

ABSTRACT
Human La antigen (Sjögren's syndrome antigen B [SSB]) is an abundant multifunctional RNA-binding protein. In the nucleoplasm, La binds to and protects from 3′ exonucleases, the ends of precursor tRNAs, and other transcripts synthesized by RNA polymerase III and facilitates their maturation, while a nucleolar isoform has been implicated in rRNA biogenesis by multiple independent lines of evidence. We showed previously that conditional La knockout (La cKO) from mouse cortex neurons results in defective tRNA processing, although the pathway(s) involved in neuronal loss thereafter was unknown. Here, we demonstrate that La is stably associated with a spliced pre-tRNA intermediate. Microscopic evidence of aberrant nuclear accumulation of 5.8S rRNA in La cKO is supported by a 10-fold increase in a pre-5.8S rRNA intermediate. To identify pathways involved in subsequent neurodegeneration and loss of brain mass in the cKO cortex, we employed mRNA sequencing (mRNA-Seq), immunohistochemistry, and other approaches. This revealed robust enrichment of immune and astrocyte reactivity in La cKO cortex. Immunohistochemistry, including temporal analyses, demonstrated neurodegeneration, followed by astrocyte invasion associated with immune response and decreasing cKO cortex size over time. Thus, deletion of La from postmitotic neurons results in defective pre-tRNA and pre-rRNA processing and progressive neurodegeneration with loss of cortical brain mass.
KEYWORDS: La protein, pre-rRNA, tRNA maturation, tRNA splicing
INTRODUCTION
La was described as a component of ribonucleoprotein particles (RNPs) targeted by autoantibodies in patients suffering from systemic lupus erythematosus and Sjögren's syndrome (1) and was later found in all free-living eukaryotes (2). La is an abundant RNA-binding protein that recognizes UUU-3′-OH, which results from transcription termination by RNA polymerase III (RNAP III) (3–5). This conserved activity functions to protect nascent RNAs from 3′ exonucleases, including the nuclear exosome Rrp6, and facilitates the ordered processing and maturation of these transcripts, the more abundant of which are the precursor tRNAs (6–12, 15; reviewed in references 13 and 14).
In addition to 3′-end protection, La also exhibits RNA chaperone activity, which serves to prevent misfolding of the pre-tRNA sequences with a propensity to form alternate structures (8, 14, 16, 17). The RNA chaperone activity works with the 3′ protection activity to assist structurally challenged pre-tRNAs that would otherwise succumb to nuclear surveillance via exosome Rrp6-mediated decay (9, 10, 15, 18, 19).
La is nonessential in the yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe (14, 137) but is essential during Drosophila melanogaster and Mus musculus development (20, 21), suggesting that its role in RNAP III transcript maturation is required in metazoa. By this reasoning, metazoa may contain an essential La-dependent RNAP III transcript(s). La proteins also interact with viral and cellular mRNAs, including 5′ terminal oligopyrimidine (TOP) mRNAs encoding ribosome subunits, to affect their translation (22–31). Furthermore, metazoan La proteins contain an atypical RNA recognition motif (RRM) (32, 33), not found in the yeast homologs, that likely extends its repertoire of RNA substrates and conveys function beyond 3′-end protection (34). Finally, as detailed below, several lines of evidence implicate La in the biogenesis of large rRNA. Thus, an alternative possibility to account for the disparity in essentiality in metazoa but not yeast that is independent of RNAP III is that La is required in higher eukaryotes for the proper metabolism or function of an RNAP II transcript and/or a pre-rRNA species produced by RNAP I.
La was estimated at 2 × 107 molecules per cell (35), comprised of multiple phosphorylated isoforms found in the nucleoplasm, nucleolus, and cytosol differentially associated with nascent RNAP III transcripts or mRNAs (12, 23, 27, 36–38). In addition to the major phosphorylation site, Ser-366, representing ∼85% of La, phosphorylation by AKT can modulate its nucleocytoplasmic distribution and the translation of specific mRNAs in mouse glial progenitor cells (39, 40).
La Ser-366 phosphoprotein is nucleoplasmic, associated with nascent pre-tRNAs (27). The Ser-366 nonphosphorylated (nonphospho) isoform comprises ∼15% of La, or about 3 × 106 molecules/cell (41). Ser-366 nonphospho-La is most concentrated in the dense fibrillar component of the nucleolus, localized with newly synthesized RNAP I transcripts, where it physically interacts with nucleolin (27, 36), a pre-rRNA-associated protein (42–44). La was shown to interact with nucleolin by fluorescence resonance energy transfer (FRET) and independently by yeast two-hybrid assays (36). It was proposed that nucleolin serves as a chaperone for pre-rRNA (42, 45). Because La exhibits RNA chaperone activity (7, 8, 10, 18), it is tempting to speculate that it may assist nucleolin in this capacity. Still, evidence that La functionally impacts rRNA biogenesis in a model system has been lacking.
We previously reported a conditional La knockout (La cKO) gene deletion in mouse cortex using CaMKIIα promoter Cre recombinase, which is activated in the hippocampus 3 to 5 weeks after birth and later more extensively in the cortex (reference 46 and references therein). La cKO brains are normal until ∼6 weeks and then fail to gain mass at the normal rate and by 12 to 13 weeks begin to lose cortical mass relative to controls (46). Defective processing of pre-tRNATyr has been documented. Specifically, La cKO cortex exhibited substantial decrease of a 3′ trailer-containing spliced pre-tRNATyr but not the nascent 3′ trailer-containing pre-tRNATyr (46), which is surprising, because both are expected to be similarly protected by La and labile in its absence (12, 15, 47). In any case, the data are consistent with reports that indicate neuronal sensitivity to faulty tRNA biogenesis (48–50; reviewed in reference 51).
Here, we show that the 3′ trailer-containing, spliced pre-tRNATyr is associated with La in the wild-type (WT) cortex, providing insight into its loss in the La cKO cortex. We also document accumulation of what appears to be a normal pre-5.8S rRNA processing intermediate in La cKO cortex. As expected, the pre-5.8S rRNA does not appear to be associated with La, suggesting that effects of La deletion on its accumulation are indirect in La cKO cortex, perhaps due to competitive inhibition of 3′ exonucleases required for 5.8S rRNA maturation by nascent pre-tRNAs and other RNAP III transcripts in the absence of 3′ binding by La. mRNA sequencing (mRNA-Seq) done on 16-week cortex revealed robust immune activation in La cKO cortex that coincides with prolific astrocytosis. Temporal analyses indicate that the onset of astrocyte invasion and neuron loss coincide, beginning at 11 to 13 weeks. The high degree of mRNA change due to immune cell reactivity overshadows direct effects of La deletion on specific mRNAs, especially since cKO cells are lost from the population, although many of the abundant TOP mRNAs were significantly decreased. The data indicate that La deletion from postmitotic neurons is associated with defects in pre-tRNA and pre-rRNA processing, progressive neuron damage and death, immune response with astrocytosis, and diminution of brain mass. The La cKO model shares features with some chronic neurodegenerative diseases involving immune activation and astrocytosis.
RESULTS
Accumulation of a pre-rRNA processing intermediate in La cKO cortex.
In addition to circumstantial evidence of La in rRNA biogenesis (see the introduction), other data implicate it in maturation of U3 and U8 snoRNAs (52, 53), which are involved in pre-rRNA processing (reviewed in references 54 and 55). Mature 28S, 18S, and 5.8S rRNAs can be derived from 47S rRNA, involving a multiplicity of activities along alternative pathways (54); a simple scheme is shown in Fig. 1A. We examined RNA from total isolated cortex from 13-week-old WT and La cKO mice by Northern blotting, using probes specific for pre-rRNA intermediates. A 30-nucleotide (nt) ITS2 probe beginning 4 nt downstream of the 5.8S rRNA sequence revealed accumulation of an rRNA in La cKO relative to WT mice (Fig. 1B). This RNA migrated slightly faster than the 300-nt SRP 7SL RNA on the same blot (data not shown) (see below), perhaps corresponding to a previously noted species of pre-5.8S rRNA (54, 56) (see Discussion). A band above the pre-5.8S rRNA that is most prominent in La cKO lanes (Fig. 1B, asterisk) may be the 12S precursor of ∼980 nt (54). Because La stabilizes its ligands, which are decreased in its absence, accumulation of an intermediate in La cKO was unexpected and is suggestive of an indirect mechanism.
FIG 1.

Pre-rRNA processing perturbation in La cKO cortex. (A) Simplified diagram of the rRNA processing pathway in mice (see the text). The locations of the ITS1 and ITS2 probes used for Northern blot analyses are indicated with dashed lines. (B) (Left) Northern blot of 13-week cortex total RNA using an ITS2 probe revealing the pre-5.8S rRNA species (see the text) (top), a probe to mature 5.8S rRNA (middle), and a probe to SRP 7SL RNA (bottom). (Right) Ethidium bromide (EtBr)-stained gel used to produce the blot. The arrowhead indicates the approximate position of the pre-5.8S rRNA relative to the 7SL RNA of 300 nt; the bands annotated with gray numbers are believed to be U2 and U3 snoRNAs. (C) Quantification of the pre-5.8S rRNA band relative to the SRP RNA bands using the following formula: La cKO(pre-5.8S/SRP)/WT(pre-5.8S/SRP), with WT set to 1. The error bars reflect two biological replicates. (D) Northern blot hybridization after agarose gel electrophoresis (EtBr-stained gel [top]) using an ITS1 probe (middle) and a GAPDH probe used as a loading control (bottom). The positions of the 28S and 18S rRNAs are indicated. (E) Analysis of La-associated RNAs by IP of mouse cortex lysate with anti-mLa or control IgG. Ten percent of inputs and supernatants were electrophoresed in a 10% TBE-urea gel, along with 100% of the bead-bound eluate RNA from the IPs, as indicated above the lanes. Each of the gels (i to v) shows the results of the same blot after hybridization with a different probe, as indicated on the right and in the text.
The same blot hybridized with a probe to the mature 5.8S rRNA revealed no significant difference in levels in WT and La cKO mice (Fig. 1B, middle left). A probe for the 300-nt 7SL RNA component of the signal recognition particle (SRP RNA) (Fig. 1B, bottom left) provided a loading control and mobility marker. The gel from which the blot was made (Fig. 1B, right) shows the position of the pre-5.8S rRNA species (arrowhead), which migrated under 7SL. Quantitation of pre-5.8S rRNA and SRP RNA revealed 10-fold more pre-5.8S rRNA in La cKO than in WT cortex (Fig. 1C). Agarose gel blot analysis with an ITS1 probe revealed no significant differences in pre-rRNA species (Fig. 1D and data not shown).
To interpret the unchanged levels of mature rRNA species, we note that La cKO cortex includes cells from which the La gene was deleted, together with cells from which La was not deleted. While ∼70% of hippocampal neurons express the CaMKIIα promoter, this is maximally only ∼35% in the larger cortex (57). At the time of RNA isolation, La cKO cortex contains fewer La+ cells than WT cortex (21), and data presented below show additional massive invasion of astrocytes. Therefore, we suppose that the majority of La+ cells contribute much of the mature rRNAs to the mixture of isolated RNAs, which also includes the pre-5.8S rRNA that does not otherwise accumulate, presumably contributed by the cells with La deleted.
