in vivo and in vitro. J Bacteriol 189:8772–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Stepanova E, Wang M, Severinov K, Borukhov S (2009) Early transcriptional arrest at Escherichia coli rplN and ompX promoters. J Biol Chem 284:35702–35713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Feng G, Lee DN, Wang D, Chan CL, Landick R (1994) GreA‐induced transcript cleavage in transcription complexes containing Escherichia coli RNA polymerase is controlled by multiple factors, including nascent transcript location and structure. J Biol Chem 269:22282–22294. [PubMed] [Google Scholar]
- 86. Hsu LM, Vo NV, Chamberlin MJ (1995) Escherichia coli transcript cleavage factors GreA and GreB stimulate promoter escape and gene expression in vivo and in vitro. Proc Natl Acad Sci USA 92:11588–11592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Borukhov S, Sagitov V, Goldfarb A (1993) Transcript cleavage factors from E. coli . Cell 72:459–466. [DOI] [PubMed] [Google Scholar]
- 88. Goldman SR, Ebright RH, Nickels BE (2009) Direct detection of abortive RNA transcripts in vivo. Science 324:927–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Skancke J, Bar N, Kuiper M, Hsu LM (2015) Sequence‐dependent promoter escape efficiency is strongly influenced by bias for the pretranslocated state during initial transcription. Biochemistry 54:4267–4275. [DOI] [PubMed] [Google Scholar]
- 90. Lerner E, Chung S, Allen BL, Wang S, Lee J, Lu SW, Grimaud LW, Ingargiola A, Michalet X, Alhadid Y, Borukhov S, Strick TR, Taatjes DJ, Weiss S. (2016) Backtracked and paused transcription initiation intermediate of Escherichia coli RNA polymerase. Proc Natl Acad Sci USA 113:E6562–E6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Duchi D, Bauer DLV, Fernandez L, Evans G, Robb N, Hwang LC, Gryte K, Tomescu A, Zawadzki P, Morichaud Z, Brodolin K, Kapanidis AN. (2016) RNA polymerase pausing during initial transcription. Mol Cell 63:939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sengupta S, Prajapati RK, Mukhopadhyay J (2015) Promoter escape with bacterial two‐component σ factor suggests retention of σ region two in the elongation complex. J Biol Chem 290:28575–28583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kapanidis AN, Margeat E, Laurence TA, Doose S, Ho SO, Mukhopadhyay J, Kortkhonjia E, Mekler V, Ebright RH, Weiss S (2005) Retention of transcription initiation factor sigma70 in transcription elongation: single‐molecule analysis. Mol Cell 20:347–356. [DOI] [PubMed] [Google Scholar]
- 94. Perdue SA, Roberts JW (2011) σ70‐dependent transcription pausing in Escherichia coli . J Mol Biol 412:782–792. [DOI] [PubMed] [Google Scholar]
- 95. Harden TT, Wells CD, Friedman LJ, Landick R, Hochschild A, Kondev J, Gelles J (2016) Bacterial RNA polymerase can retain σ 70 throughout transcription. Proc Natl Acad Sci USA 113:602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Vvedenskaya IO, Vahedian‐Movahed H, Bird JG, Knoblauch JG, Goldman SR, Zhang Y, Ebright RH, Nickels BE (2014) Interactions between RNA polymerase and the core recognition element counteract pausing. Science 344:1285–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sekine S, Murayama Y, Svetlov V, Nudler E, Yokoyama S (2015) Ratcheting of RNA polymerase toward structural principles of RNA polymerase operations. Transcription 6:56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kühlbrandt W (2014) The resolution revolution. Science 343:1443–1444. [DOI] [PubMed] [Google Scholar]
- 99. Callaway E (2015) The revolution will not be crystallized: a new method sweeps through structural biology. Nat News 525:172. [DOI] [PubMed] [Google Scholar]