Mice with systemic lupus erythematosus generate autoimmunity to 5.8S rRNA, from which monoclonal antibodies (Abs) have been generated for use by biologists (58). We examined cells by direct immunofluorescence using a previously characterized monoclonal anti-5.8S rRNA antibody, Y10b. We characterized Y10b prior to use on our mouse brain sections. The results showed clearly that the fluorescence signal from Y10b was ablated if the cells were first treated with RNase, washed, and further processed for anti-actin, which showed robust signal in both RNase-treated and mock-treated cells, whereas only the mock-treated cells showed Y10b staining (not shown). Use of Y10b on brain sections revealed a distinctive pattern of staining in La cKO cortex reflective of aberrant accumulation of nuclear 5.8S rRNA, whereas WT cortex showed less staining, with a diffuse ring pattern (Fig. 2).
FIG 2.

Aberrant accumulation of RNA in La cKO cortex cell nuclei that is recognized by anti-5.8S rRNA monoclonal antibody. Brain block sections were stained for 5.8S rRNA with Y10b fluorescent antibody and DAPI. Shown are representative images from 11-week WT and La cKO cortices.
No evidence of stable binding of pre-5.8S rRNA or pre-U3 or pre-U8 snoRNA to La in cortex.
We examined La-associated RNAs by anti-mouse La (mLa) immunoprecipitation (IP) of lysate made from 13-week wild-type cortex, followed by Northern blotting. The inputs, supernatants from the IPs, anti-mLa IP, and control IP with nonimmune IgG, were analyzed. There was no attempt at anti-La IP from La cKO cells due to concern about reassociation of La molecules from La+ cells with pre-5.8S rRNA from La cKO cells after cell lysis (59). The ITS2 probe used to detect the pre-5.8S rRNA (Fig. 1B) revealed no signal in any lanes despite multiple attempts and long exposure (not shown). The pre-5.8S rRNA does not appear to be stable in WT lysate containing La, and the cumulative data provide no evidence that La stabilizes it. We were unable to detect a previously identified mouse 3′-extended pre-U8 snoRNA (53) using the 3′ probe (not shown). This was not surprising, due to the requirement for preenrichment prior to detection (53). Mammalian pre-U3 snoRNAs ending in U-rich sequences are found in large complexes and are immunoprecipitable by anti-La antibody (52). The 3′ trailers of pre-U3 snoRNAs are ∼8 nt, extending them to ∼225 nt (52, 60, 61). The blot used for pre-5.8S rRNA and pre-U8 snoRNA was reprobed with a 3′ trailer probe designed to detect two of the four mouse pre-U3 snoRNAs (52). This detected a band of approximately the expected size in the input and supernatants (Fig. 1E, i, lanes 1 to 3) but with no evidence of enrichment of any species by anti-mLa (lane 5). A probe for mature 5.8S rRNA readily detected a band of the expected size with no evidence of the longer pre-5.8S rRNA (Fig. 1E, ii, and data not shown). Note that the 5.8S rRNA signal with IgG, and considerably more with anti-La (lanes 4 and 5), was less than in the input and supernatants (lanes 1 to 3). We note that failure to detect the pre-58S rRNA in the lysate used for the IP suggests the possibility that it was processed or otherwise diminished during lysate preparation, whereas detection in Fig. 1B was from total RNA isolated directly from the brain using denaturing TriReagent (see Discussion). In the next paragraph is described the positive control for this IP.
A 3′ trailer-containing spliced pre-tRNATyr intermediate is stably bound by La.
Pre-tRNA processing involves multiple activities that include endo- and exonucleases that remove the 3′ trailer prior to CCA addition to the matured end (47, 62). For pre-tRNAs that contain an intron, splicing must also occur. The above-described blot was also examined with a probe to the 3′ trailer of pre-tRNATyr (Fig. 1E, iii); this and separate probes to the intron and splice junction revealed the upper band in Fig. 1E, iii, to be nascent pre-tRNATyr that contained a 3′ trailer and intron (denoted “a”) and the lower (“b”) band to be a relatively abundant 3′ trailer-containing spliced pre-tRNATyr intermediate, in good agreement with previous data (21). IP showed that these 3′ trailer-containing pre-tRNATyr species were specifically associated with La (Fig. 1E, iii, lane 5), not the control IgG (lane 4). Unlike mature 5.8S rRNA, these 3′ trailer-containing pre-tRNATyr species were more enriched by anti-La than the input and supernatants. A faster-migrating, 3′ trailer-containing minor species that was barely detectable in the input and supernatants was enriched with La (Fig. 1E, iii, lane 5, c) and relatively nonreactive with the intron probe (Fig. 1E, iv). A probe spanning the splice sites of the products of the 10 mouse tRNATyr genes showed that mature tRNATyr was not enriched with La as expected (Fig. 1E, iv) (expected size, 76 nt; faster than band c [not shown]). The data indicate that La is stably bound to the 3′ trailer-containing, intron-containing tRNATyr prior to splicing and to the 3′ trailer-containing spliced product (see Discussion).
RNA-Seq reveals immune activation in the La cKO cortex.
Conditional deletion of La by CaMKIIα-Cre revealed that brain mass begins to decline at 12 to 13 weeks of age and continues for several weeks thereafter, with the first evidence of altered brain morphology at 16 weeks (46). We performed transcriptome sequencing (RNA-Seq) on 16-week cortices from WT and La cKO mice. A list of the top 30 mRNAs that were increased and decreased in cKO relative to WT is provided in Table 1 (a list of ∼1,800 mRNAs is provided in the supplemental material). The magnitudes of increase of several of the mRNAs ranged from 40- to 7-fold elevated relative to the WT (Table 1).
TABLE 1.
mRNAs increased or decreased in La cKO relative to WT cortex
| mRNA | Product | Function | Fold change |
|---|---|---|---|
| Increased | |||
| Itgax | Integrin subunit alpha X | Leukocyte-specific integrin | 47.1 |
| Clec7a | C-type lectin domain family 7 member A | Involved in antifungal immunity | 43.1 |
| Bfsp2 | Beaded filament structural protein 2 | Found in a structurally unique cytoskeletal element that is referred to as the beaded filament (BF) | 23.1 |
| Fgr | FGR proto-oncogene Src family tyr kinase | Negative regulator of cell migration and adhesion | 22.5 |
| Pttg1 | Pituitary tumor-transforming 1 | Anaphase-promoting complex (APC) substrate that associates with a separin until activation of APC | 15.8 |
| Ly9 | Lymphocyte Ag 9 | SLAM family of immunomodulatory receptors | 15.1 |
| Dgkk | Diacylglycerol kinase kappa | Enzyme that phosphorylates diacylglycerol | 13.1 |
| Dlk1 | Delta-like 1 homolog | Notch family homolog involved in differentiation | 12.8 |
| Tnni1 | Troponin I type 1 (skeletal) | Tissue-specific subtype of troponin I | 12.7 |
| Lilrb4 | Leukocyte Ig-like receptor B4 | Binds major histocompatibility comples (MHC) class I on Ag-presenting cells | 12.0 |
| Cybb | NADPH oxidase 2 | Mediates electron transfer to generate superoxide | 11.9 |
| Irs4 | Insulin receptor substrate 4 | Phosphorylated upon receptor stimulation | 11.7 |
| Ctse | Cathepsin E | Aspartic endopeptidase; in neurodegeneration | 11.2 |
| C4b | Complement component 4B | Interaction Ag-Ab complex and complement | 10.9 |
| Lhx8 | LIM homeobox 8 | Transcription factor; involved in neuron differentiation | 10.1 |
| Ccl6 | Chemokine (C-C motif) ligand 6 | Chemokine macrophage and B cell chemoattractant | 10.0 |
| Cd74 | HLA class II gamma chain | Binds/facilitates MHC class II export from the endoplasmic reticulum (ER) | 9.8 |
| Drd3 | Dopamine receptor D3 | D3 subtype receptor | 9.6 |
| Arhgap36 | Rho GTPase activated protein 36 | GTPase activator for the Rho-type GTPases | 8.3 |
| Pdcd1 | Programmed cell death 1 | Cell surface protein of the Ig superfamily | 8.2 |
| AI606473 | Expressed sequence | NcRNA immediately adjacent to Lhx8 | 8.1 |
| Ccl9 | Chemokine (C-C motif) ligand 9 | Attracts dendritic cells with exposed Cd11b(ITGAM) and CCR1 | 8.0 |
| Gpr101 | G protein-coupled receptor 101 | Orphan G protein-coupled receptor | 7.9 |
| Gabre | Gamma-aminobutyric acid (GABA) receptor subunit epsilon | GABA A receptor-multisubunit chloride channel | 7.7 |
| Galr1 | Galanin receptor 1 | GALR1 inhibits adenylyl cyclase via G protein | 7.7 |
| Siglec5 | Sialic acid binding Ig-like lectin (Siglec) 5 | Member of the Siglec family | 7.5 |
| Baiap3 | BAI1-associated protein 3 | Secretin receptor family member; p53 target gene encoding brain-specific angiogenesis inhibitor | 7.4 |
| Gbx1 | Gastrulation brain homeobox 1 | Homeodomain transcription factor involved in neuronal subpopulation specification | 7.3 |
| Bcl2a1b | B cell leukemia/lymphoma 2-related protein A1b | Reduces release of proapoptotic cytochrome c from mitochondria and blocks caspase activation | 7.1 |
| H2-Aa | Histocompatibility 2, class II antigen A, alpha | MHC class II heterodimer of α/β; central role in immune system; expressed in Ag-presenting cells | 7.0 |
| Decreased | |||
| Lct | Lactase | Hydrolysis of lactose | 0.07 |
| Dsp | Desmoplakin | Desmosome formation; anchors intermediate filaments | 0.08 |
| Clcnka | Chloride channel voltage-sensitive Ka | K+ recycling | 0.11 |
| Irx2 | Iroquois homeobox 2 | Homeodomain transcription factor | 0.11 |
| Wnt9b | Wingless-type mouse mammary tumor virus (MMTV) integration member 9B | Secreted signaling molecule | 0.12 |
| Iyd | Iodotyrosine deiodinase | Iodide scavenger | 0.13 |
| Pkp1 | Desmosome formation | Links cadherin to intermediate filaments | 0.13 |
| Gpr151 | GalRL | G-protein coupled receptor; homoly to galanin receptors | 0.15 |
| Tdo2 | Tryptophan 2,3-dioxygenase | Catalyzes the first and rate-limiting steps of tryptophan degradation along the kynurenine pathway | 0.18 |
| Lhx9 | LIM homeobox 9 | Transcription factor involved in development | 0.19 |
| Nhlh2 | Nescient HLH 2 | Transcription factor, helix-loop-helix type | 0.19 |
| Slc26a7 | Solute carrier family 26 member 7 | Anion antiporter | 0.20 |
| Slc9a4 | Solute carrier family 9 member A4 | Sodium proton antiporter | 0.22 |
| Mpzl2 | Myelin protein zero-like 2 | Mediates cell adhesion and T cell maturation | 0.22 |
| Nhlh1 | Nescient HLH 1 | Transcription factor, helix-loop-helix type | 0.23 |
| Spink8 | Serine peptidase inhibitor, Kazal type 8 | Serine protease inhibitor | 0.24 |
| Crybb1 | Crystallin beta B1 | Structural lens component | 0.25 |
| Irx1 | Iroquois homeobox 1 | Homeodomain transcription factor; development patterning | 0.25 |
| Il12a | Interleukin-12 subunit α | Required for T-cell-independent induction of gamma interferon (IFN-γ) | 0.25 |
| Smoc2 | SPARC-related modular calcium binding 2 | Promotes matrix assembly, endothelial cell proliferation, and migration; angiogenic activity | 0.25 |
| Eomes | Eomesodermin | Transcription factor; differentiation effector CD8+ T cells | 0.25 |
| Lefty1 | Left-right determination factor 1 | Transforming growth factor β involved in left-right axis patterning | 0.26 |
| Chrnb4 | Neuronal acetylcholine receptor subunit beta 4 | Acetylcholine receptor component | 0.27 |
| Ovol2 | Ovo-like zinc finger 2 | Transcription factor involved in differentiation | 0.28 |
| Cd6 | Cluster differentiation 6 | Lymphocyte glycoprotein involved in T cell activation | 0.28 |
| Rab37 | RAB37, member RAS oncogene family | GTPase involved in vesicle trafficking | 0.29 |
| Chrna3 | Neuronal acetylcholine receptor subunit alpha 3 | Acetylcholine receptor component | 0.29 |
| Crlf1 | Cytokine receptor-like factor 1 | Acts on cells expressing ciliary neurotrophic factor receptors. Promotes survival of neuronal cells. | 0.29 |
| Sytl1 | Synaptotagmin-like protein 1 | Binds phosphatidylinositol 3,4,5-trisphosphate and acts as a RAB27A effector protein | 0.30 |
| Scgn | Secretagogin, EF hand calcium binding protein | Involved in KCl-stimulated calcium flux and cell proliferation | 0.30 |
To determine the biological processes most represented by La cKO cortex mRNAs, we examined those with 2-fold increase (353 mRNAs) or decrease (152 mRNAs) relative to the WT using three gene ontology (GO) analysis programs: PANTHER, DAVID, and network ontology analysis (NOA), each of which uses a different algorithm (see Materials and Methods). Table 2 shows the top 5 enriched GO terms for increased and decreased mRNAs in La cKO cortex relative to the WT. While there are differences among the terms identified for increased mRNAs, each program classified immune-related processes in La cKO cortex as enriched to a highly statistically significant degree. The results suggest that the pathology of La cKO 16-week cortex involves robust immune activation and, as the data below indicate, activation of astrocytes, hallmarks of inflammation in the central nervous system (CNS) (63). Many of the mRNAs increased in La cKO cortex overlap those in other neurodegenerative models, and their increase can be attributed to astrocyte activation (see Discussion). Decreased mRNAs were enriched in neuron-specific transcripts in La cKO cortex (Table 2). The increased mRNAs reflect immune and invading proliferative astrocyte reactivity, as substantiated below, whereas the decreased mRNAs reflect loss of excitatory neurons, consistent with CamKII-Cre-mediated La deletion (64).
TABLE 2.
GO analysis of mRNAs increased or decreased in La cKO relative to WT cortex
| Term | Function | P value |
|---|---|---|
| Increased | ||
| PANTHER | Response to stimulus | 1.21E−19 |
| Immune response | 4.79E−19 | |
| Immune system process | 5.34E−17 | |
| Response to organic substance | 1.68E−16 | |
| Positive regulation of biological process | 1.41E−15 | |
| DAVID | Immune response | 2.40735E−15 |
| Defense response | 4.20214E−13 | |
| Inflammatory response | 1.11918E−09 | |
| Response to wounding | 6.2233E−09 | |
| Innate immune response | 9.89764E−09 | |
| NOA | Response to stimulus | 3.30E−31 |
| Biological regulation | 1.80E−28 | |
| Immune system process | 1.50E−27 | |
| Regulation of biological process | 2.30E−27 | |
| Cellular process | 2.50E−25 | |
| Decreased | ||
| PANTHER | Generation of neurons | 3.36E−04 |
| Regulation of localization | 3.55E−04 | |
| Ion transport | 1.05E−03 | |
| Nervous system development | 7.10E−03 | |
| Regulation of apoptotic process | 2.15E−02 | |
| DAVID | Wnt receptor signaling pathway | 4.30E−04 |
| Ion transport | 8.50E−04 | |
| Cell-cell signaling | 1.50E−03 | |
| Neuron differentiation | 3.10E−03 | |
| Behavioral defense response | 4.80E−03 | |
| NOA | Cell junction | 4.20E−07 |
| Regulation of metabolic process | 5.80E−06 | |
| Ion channel activity | 3.20E−06 | |
| Behavioral fear response | 4.80E−03 | |
| Transmembrane transporter activity | 1.40E−04 |
mRNAs found to be increased in La cKO cortex and other neurodegenerative models were selected for use to validate our RNA-Seq results by quantitative reverse transcription (qRT)-PCR: CD74, C4b, H2-Aa, and H2-Ab (histocompatibility 2, class II antigen [Ag] A, α and β, respectively) are directly involved in antigen presentation (65–69), and interleukin 1β (IL-1β) is associated with neurodegenerative and inflammatory phenotypes (70–72). We compared the levels of these mRNAs to that of GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA, used as a control in the same samples, which itself showed no significant differences among the four samples (two biological replicates each of WT and La cKO cells) by independent quantitative PCR (qPCR) or by analysis of the RNA-Seq data (not shown). For each mRNA examined, the level of increase detected by RNA-Seq agreed well with the qRT-PCR results obtained from independent cortex samples (Fig. 3).
FIG 3.
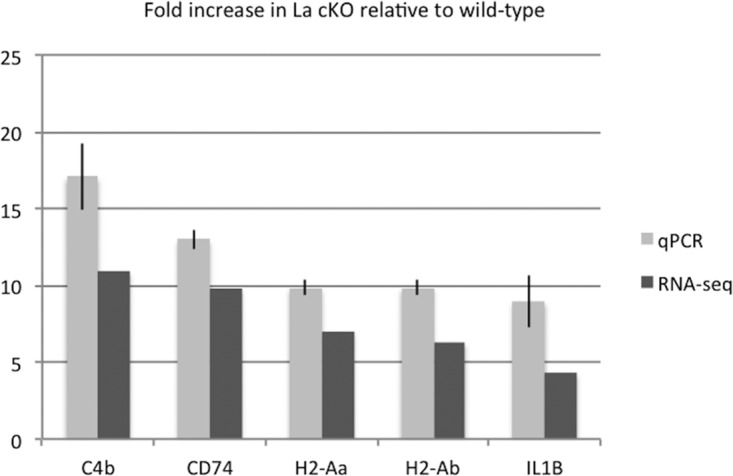
Agreement between qPCR and RNA-Seq results. Table 1 lists the mRNAs found by RNA-Seq to be significantly altered in abundance in La cKO relative to WT cortex. To validate and determine the accuracy of the RNA-Seq data, 5 mRNAs were chosen that were either directly involved in immune cell activation (C4b, H2-Aa, and H2-Ab) or implicated in neurodegenerative pathologies (CD74 and IL-1β). qPCR was performed on two La cKO and two WT biological replicate samples. Threshold cycle (CT) values were normalized to GAPDH, and the fold change was calculated relative to the WT samples; relative expression levels were calculated using the 2−ΔΔCT method (157). The error bars indicate 95% confidence intervals for both RNA-Seq and qRT-PCR data. For each mRNA, a negative-control reaction omitting reverse transcriptase showed negligible amplification (≤2%), indicating dependence on input RNA (not shown).
According to a model of cellular reactivity, the majority of mRNAs with the highest-magnitude changes would not reflect primary or direct interactions between La and specific mRNAs but rather transcripts in La+ immune cells and proliferative astrocytes invading the La cKO but not the WT cortex. We suspected that lower-magnitude changes might exist and attempted to find them among mRNAs with previously known association with La. We focused on mRNAs that contain the 5′ TOP sequence motif, which represents a significant group of ∼92 mRNAs that include ≥95% of the mRNAs that encode the 80 ribosomal proteins (73, 74), many of which have been documented to be bound by La in various animal cell models (24, 26, 27, 30, 75, 76). We found in our biological-replicate mRNA-Seq data set a statistically significant decrease of ∼30% in 27 ribosomal protein mRNAs in La cKO relative to WT cortex (Table 3). Significantly, all of these changes were in the same direction, decreased in La cKO. We also performed independent GO analysis on all mRNAs that decreased to a statistically significant level (26% decreased; P < 0.006) in La cKO relative to WT cortex and found the enrichment of the term “ribosome” with a P value of 2.40E−13.
TABLE 3.
mRNAs in DAVID term “ribosome” that include TOP sequence mRNAs that are decreased by at least 27% in La cKO relative to WT cortex
| mRNA | log2 (fold change) | % decrease (relative to WT) |
|---|---|---|
| rps17 | −0.46 | 0.273013741 |
| rps8 | −0.46 | 0.273013741 |
| rpl23a | −0.47 | 0.278035402 |
| rpl27a | −0.47 | 0.278035402 |
| rps25 | −0.48 | 0.283022376 |
| rps29 | −0.48 | 0.283022376 |
| rps10 | −0.49 | 0.287974902 |
| rps27a | −0.49 | 0.287974902 |
| rpl27 | −0.49 | 0.287974902 |
| rpl37a | −0.49 | 0.287974902 |
| rpl9 | −0.49 | 0.287974902 |
| rpl35a | −0.49 | 0.287974902 |
| rps13 | −0.5 | 0.292893219 |
| rpl17 | −0.5 | 0.292893219 |
| rpl37 | −0.5 | 0.292893219 |
| rpl14 | −0.51 | 0.297777562 |
| rpl11 | −0.52 | 0.302628167 |
| rpl19 | −0.52 | 0.302628167 |
| rpl34 | −0.55 | 0.316979872 |
| rpl36a1 | −0.56 | 0.321697836 |
| rps16 | −0.57 | 0.326383212 |
| rpl41 | −0.57 | 0.326383212 |
| rpl29 | −0.61 | 0.344803298 |
| rpl39 | −0.62 | 0.349329072 |
| rps21 | −0.63 | 0.353823585 |
| rps14 | −0.65 | 0.362719686 |
| rpl22l1 | −0.71 | 0.388679861 |
Neuronal death in La cKO cortex initiates early.
To examine the time course of neuronal damage, we collected brains at 11, 13, 16, and 20 weeks from three La cKO and three WT mice. The preserved brains were set in a solid matrix in triplicate and sectioned using MultiBrain technology (NeuroScience Associates [NSA], Knoxville, TN). A single set of each is shown in Fig. 4. To characterize the progression of neuronal death, de Olmos amino silver cupric (Ag-Cu) staining for neuron degeneration (77, 78) was performed. Minimal staining was seen at all time points in WT brains, in contrast to distinctive staining at 11 weeks and progressively thereafter in La cKO brains (Fig. 4A). Neuronal degeneration in La cKO cortex appeared to progress to the latest time examined, when staining extended prominently to the striatum (Fig. 4A, 20 weeks, arrows). Higher magnification of the stained sections (Fig. 4B) revealed cell body degeneration first apparent in cortical layers II and III at 11 weeks in La cKO cortex that extended to dendritic and axonal processes in other layers at later time points. This layer pattern fits well with previously determined expression by the CaMKIIα promoter (57). Cortical layers II and III were impacted earliest and most dramatically, followed by white matter tracts (Fig. 4B, WM). The data suggest damage to neuronal bodies occurs before damage to axons or dendrites.
FIG 4.

Neurodegeneration is apparent at 11 weeks and increases thereafter in the La cKO cortex. Brain slices (30 μm) from WT and La cKO mice 11 to 20 weeks old were stained with Ag-Cu to detect degenerating neurons. (A) Phase-contrast images (×10 magnification) of whole brain were tiled with a Zeiss 880 confocal microscope. Hatched bars of the same size show progressive shrinking of the La cKO cortex relative to WT. (B) Representative brain block images under phase-contrast microscopy at ×10 magnification to reveal finer structural detail. The cortical layers are annotated with Roman numerals I to VI. WM, white matter.
Dramatic astrocyte proliferation in La cKO cortex.
The glial fibrillary acidic protein (GFAP) mRNA was increased 5.8-fold in La cKO cortex by RNA-Seq (see Data Set S1 in the supplemental material). GFAP is produced by proliferative astrocytes in response to neuronal damage of various etiologies (79–81). We examined GFAP and La protein by confocal microscopy in 16-week cortices. La was readily detected in nuclei throughout WT cortex but was very much reduced in La cKO cortex (Fig. 5, α-La), in good agreement with prior results (46). A relatively low level of GFAP was observed in WT cortex but was more extensive in La cKO cortex (Fig. 5, α-GFAP), consistent with the RNA-Seq data. We noted a trend of preferential astrocyte localization in areas with relatively low levels of La (Fig. 5, upper part of bottom row) compared to areas where loss of La was relatively small and La appeared to remain more concentrated (Fig. 5, bottom, outlined in white). This suggests that cells degenerating due to decreasing levels of La may be preferential targets of astrocytes.
FIG 5.
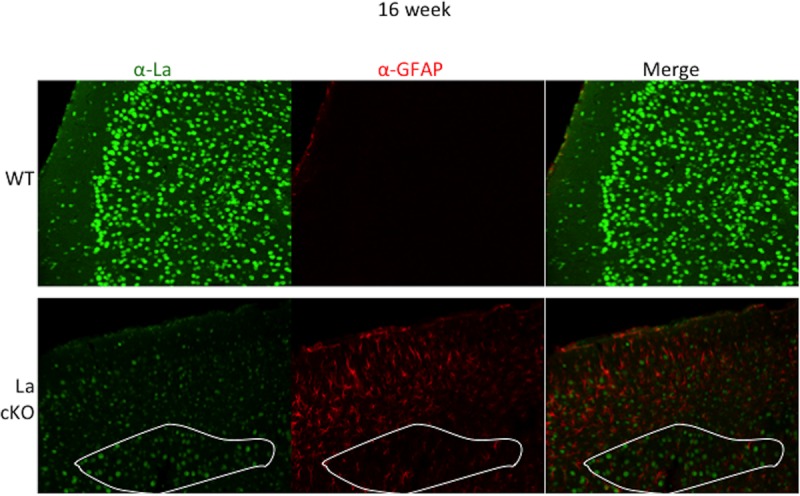
Astrocytes invade the La cKO cortex, evidence of astrogliosis. Brain slices (30 μm) from 16-week-old mice were incubated with antibodies directed against GFAP and La. Confocal images were taken with the same laser settings at ×20 magnification. (Bottom) A demarcated region of generally decreased La staining in the cKO cortex, outlined in white (left), corresponds to relatively low levels of GFAP (middle); note that in areas with relatively reduced La staining above the demarcated area, the relatively high levels of GFAP suggest that astrocytes migrated to cells that have lost, or are losing, La protein (see the text).
Imaging the progression of astrocyte proliferation.
Astrocytes and microglia are major immune effector cells in the CNS (82, 83). Slices from the same time course samples used in Fig. 4A were immunostained for GFAP (an astrocyte marker) and Iba1 (ionized calcium binding adaptor molecule 1) (a microglial marker) (Fig. 6B and C, respectively). WT and La cKO showed low GFAP reactivity at 11 weeks (Fig. 6A). Robust GFAP reactivity was observed in all three cKO triplicates at 13 weeks and reproducibly intensified thereafter, whereas WT cells showed no significant GFAP over the same time (Fig. 6A, replicates not shown). Importantly, Fig. 6A revealed that the onset of GFAP reactivity roughly coincided with the onset of loss of La cKO cortical mass previously reported (46) and with the neuronal degeneration seen in Fig. 4. Higher magnification of GFAP-stained La cKO cortex (Fig. 6B) revealed astrocytes localized in the same layers as degenerated neurons detected by Ag-Cu stain (Fig. 4A and B).
FIG 6.

La deletion is followed by progressive astrocyte reaction and cortical loss with relatively moderate microglial reactivity. Triplicate WT and La cKO cortices at 11, 13, 16, and 20 weeks of age (24 brains) were embedded in a block and sectioned by NSA using MultiBrain technology. NSA performed staining for Iba1 and GFAP. The stained sections were then photographed. (A) Representative age progression of WT and La cKO cortices stained for GFAP (the boxed regions indicate the approximate areas imaged at higher magnification in panel B). (B) Phase-contrast images (×10 magnification) of GFAP-stained sections from WT and La cKO cortices; the cortical layers are indicated by Roman numerals. (C) Iba1 (microglial) staining. WT and La cKO cortices from 11 to 20 weeks. (D) Representative age progression of WT and La cKO cortices stained for DAPI and the interneuron marker parvalbumin (×20 magnification).
Iba1 is a protein marker used to stain microglia (84, 85). Astrocytes and microglia sometimes respond to brain insults in a coordinated way (82, 83, 86, 87). However, our RNA-Seq data showed a 5.8-fold difference in GFAP but no significant difference in Iba1 mRNA levels between WT and La cKO cortices (not shown). Nonetheless, we examined microglia. Consistent with RNA-Seq, microglial changes as monitored by Iba1 were modest in La cKO cortex (Fig. 6C). Close inspection revealed somewhat more hypertrophied microglial cell bodies and more extensions beginning at 13 weeks in La cKO cortex, possibly reflecting “activated” morphology (Fig. 6C) (88). The data indicate that astrocytes proliferate more than microglia in response to La deletion from cortical neurons.
Activation of immune cells, particularly microglia, often leads to reactive oxygen species and proinflammatory cytokines, which can damage neighboring healthy neurons, leading to death (89, 90). Therefore, we examined La+ neurons in La cKO cortex by immunofluorescence staining for parvalbumin, a marker for inhibitory interneurons that are not subjected to CamKIIα promoter activity and therefore La deletion (64, 91, 92). There was no reproducible difference in parvalbumin-positive staining (Fig. 6D), suggesting that loss of cortical mass is largely restricted to La cKO neurons.
Evidence of limited apoptosis in La cKO cortex.
Others have noted links between La and apoptosis (93–96). We performed immunostaining using antibody to cleaved caspase 3 (CC3), which initiates the final stages of apoptosis (97). We found more CC3-reactive cells in La cKO 20-week cortex than in WT cortex (Fig. 7A). Further examination revealed La cKO nuclei to be smaller than WT nuclei (Fig. 7B), consistent with apoptosis (98). Surprisingly however, thorough examinations of La cKO cortex at earlier time points, when cortical mass was in sharp decline (46), revealed very little CC3 staining (not shown). Due to technical limitations, we could not readily determine if the CC3-positive cells were derived from La cKO neurons or other cell types.
FIG 7.

Evidence of limited apoptosis in La cKO cortex. Brain block sections were stained for CC3. (A) Representative images from 20-week WT and La cKO cortices. (B) Quantification of nuclear area determined from DAPI staining and using the ImageJ NII plugin (see Materials and Methods); standard errors of the mean are plotted based on measurements of 70 nuclei each from WT and La cKO cells.
We examined the Seq data for mRNAs indicative of apoptosis and found no significant differences for La cKO and WT cells, including p53 (not shown), or for MDM2, XIAP, or Bcl2, other apoptosis factors whose mRNAs were found to be associated with La (99–102). Thus, while our data document substantial loss of La cKO cortical neurons, apoptosis cannot fully account for this, and involvement of other pathways known for mature neurons should be suspected (103).
DISCUSSION
Mouse La protein is essential for development prior to embryo implantation and is required for establishment or survival of embryonic stem (ES) cells (21). Premitotic immune B cells also fail to develop or survive in the absence of La (46). CaMKIIα-driven Cre activity begins at ∼3 weeks postbirth in the CA1 cells of the hippocampus (104) and is later activated in the outer cortex (46, 57; http://www.informatics.jax.org/allele/key/6462?recomRibbon=open). Thus, within 2 to 3 weeks following La deletion, the cKO brain stops growing while WT brains continue to grow, and then at weeks 12 to 13 it begins to lose cortical mass (46). The data indicate that postmitotic cortical neurons die following deletion of La. The collective data from this and previous reports (21, 46) and known ubiquitous expression of La argue that it serves an essential function in many if not most or all mammalian cells.
Evidence of a pre-rRNA processing deficiency in La cKO cortex.
We showed that a pre-5.8S rRNA intermediate accumulates in La cKO cortex 10-fold more than in WT cortex (Fig. 1C). The estimated size of the transcript, based on mobility relative to the 7SL SRP RNA, is ∼250 nt (Fig. 1B and data not shown), close to that described, after agarose gel electrophoresis, as an ∼7S species derived from a 12S species by 3′ exonucleolytic processing (54). Aberrant accumulation in La cKO cortex was corroborated by microscopy using an anti-5.8S rRNA antibody (Fig. 2).
The pre-5.8S rRNA processing deficiency in the subset of La cKO cells does not lead to an apparent decrease in overall levels of mature 5.8S rRNA in the whole cortex. The 10-fold accumulation occurred in cortex in which La was deleted but that also contained neuronal and nonneuronal cells from which La was not deleted. In addition to the resident La+ neurons and other integral CNS cells, there was extensive invasion by proliferative astrocytes at the time of RNA analysis. At this time, cortical brain mass was declining, indicating that cells with La deleted were being lost from the population. Accordingly, much of the RNA would be from WT cells with normal, mature 18S, 28S, and 5.8S rRNA levels that may mask a decrease in these RNA species that was due to defective processing in La cKO cells while allowing detection above background of accumulated pre-5.8S rRNA. We suspect that the presumed defect that leads to pre-5.8S rRNA accumulation would decrease the levels of mature 5.8S rRNA in the subset of La cKO cells if they could be isolated and checked, although doing so is beyond the scope of this study.
The mechanism of pre-5.8S rRNA accumulation is likely indirect.
Because La binding to RNA 3′ ends normally protects against exonucleases and stabilizes transcripts, accumulation of pre-5.8S rRNA in La cKO cortex was unexpected and suggested an indirect mechanism. Processing of the 47S rRNA transcript is complex and includes alternative pathways, dozens of intermediates, and different enzymes that can work on a given intermediate to generate the same product, as well as enzymes and complexes that work on multiple intermediates (105). Consistent with the indirect-effect hypothesis, we could find no evidence in the history of La of its association with pre-5.8S rRNA (14, 17, 47, 106–108). Because La was reported to be associated with pre-U8 and pre-U3 snoRNAs, it was plausible that its depletion might destabilize them and indirectly lead to pre-5.8S rRNA accumulation. However, we did not find La association with pre-5.8S rRNA or pre-U8 or pre-U3 snoRNA in mouse cortex lysate by IP despite clear association with pre-tRNATyr. The data and observations argue that pre-5.8S rRNA accumulation is not due to a direct effect of La. Below, we consider possibilities and factors that may affect its accumulation as a result of indirect effects of La deletion.
Certain ribosome proteins themselves contribute to ribosome assembly, and their deficiency can inhibit rRNA processing, as in some cases of Diamond-Blackfan anemia, a ribosomeopathy associated with a plethora of mutations in nine ribosome protein genes (109). Relevant here is Rpl35a, encoded by one of the 27 TOP mRNAs that are decreased in La cKO cortex (Table 3) and whose decrease was shown to negatively affect 7S rRNA accumulation in pre-5.8S rRNA processing (56). Although we believe that the deficiency of pre-5.8S rRNA accumulation described by Farrar et al. (56) is not consistent with a simple explanation of our results, it serves as an example showing that deficiency in one or another ribosomal protein alone or in combination could alter pre-5.8S rRNA accumulation in La cKO cortex.
Although ITS1 probing revealed no aberrant accumulation of a pre-18S rRNA species (Fig. 1D), it is possible that a defect in a general ribosome assembly factor causes pre-5.8S rRNA accumulation. Therefore, we examined our RNA-Seq data for alterations in mRNAs encoding the ∼70 components of the ribosome small subunit processome (SSU), some of which play multiple roles in rRNA processing (110). However, we found no significant changes in SSU protein mRNAs or in nucleolin or fibrillarin (not shown).
The pathway that generates mature 5.8S rRNA is known to involve multiple exonucleolytic digestions, although all of the individual events are not precisely known in animal cells (105). A 7S transcript that corresponds to a pre-5.8S rRNA is derived from 12S RNA (Fig. 1A) by exonucleolytic digestion (54). Our ITS2 probe revealed RNAs longer than pre-5.8S rRNA in La cKO cortex (Fig. 1B), suggesting inefficient exonucleolytic cleavages earlier in the pathway. The La cKO pre-5.8S rRNA of ∼250 nt would have a 3′ extension of ∼90 nt, suggesting this is a more prominent processing deficiency relatively late in 5.8S rRNA processing (Fig. 1A) (54, 105, 111). Studies of yeast and human cells indicate that relatively late processing of 3′-extended pre-5.8S rRNA to mature 5.8S rRNA requires exonucleases, including the nuclear exosome Rrp6 (111, 112). In summary, 3′ exonucleolytic processing of a relatively late pre-5.8S rRNA intermediate appears to be most inhibited in La cKO.
We suspect that this apparent block in pre-5.8S rRNA 3′ exonucleolytic processing in La cKO cortex may be caused at least in part by competitive inhibition by nascent pre-tRNAs whose 3′ ends are unprotected in the absence of La. Production of 12- to 15-fold molar excess of tRNAs over ribosomes (113) begins with synthesis of nascent pre-tRNAs whose ends are bound by the abundant La protein (see the introduction). 3′ exonucleases, including exosome Rrp6, compete with La for pre-tRNA, pre-5S rRNA, and pre-SRP RNA 3′ ends (19). In yeast lacking La, the nuclear exosome Rrp6 and other 3′ exonucleases engage these 3′ ends (10, 19, 114). This is consistent with the nucleoplasmic localization of the nuclear exosome Rrp6 and the late-stage pre-5.8S rRNA intermediates (110, 112, 115). In animal cells, La binds to precursors to tRNAs, 5S rRNA, SRP, and others, including transcripts from B1-Alu repeat elements. Thus, we should consider the possibility that the La cKO brain accumulates pre-5.8S rRNA by an indirect mechanism, in part because, in the absence of La, a massive number of free 3′ ends are available and may become competitive inhibitors of the 3′ exonucleases that act as part of its normal maturation pathway. Future experiments may test this.
Temporal analysis of the innate immune response to loss of neuronal La.
When expression of CaMKIIα promoter-driven Cre begins at ∼3 weeks postbirth, its activity is limited to the CA1 cells of the hippocampus (104) and is later activated in the outer cortex (46; http://www.informatics.jax.org/allele/key/6462?recomRibbon=open). The first evidence of neuronal degeneration is apparent at 11 weeks via Ag-Cu staining. By 13 weeks, astrocyte infiltration of La cKO cortex is evident, and it progresses thereafter. At 16 weeks, La expression is much reduced, and astrocyte invasion is robust (Fig. 5). More astrocyte reactivity was apparent in regions with relatively low La expression, whereas less astrocyte reactivity was observed in regions containing cells with more La (Fig. 5).
The Ag-Cu data suggest that by 20 weeks, axons extending from the cortical neuronal bodies to internal striatal regions undergo degeneration (Fig. 4A, 20 weeks, arrows). The cumulative data lead to a plausible pathway by which the La cKO cortex loses mass, which nicely coincides with the onset of brain mass loss documented previously. Following decreasing La levels, neurons undergo progressive degeneration, activating an immune response via astrocytes with ensuing loss of the neurons and a large decrease in cortical mass. We note that with mRNAs such as Cd11c, Clec7a, Fgr, and CD74, induced in La cKO cortex, the RNA-Seq data might suggest a myeloid dendritic cell response. However, reproducible evidence of a compromised blood-brain barrier in La cKO cortex was not observed (not shown).
The La cKO cortex as a neurodegenerative model.
Neurodegeneration can be initiated by diverse mechanisms, such as trauma (116); infection (117); gene mutations, including those that cause protein misfolding; aberrant trafficking of aggregate-prone proteins (118); and other mechanisms, including RNA- and protein-induced toxicity (119). While neurodegenerative disorders may arise from disparate mechanisms, they share a gene expression immune response signature that reflects activation of astrocytes and microglia (120–123). Our GO analysis agrees well with this signature. In some cases, astrocyte activation can be chronic and can contribute to pathogenesis (121).
Remarkably, no overt phenotypic difference in gait or other behavior was apparent in La CamKIIα cKO mice as they aged, even at 65 weeks and despite massive loss of cortex (46). Thus, the work described here suggests that these mice represent a model of neurodegeneration accompanied by robust astrocyte proliferative activation that is not associated with mortality or apparent morbidity. An important issue that may be addressed by this model is distinguishing activation of CNS immune cell responses that are helpful or protective from those that are pathological (124).
In the healthy brain, astrocytes are supportive of neuronal function, such as clearance of excess glutamate from synaptic clefts, preventing excitotoxicity, delivering neurotrophic factors, and other activities (125). Astrocytes play additional positive roles during recovery from insults (126). Astrocytes in La cKO cortex may be protective, perhaps contributing to the relative lack of phenotype despite massive loss of cortex. A recent report found that axon repair, mediated by a beneficial immune response, requires dectin-1 and, to a lesser extent, Toll-like receptor 2 (TLR2) to facilitate axon regrowth (127). Examination of our RNA-Seq data revealed that TLR2 and dectin-1 (Clec7a) were increased 6-fold and 42-fold, respectively (Table 1; see the supplemental material). Perhaps some feature specific to the La cKO cortex, possibly related to misprocessed pre-tRNA or rRNAs, affects CNS immune cells via distinct pathways to support a protective role.
Primary and secondary effects of La depletion on complex phenotypes.
The massive increase in mRNAs in La cKO cortex reflects proliferative cellular reactivity as an effect secondary to the preceding neurodegeneration. The temporal and other analyses, as well as prior data, indicate that pre- or postmitotic cells depleted of the essential functions of La fail to survive, undergo degeneration, and die. We found an unexpected increase in a pre-5.8S rRNA intermediate in La cKO cortex, which was surprising, because most ligands of La decrease upon its depletion, suggesting this was another indirect or secondary effect. While this most highly accumulated pre-5.8S rRNA species represents a late event in 5.8S rRNA production, the ITS2 probe also detected larger precursors in La cKO cortex (Fig. 1B) that suggest deficiencies earlier in rRNA processing. Thus, different types of observations, from mRNA-Seq and rRNA analysis, coupled with temporal pathology progression, indicate that most of what was observed in La cKO mouse cortex after initiating deletion of the La gene at 3 to 5 weeks after birth was secondary, resulting from deficiency of a primary activity(ies). The demonstration of La-associated, spliced pre-tRNA intermediates whose levels are decreased in La-depleted cKO cortex very likely reflects a primary deficiency in an essential function, tRNA production. We have not identified a primary activity of La in rRNA biogenesis. Evidence that a specific isoform of La associates with nucleolin in the dense fibrillar component of nucleoli (see the introduction) suggests involvement in a very early step in pre-rRNA biogenesis, which may have been obscured in this study due to the complexity of the system at hand. We believe that these findings are nonetheless valuable, not only because they further document the importance of La in the animal and demonstrate the mechanisms involved in the associated pathology, but because they should encourage future studies of tRNA and rRNA biogenesis in simpler systems, including cultured cells.
Perturbation of pre-tRNA processing in La cKO cortex.
In a previous study and here, we examined tRNATyr (46). All of the genes encoding major tRNATyr in prevalent model organisms for yeast, worms, flies, mice, and humans contain introns, including in Caenorhabditis elegans and D. melanogaster, in which only 4 and 3 different tRNA isoacceptors have introns (128). In the phylogenetically simplest eukaryotes, Plasmodium and Leishmania, which contain 35 to 90 tRNA genes (129, 130), the only one with introns is tRNATyr, which is also true for Dictyostelium species, which have ∼400 tRNA genes (131, 132). The first processing pathway dissected for a tRNA in an animal was for pre-tRNATyr (133, 134). Our data on mouse pre-tRNATyr are consistent with this pathway.
It had been shown that the 3′ trailer-containing, spliced pre-tRNATyr intermediate is decreased in abundance in La cKO relative to WT cortex (46). Although we are unaware of other animal evidence that La functionally impacts pre-tRNA metabolism, it is accepted that 3′ trailer-containing pre-tRNAs are destabilized in yeast with La deleted and stabilized by its overexpression (11, 15, 135–137). Thus, by analogy, it might be expected that the observed decrease in 3′ trailer-containing, spliced pre-tRNATyr in cKO cortex (46) was due to loss of La binding. However, yeast spliced pre-tRNAs are not La ligands, because pre-tRNA splicing occurs in the cytoplasm of yeast but in cell nuclei of animals (11, 62, 138–141). Thus, yeast pre-tRNAs are spliced only after end maturation and nuclear export (62), although a tiny fraction of end-containing species are found in the cytoplasm due to erroneous nuclear export (142). Only in yeast mutants in which La is aberrantly exported to the cytoplasm with its bound pre-tRNAs was it found bound to 3′ trailer-containing spliced pre-tRNAs (6, 11).
The new data reported here showing that La is bound to pre-tRNATyr intermediates in the splicing pathway are significant in multiple ways. They provide insight into the observed decrease in the 3′ trailer-containing spliced pre-tRNA in La cKO cortex (46). Accordingly, it seems that the 3′ trailer-containing pre-tRNA is destabilized in cells depleted of La. However, it is notable that here it applies to substrates and products of splicing not applicable to tRNA maturation in yeast. This indicates differential intranuclear handling of pre-tRNAs; intron-containing species are removed from yeast nuclei by the tRNA nuclear export factor Los1, which binds to 3′ ends (143), whereas in animal cell nuclei they would be exposed to the nuclear exosome Rrp6 and other nuclear exonucleases in the absence of La. These data also highlight another contrast with La-depleted yeast, in which the nascent pre-tRNAs are destabilized and decreased in abundance but with less, if any, effect on spliced intermediates, whereas in La cKO cortex the nascent pre-tRNA (band a) is relatively unaffected compared to reduction in the spliced pre-tRNA (46). The IP data (Fig. 1E, iii) are consistent with and provide insight into this. Quantitation suggests that the spliced product (b) is more enriched by La than the nascent transcript (a) (not shown), and although several mechanistic possibilities might account for this, it is consistent with a lack of protection of this species in La cKO (46).
The findings suggest an element of heretofore unknown intricacy in the splicing branch of a pre-tRNA production pathway in animals. From a biochemical view, because La is bound to the intron-containing substrate of splicing and is also bound to the spliced product, the data suggest that La can be accommodated during splicing by the tRNA splicing endonuclease (references 144, 145, and 146 provide docking models). From a cell biology view, the findings expand the fundamental difference in the tRNA maturation pathways in yeast and metazoa due to different locations of tRNA splicing and nuclear cytoplasmic transport.
Mutations in the tRNA splicing-associated factor CLP1 result in motor neuron loss in mice (48) and reduction of cortical and/or cerebellar brain mass in patients with microcephaly and other severe neurological impairments (48, 147, 148). The splicing deficiency leads to toxic tRNA fragments derived from pre-tRNATyr (48), and these and/or other pre-tRNA fragments were found in patients with mutations in CLP1 (147, 148). Mutations in genes encoding two subunits of the tRNA splicing endonuclease also cause neurodegenerative microcephalies (149, 150). We suspect that the decrease in the La-associated spliced pre-tRNA in La-depleted mouse brain (46) would contribute to the associated neurodegeneration and ensuing pathology. As it is expected that the La-bound spliced pre-tRNA would go on to be efficiently matured in WT brain, its destabilization in the absence of La may decrease the efficiency of mature-tRNA production, although future work will be necessary to investigate this.
Finally, the new data and observations should be considered with regard to the disparity in essentiality in metazoa but not yeast (see the introduction). Specifically, we should consider to what extent La essentiality in animals is related to its role in the pre-tRNA splicing pathway, as this appears to apply to animals but not yeast.
MATERIALS AND METHODS
All mouse housing, husbandry, handling, and studies were done at the NIH under protocol ASP 10-005, approved by the IACUC of the NICHD.
RNA isolation.
Two WT females and 2 La cKO mice (1 male and 1 female) were sacrificed by cervical dislocation; their brains were removed and washed three times in 1× Hanks' balanced salt solution (HBSS) plus 40 U/ml RNasin. The La cKO mice bearing PCR-verified genotypes, Lax/- CreCaMKII, were as described previously (46). WT mice had the La+ allele (46). Samples were handled independently to produce two biological duplicates for WT and cKO cortices. Cortices were isolated, transferred to a glass Dounce homogenizer, homogenized in TriReagent, combined with 200 μl each of double-distilled water (ddH2O) and chloroform, and vortexed. After phase extraction, RNA was precipitated with 1/10 volume 3 M sodium acetate (NaOAc) plus 3 volumes ethanol (EtOH). RNA was subjected to bioanalyzer analysis to ensure sample uniformity and high quality. RNA samples were transferred to the NIH Intramural Sequencing Center (NISC), Rockville, MD, for poly(A)+ selection and Illumina sequencing. RNAs for Northern blotting were isolated in the same way but from different biological duplicates.
RNA-Seq data analysis.
Each WT and LaKO sample was analyzed independently as one of two biological duplicates. Gene ontology analysis was performed using three independent, widely used methods: DAVID (151), PANTHER (152, 153), and network ontology analysis (154). GO analysis was performed on mRNAs that were altered to a statistically significant degree (P < 0.06) and found to be 2-fold increased or decreased in the La cKO cortex. Redundant and overly general GO terms were omitted. The terms are listed with their respective P values to indicate the statistical power of each enriched term.
TOP mRNA analysis.
All mRNAs found to be decreased to a statistically significantly level (26% cutoff) in La cKO relative to WT cortex and subjected to DAVID GO analysis that revealed enrichment of the term “ribosome” are listed in Table 3.
qRT-PCR.
Isolated RNA was DNase treated, extracted with phenol-chloroform, and precipitated. cDNAs were transcribed using gene-specific primers with SuperScript III reverse transcriptase (Life Technologies; 18080400) according to the manufacturer's protocol. qPCR was performed (Sybr green PCR master mix; Applied Biosystems; 4309155) with 2 μl of each cDNA reaction mixture, using an ABI Prism 7000 sequence detection system. Amplification efficiency and melting curves were determined for each primer set to ensure accurate qPCR results. Relative mRNA abundance was calculated via the ΔΔCT method from the biological-duplicate samples using GAPDH mRNA for normalization. The qRT-PCR RNA samples were prepared from different mice than the samples that the RNA-Seq was performed on.
qRT-PCR primers.
The qRT-PCR primers used were as follows: C4b forward (5′-CGCCTGCCCATCTCCATC), C4b reverse (5′-TGGACACTCACAGCCACATC), CD74 forward (5′-GGCTCCACCTAAAGTACTGACC), CD74 reverse (5′-TACCGTTCTCGTCGCACTTG), H2-Aa forward (5′-TTTGACCCCCAAGGTGGACT), H2-Aa reverse (5′-GCTTGAGGAGCCTCATTGG-TA), H2-Ab forward (5′-CAGGAGTCAGAAAGGACCTCG), H2-Ab reverse (5′-ACTGGCAGTCAGGAATTCGG), IL1b forward (5′-TGCCACCTTTTGACAGTGATG), IL1b reverse (5′-TGATGTGCTGCTGCGAGATT), GAPDH forward (5′-GCTCTCAATGACAACTTTGTCAAGCTCATTTC), and GAPDH reverse (5′-TAGGGCCTCTCTTGCTCAGTGTCCT).
Northern blot analysis.
Twenty micrograms of RNA isolated from mouse cortex was denatured and separated in either 8% or 10% (IP) Tris-borate-EDTA (TBE)–urea–polyacrylamide gels (ITS2 analysis) or 1.2% denaturing formaldehyde–agarose gels (ITS1 analysis). After electrophoresis, the RNA was transferred to a nylon membrane with an iBlot transfer apparatus for polyacrylamide gels (Thermo-Fisher) or by capillary action for agarose gels, UV cross-linked, and vacuum baked at 80°C. Antisense oligonucleotides were 5′ labeled with [γ-32P]ATP with T4 polynucleotide kinase. GAPDH–[α-32P]UTP-labeled riboprobe was produced from the GAPDH PCR product using a MaxiScript T7 transcription kit (Thermo-Fisher; AM1324) and hybridized overnight. Blots were prehybridized for 2 h in 15 ml hybridization buffer (6× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 0.5% SDS, 2× Denhardt's solution [catalog number 750018; Life Technologies], 100 μg/ml S. cerevisiae total RNA) at the incubation temperature (Ti) [Ti = Tm (melting temperature) − 15°C, where Tm is equal to 16.6 log(M) + 0.41(Pgc) + 81.5 − Pm − (B/L), where M is the molar salt concentration up to a maximum of 0.5; Pgc is the percent G+C content in the oligonucleotide DNA probe; Pm is the percentage of mismatched bases, if any; B is 675; and L is the oligonucleotide DNA probe length]. The blots were washed 4 times for 15 min each time at room temperature before being exposed to a phosphorimager screen. Northern blots were washed 3 times for 10 min each time at room temperature with 2× SSC plus 0.1% SDS and then 2 times for 15 min each time at 15°C below the Ti. For additional probes, blots were stripped with 0.1× SSC and 0.1% SDS at 95°C, monitored to validate removal of radioactivity, and then reprobed.
Probes.
The probes used were as follows: ITS2 (pre-5.8S rRNA), 5′-ACCCACCGCAGCGGGTGACGCGATTGATCG (nt 2263 to 2292 of rRNA, beginning 3 nt 3′ of the 5.8S rRNA and extending for 30 nt); ITS1, 5′-CTCTCACCTCACTCCAGACACCTCGCTCCA; 5.8S rRNA mature, 5′-AAGTGCATTTGAAGTGTCAATGATCAATGTGTCCTGCAGTT; SRP RNA, 5′-CGAGTAGCTGGGACTACAGGCGGTG; pre-U3 snoRNA, 5′-AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA; pre-U8 snoRNA, 5′-AAGTGTGATCGCCAGGGAATCAGATAGGAGCAA; pre-tRNATyr intron, 5′-TACCATATTGTACTGACTACAGTCC; pre-tRNATyr 3′ trailer, 5′-AAGTAGTGCACGAAGTCCTTCG; and tRNATyr spliced, 5′-AGCGACCTAAGGATCTACAGT. These are to the mouse tRNA-Tyr-GTA-2-1 gene, chr5.trna45-TyrGTA (128).
RNA immunoprecipitation.
Thirteen-week-old mice were euthanized by cervical dislocation, and their brains were removed and washed 3 times in 1× PBS plus 40 U/ml SupeRasin. The cortex was isolated and Dounce homogenized in IP lysis buffer (IPLB) (50 mM HEPES, pH 7.0, 150 mM NaCl, 0.05% NP-40, EDTA-free protease inhibitor tablets [Roche], 1 mM EDTA, 40 U/ml SupeRasein [ThermoFisher]). Extracts were centrifuged for 10 min at 3,000 × g at 4°C, and the supernatant was isolated and clarified by centrifugation for 10 min at 8,000 × g. Eight micrograms of anti-mLa IgG (21) or mouse IgG as a control was immobilized on protein G Dynabeads. The beads were preincubated with 250 μg/ml bovine serum albumin (BSA), washed 3 times with 500 μl IPLB, and then incubated with 1 mg extract in 1 ml IPLB for 2 h at 4°C. The supernatant was removed, and the beads were washed 5 times with 700 μl IPLB. After the final wash, the beads were resuspended in 50 μl H2O, and RNA was extracted with acid phenol-chloroform. The RNA was separated on a 10% polyacrylamide gel containing TBE-urea. The RNA was transferred to a nylon membrane and hybridized with the indicated oligonucleotide probes.
Perfusion/fixation and brain sectioning.
Mice were deeply anesthetized with isoflurane. Transcardial perfusion was performed via gravity flow using sterile 1× PBS, followed by 4% paraformaldehyde (PFA)-1× PBS. The brains were then removed and postfixed in 4% PFA for 4 h. The brains were then cryoprotected in graded sucrose solutions containing 156 mM NaH2PO4, 107 mM NaOH, 0.01% thimerosal. The brains were placed in 4% sucrose for 4 h, then in 20% sucrose for 12 h, and then in 30% sucrose for 24 h. Thirty-micrometer-thick floating sagittal brain sections were made using a Leica SM2000R sliding microtome. For Neuroscience Associates brain block preparation, 24 mice were perfused as described above and then postfixed for 24 h in 15% sucrose, 1× PBS, 4% paraformaldehyde. The sex distribution for wild-type mice was as follows: 11 weeks, 2 females and 1 male; 13 weeks, 1 female and 2 males; 16 weeks, 2 females and 1 male; 20 weeks, 2 males and 1 female. The sex distribution for La cKO mice was as follows: 11 weeks, 2 females and 1 male; 13 weeks, 1 female and 2 males; 16 weeks, 3 females; 20 weeks, 2 females and 1 male. The brains were shipped to NSA. NSA embedded all 24 fixed brains into one gelatin block so that after sectioning, each 30-μm section contained a slice of all 24 samples in one immobilized block section. The sections were stored at −20°C in cryoprotectant solution.
Tiled images of brain sections.
Tiled ×10 magnification images were of entire brain section slices from WT and La cKO mice at 11 to 20 weeks. Tile stitching was performed using Fiji Image J software and the stitching grid collection plugin (155). Equal pixel areas were centered over the brain sections and cropped to preserve scale. Images from biological replicates (not shown) were consistent with those depicted.
Confocal immunofluorescence.
Antibodies were titrated to determine the optimal signal-to-background conditions. Antigen retrieval was performed on floating sections in citrate buffer, pH 6.0, using a Pelco BioWave Pro. After antigen retrieval, the sections were blocked in 1× PBS, 20% normal goat serum, 0.5% BSA, 0.3% Triton X-100 for 1 h. Antibodies were then added at the indicated concentrations and incubated with the sections overnight at 4°C with shaking. The sections were washed 3 times for a total of 30 min, and then appropriate secondary Abs were added and incubated for 1 h at 25°C. The sections were washed 2 times for a total of 20 min and then transferred to 1× PBS solution for 5 min. The sections were transferred to gelatin-coated slides, and coverslips were mounted with mowiol. Imaging was performed with a Zeiss LSM780 confocal microscope. NSA performed immunohistochemical staining for GFAP and Iba1 according to their standard optimized protocols (references 77 and 78 show published images of NSA staining for Iba1 and GFAP).
Antibodies.
The antibodies used were as follows: chicken anti-GFAP 1:5,000 (ab4674), rabbit anti-mLa 1:5,000 (polyclonal antibody against recombinant mouse La) (21); goat anti-chicken antibody–Alexa Fluor 647 (1:1,000) (ab150175), donkey anti-rabbit antibody–Alexa Fluor 488 (1:1,000) (Life Technologies; A-21206), Y10b anti-5.8S rRNA 1:2,000 (Thermo-Fisher; MA5-16064), anti-gamma actin (Thermo Fisher; PA1-16890), and rat antiparvalbumin (Sigma-Aldrich; SAB2500752).
Nuclear size.
Confocal images (×40) of DAPI (4′,6-diamidino-2-phenylindole)-stained WT and La cKO cortices were subjected to nuclear morphometric analysis (98) using the NII plug-in for ImageJ (156).
Supplementary Material
ACKNOWLEDGMENTS
We thank Lynne Holtzclaw for expertise and training in perfusion and immunofluorescence, Vincent Schram for imaging, Bob Switzer of Neuroscience Associates for discussion and expertise in brain immunology, Brenda Peculis for discussion, and anonymous reviewers for helpful comments.
This work was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH.
We declare no competing financial interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00588-16.
REFERENCES
- 1.Lerner MR, Boyle JA, Hardin JA, Steitz JA. 1981. Two novel classes of small ribo-nucleoproteins detected by antibodies associated with lupus erythematosus. Science 211:400–402. doi: 10.1126/science.6164096. [DOI] [PubMed] [Google Scholar]
- 2.Bousquet-Antonelli C, Deragon JM. 2009. A comprehensive analysis of the La-motif protein superfamily. RNA 15:750–764. doi: 10.1261/rna.1478709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rinke J, Steitz JA. 1982. Precursor molecules of both human 5S ribosomal RNA and tRNAs are bound by a cellular protein reactive with anti-La lupus antibodies. Cell 29:149–159. doi: 10.1016/0092-8674(82)90099-X. [DOI] [PubMed] [Google Scholar]
- 4.Stefano JE. 1984. Purified lupus antigen La recognizes an oligouridylate stretch common to the 3′ termini of RNA polymerase III transcripts. Cell 36:145–154. doi: 10.1016/0092-8674(84)90083-7. [DOI] [PubMed] [Google Scholar]
- 5.Teplova M, Yuan Y-R, Ilin S, Malinina L, Phan AT, Teplov A, Patel DJ. 2006. Structural basis for recognition and sequestration of UUU-OH 3′-termini of nascent RNA pol III transcripts by La, a rheumatic disease autoantigen. Mol Cell 21:75–85. doi: 10.1016/j.molcel.2005.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bayfield MA, Kaiser TE, Intine RV, Maraia RJ. 2007. Conservation of a masked nuclear export activity of La proteins and its effects on tRNA maturation. Mol Cell Biol 27:3303–3312. doi: 10.1128/MCB.00026-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bayfield MA, Yang R, Maraia R. 2010. Conserved and divergent features of structure and function of La and La-related proteins (LARPs). Biochim Biophys Acta 1799:365–378. doi: 10.1016/j.bbagrm.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakshusmathi G, Kim SD, Rubinson DA, Wolin SL. 2003. A La protein requirement for efficient pre-tRNA folding. EMBO J 22:6562–6572. doi: 10.1093/emboj/cdg625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Copela LA, Chakshusmathi G, Sherrer RL, Wolin SL. 2006. The La protein functions redundantly with tRNA modification enzymes to ensure tRNA structural stability. RNA 12:644–654. doi: 10.1261/rna.2307206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Y, Bayfield MA, Intine RV, Maraia RJ. 2006. Separate RNA-binding surfaces on the multifunctional La protein mediate distinguishable activities in tRNA maturation. Nat Struct Mol Biol 13:611–618. doi: 10.1038/nsmb1110. [DOI] [PubMed] [Google Scholar]
- 11.Intine RV, Dundr M, Misteli T, Maraia RJ. 2002. Aberrant nuclear trafficking of La protein leads to disordered processing of associated precursor tRNAs. Mol Cell 9:1113–1123. doi: 10.1016/S1097-2765(02)00533-6. [DOI] [PubMed] [Google Scholar]
- 12.Intine RVA, Sakulich AL, Koduru SB, Huang Y, Pierstorrf E, Goodier JL, Phan L, Maraia RJ. 2000. Control of transfer RNA maturation by phosphorylation of the human La antigen on serine 366. Mol Cell 6:339–348. doi: 10.1016/S1097-2765(00)00034-4. [DOI] [PubMed] [Google Scholar]
- 13.Maraia RJ. 2001. La protein and the trafficking of nascent RNA polymerase III transcripts. J Cell Biol 153:F13–F17. doi: 10.1083/jcb.153.4.F13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolin SL, Cedervall T. 2002. The La protein. Annu Rev Biochem 71:375–403. doi: 10.1146/annurev.biochem.71.090501.150003. [DOI] [PubMed] [Google Scholar]
- 15.Yoo CJ, Wolin SL. 1997. The yeast La protein is required for the 3′ endonucleolytic cleavage that matures tRNA precursors. Cell 89:393–402. doi: 10.1016/S0092-8674(00)80220-2. [DOI] [PubMed] [Google Scholar]
- 16.Kucera NJ, Hodsdon ME, Wolin SL. 2011. An intrinsically disordered C terminus allows the La protein to assist the biogenesis of diverse noncoding RNA precursors. Proc Natl Acad Sci U S A 108:1308–1313. doi: 10.1073/pnas.1017085108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolin SL, Wurtmann EJ. 2006. Molecular chaperones and quality control in noncoding RNA biogenesis. Cold Spring Harbor Symp Quant Biol 71:505–511. doi: 10.1101/sqb.2006.71.051. [DOI] [PubMed] [Google Scholar]
- 18.Bayfield MA, Maraia RJ. 2009. Precursor-product discrimination by La protein during tRNA metabolism. Nat Struct Mol Biol 16:430–437. doi: 10.1038/nsmb.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Copela LA, Fernandez CF, Sherrer RL, Wolin SL. 2008. Competition between the Rex1 exonuclease and the La protein affects both Trf4p-mediated RNA quality control and pre-tRNA maturation. RNA 14:1214–1227. doi: 10.1261/rna.1050408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bai C, Tolias PP. 2000. Genetic analysis of a La homolog in Drosophila melanogaster. Nucleic Acids Res 28:1078–1084. doi: 10.1093/nar/28.5.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park JM, Kohn MJ, Bruinsma MW, Vech C, Intine RV, Fuhrmann S, Grinberg A, Mukherjee I, Love PE, Ko MS, DePamphilis ML, Maraia RJ. 2006. The multifunctional RNA-binding protein La is required for mouse development and for the establishment of embryonic stem cells. Mol Cell Biol 26:1445–1451. doi: 10.1128/MCB.26.4.1445-1451.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ali N, Pruijn GJ, Kenan DJ, Keene JD, Siddiqui A. 2000. Human La antigen is required for the hepatitis C virus internal ribosome entry site (IRES)-mediated translation. J Biol Chem 275:27531–27540. [DOI] [PubMed] [Google Scholar]
- 23.Brenet F, Dussault N, Borch J, Ferracci G, Delfino C, Roepstorff P, Miquelis R, Ouafik L. 2005. Mammalian peptidylglycine alpha-amidating monooxygenase mRNA expression can be modulated by the La autoantigen. Mol Cell Biol 25:7505–7521. doi: 10.1128/MCB.25.17.7505-7521.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cardinali B, Carissimi C, Gravina P, Pierandrei-Amaldi P. 2003. La protein is associated with terminal oligopyrimidine mRNAs in actively translating polysomes. J Biol Chem 278:35145–35151. doi: 10.1074/jbc.M300722200. [DOI] [PubMed] [Google Scholar]
- 25.Costa-Mattioli M, Svitkin Y, Sonenberg N. 2004. La autoantigen is necessary for optimal function of the poliovirus and hepatitis C virus internal ribosome entry site in vivo and in vitro. Mol Cell Biol 24:6861–6870. doi: 10.1128/MCB.24.15.6861-6870.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crosio C, Boyl PP, Loreni F, Pierandrei-Amaldi P, Amaldi F. 2000. La protein has a positive effect on the translation of TOP mRNAs in vivo. Nucleic Acids Res 28:2927–2934. doi: 10.1093/nar/28.15.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Intine RV, Tenenbaum SA, Sakulich AS, Keene JD, Maraia RJ. 2003. Differential phosphorylation and subcellular localization of La RNPs associated with precursor tRNAs and translation-related mRNAs. Mol Cell 12:1301–1307. doi: 10.1016/S1097-2765(03)00429-5. [DOI] [PubMed] [Google Scholar]
- 28.Meerovitch K, Svitkin YV, Lee HS, Lejbkowicz F, Kenan DJ, Chan EK, Agol VI, Keene JD, Sonenberg N. 1993. La autoantigen enhances and corrects aberrant translation of poliovirus RNA in reticulocyte lysate. J Virol 67:3798–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pudi R, Abhiman S, Srinivasan N, Das S. 2003. Hepatitis C virus internal ribosome entry site-mediated translation is stimulated by specific interaction of independent regions of human La autoantigen. J Biol Chem 278:12231–12240. doi: 10.1074/jbc.M210287200. [DOI] [PubMed] [Google Scholar]
- 30.Schwartz E, Intine RV, Maraia RJ. 2004. CK2 is responsible for phosphorylation of human La protein serine-366 and can modulate 5′TOP mRNA metabolism. Mol Cell Biol 24:9580–9591. doi: 10.1128/MCB.24.21.9580-9591.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Svitkin YV, Ovchinnikov LP, Dreyfuss G, Sonenberg N. 1996. General RNA binding proteins render translation cap dependent. EMBO J 15:7147–7155. [PMC free article] [PubMed] [Google Scholar]
- 32.Ohndorf UM, Steegborn C, Knijff R, Sondermann P. 2001. Contributions of the individual domains in La protein to its RNA 3′-end binding activity. J Biol Chem 276:27188–27196. doi: 10.1074/jbc.M102891200. [DOI] [PubMed] [Google Scholar]
- 33.Yoo CJ, Wolin SL. 1994. La proteins from Drosophila melanogaster and Saccharomyces cerevisiae: a yeast homolog of the La autoantigen is dispensable for growth. Mol Cell Biol 14:5412–5424. doi: 10.1128/MCB.14.8.5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martino L, Pennell S, Kelly G, Bui TT, Kotik-Kogan O, Smerdon SJ, Drake AF, Curry S, Conte MR. 2012. Analysis of the interaction with the hepatitis C virus mRNA reveals an alternative mode of RNA recognition by the human La protein. Nucleic Acids Res 40:1381–1394. doi: 10.1093/nar/gkr890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gottlieb E, Steitz JA. 1989. The RNA binding protein La influences both the accuracy and the efficiency of RNA polymerase III transcription in vitro. EMBO J 8:841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Intine RV, Dundr M, Vassilev A, Schwartz E, Zhao Y, Depamphilis ML, Maraia RJ. 2004. Nonphosphorylated human La antigen interacts with nucleolin at nucleolar sites involved in rRNA biogenesis. Mol Cell Biol 24:10894–10904. doi: 10.1128/MCB.24.24.10894-10904.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sommer G, Dittmann J, Kuehnert J, Reumann K, Schwartz PE, Will H, Coulter BL, Smith MT, Heise T. 2011. The RNA-binding protein La contributes to cell proliferation and CCND1 expression. Oncogene 30:434–444. doi: 10.1038/onc.2010.425. [DOI] [PubMed] [Google Scholar]
- 38.Tang J, Zhang ZH, Huang M, Heise T, Zhang J, Liu GL. 2013. Phosphorylation of human La protein at Ser 366 by casein kinase II contributes to hepatitis B virus replication and expression in vitro. J Viral Hepat 20:24–33. doi: 10.1111/j.1365-2893.2012.01636.x. [DOI] [PubMed] [Google Scholar]
- 39.Brenet F, Socci N, Sonenberg N, Holland E. 2009. Akt phosphorylation of La regulates specific mRNA translation in glial progenitors. Oncogene 28:128–139. doi: 10.1038/onc.2008.376. [DOI] [PubMed] [Google Scholar]
- 40.Kuehnert J, Sommer G, Zierk AW, Fedarovich A, Brock A, Fedarovich D, Heise T. 2015. Novel RNA chaperone domain of RNA-binding protein La is regulated by AKT phosphorylation. Nucleic Acids Res 43:581–594. doi: 10.1093/nar/gku1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan H, Sakulich AL, Goodier JL, Zhang X, Qin J, Maraia RJ. 1997. Phosphorylation of the human La antigen on serine 366 can regulate recycling of RNA polymerase III transcription complexes. Cell 88:707–715. doi: 10.1016/S0092-8674(00)81913-3. [DOI] [PubMed] [Google Scholar]
- 42.Allain FH, Bouvet P, Dieckmann T, Feigon J. 2000. Molecular basis of sequence-specific recognition of pre-ribosomal RNA by nucleolin. EMBO J 19:6870–6881. doi: 10.1093/emboj/19.24.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ginisty H, Amalric F, Bouvet P. 1998. Nucleolin functions in the first step of ribosomal RNA processing. EMBO J 17:1476–1486. doi: 10.1093/emboj/17.5.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ginisty H, Sicard H, Roger B, Bouvet P. 1999. Structure and functions of nucleolin. J Cell Sci 112:761–772. [DOI] [PubMed] [Google Scholar]
- 45.Gerbi SA, Borovjagin AV, Lange TS. 2003. The nucleolus: a site of ribonucleoprotein maturation. Curr Opin Cell Biol 15:318–325. doi: 10.1016/S0955-0674(03)00049-8. [DOI] [PubMed] [Google Scholar]
- 46.Gaidamakov S, Maximova OA, Chon H, Blewett NH, Wang H, Crawford AK, Day A, Tulchin N, Crouch RJ, Morse HC III, Blitzer RD, Maraia RJ. 2014. Targeted deletion of the gene encoding the La protein SSB autoantigen in B cells or cerebral cortex causes extensive tissue loss. Mol Cell Biol 34:123–131. doi: 10.1128/MCB.01010-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maraia RJ, Lamichhane TN. 2011. 3′ Processing of eukaryotic precursor tRNAs. WIRES RNA 2:362–375. doi: 10.1002/wrna.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hanada T, Weitzer S, Mair B, Bernreuther C, Wainger BJ, Ichida J, Hanada R, Orthofer M, Cronin SJ, Komnenovic V, Minis A, Sato F, Mimata H, Yoshimura A, Tamir I, Rainer J, Kofler R, Yaron A, Eggan KC, Woolf CJ, Glatzel M, Herbst R, Martinez J, Penninger JM. 2013. CLP1 links tRNA metabolism to progressive motor-neuron loss. Nature 495:474–480. doi: 10.1038/nature11923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishimura R, Nagy G, Dotu I, Zhou H, Yang XL, Schimmel P, Senju S, Nishimura Y, Chuang JH, Ackerman SL. 2014. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 345:455–459. doi: 10.1126/science.1249749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thiffault I, Wolf NI, Forget D, Guerrero K, Tran LT, Choquet K, Lavallee-Adam M, Poitras C, Brais B, Yoon G, Sztriha L, Webster RI, Timmann D, van de Warrenburg BP, Seeger J, Zimmermann A, Mate A, Goizet C, Fung E, van der Knaap MS, Fribourg S, Vanderver A, Simons C, Taft RJ, Yates JR III, Coulombe B, Bernard G. 2015. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun 6:7623. doi: 10.1038/ncomms8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arimbasseri AG, Maraia RJ. 2016. RNA Polymerase III advances: structural and tRNA functional views. Trends Biochem Sci 41:546–559. doi: 10.1016/j.tibs.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Watkins NJ, Lemm I, Ingelfinger D, Schneider C, Hossbach M, Urlaub H, Luhrmann R. 2004. Assembly and maturation of the U3 snoRNP in the nucleoplasm in a large dynamic multiprotein complex. Mol Cell 16:789–798. doi: 10.1016/j.molcel.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 53.Watkins NJ, Lemm I, Luhrmann R. 2007. Involvement of nuclear import and export factors in U8 box C/D snoRNP biogenesis. Mol Cell Biol 27:7018–7027. doi: 10.1128/MCB.00516-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mullineux ST, Lafontaine DL. 2012. Mapping the cleavage sites on mammalian pre-rRNAs: where do we stand? Biochimie 94:1521–1532. doi: 10.1016/j.biochi.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 55.Peculis BA, Steitz JA. 1993. Disruption of U8 nucleolar snRNA inhibits 5.8S and 28S rRNA processing in the Xenopus oocyte. Cell 73:1233–1245. doi: 10.1016/0092-8674(93)90651-6. [DOI] [PubMed] [Google Scholar]
- 56.Farrar JE, Nater M, Caywood E, McDevitt MA, Kowalski J, Takemoto CM, Talbot CC Jr, Meltzer P, Esposito D, Beggs AH, Schneider HE, Grabowska A, Ball SE, Niewiadomska E, Sieff CA, Vlachos A, Atsidaftos E, Ellis SR, Lipton JM, Gazda HT, Arceci RJ. 2008. Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood 112:1582–1592. doi: 10.1182/blood-2008-02-140012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X, Zhang C, Szabo G, Sun QQ. 2013. Distribution of CaMKIIalpha expression in the brain in vivo, studied by CaMKIIalpha-GFP mice. Brain Res 1518:9–25. doi: 10.1016/j.brainres.2013.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lerner EA, Lerner MR, Janeway CA Jr, Steitz JA. 1981. Monoclonal antibodies to nucleic acid-containing cellular constituents: probes for molecular biology and autoimmune disease. Proc Natl Acad Sci U S A 78:2737–2741. doi: 10.1073/pnas.78.5.2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mili S, Steitz JA. 2004. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA 10:1692–1694. doi: 10.1261/rna.7151404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mazan S, Bachellerie JP. 1988. Structure and organization of mouse U3B RNA functional genes. J Biol Chem 263:19461–19467. [PubMed] [Google Scholar]
- 61.Tyc K, Steitz JA. 1992. A new interaction between the mouse 5′ external transcribed spacer of pre-rRNA and U3 snRNA detected by psoralen crosslinking. Nucleic Acids Res 20:5375–5382. doi: 10.1093/nar/20.20.5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Phizicky EM, Hopper AK. 2010. tRNA biology charges to the front. Genes Dev 24:1832–1860. doi: 10.1101/gad.1956510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Walsh JG, Muruve DA, Power C. 2014. Inflammasomes in the CNS. Nat Rev Neurosci 15:84–97. doi: 10.1038/nrn3638. [DOI] [PubMed] [Google Scholar]
- 64.Liu XB, Jones EG. 1996. Localization of alpha type II calcium calmodulin-dependent protein kinase at glutamatergic but not gamma-aminobutyric acid (GABAergic) synapses in thalamus and cerebral cortex. Proc Natl Acad Sci U S A 93:7332–7336. doi: 10.1073/pnas.93.14.7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arvieux J, Yssel H, Colomb MG. 1988. Antigen-bound C3b and C4b enhance antigen-presenting cell function in activation of human T-cell clones. Immunology 65:229–235. [PMC free article] [PubMed] [Google Scholar]
- 66.Bryan KJ, Zhu X, Harris PL, Perry G, Castellani RJ, Smith MA, Casadesus G. 2008. Expression of CD74 is increased in neurofibrillary tangles in Alzheimer's disease. Mol Neurodegener 3:13. doi: 10.1186/1750-1326-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lotteau V, Teyton L, Peleraux A, Nilsson T, Karlsson L, Schmid SL, Quaranta V, Peterson PA. 1990. Intracellular transport of class II MHC molecules directed by invariant chain. Nature 348:600–605. doi: 10.1038/348600a0. [DOI] [PubMed] [Google Scholar]
- 68.Morris P, Shaman J, Attaya M, Amaya M, Goodman S, Bergman C, Monaco JJ, Mellins E. 1994. An essential role for HLA-DM in antigen presentation by class II major histocompatibility molecules. Nature 368:551–554. doi: 10.1038/368551a0. [DOI] [PubMed] [Google Scholar]
- 69.Roche PA, Cresswell P. 1990. Invariant chain association with HLA-DR molecules inhibits immunogenic peptide binding. Nature 345:615–618. doi: 10.1038/345615a0. [DOI] [PubMed] [Google Scholar]
- 70.Durrenberger PF, Fernando FS, Kashefi SN, Bonnert TP, Seilhean D, Nait-Oumesmar B, Schmitt A, Gebicke-Haerter PJ, Falkai P, Grunblatt E, Palkovits M, Arzberger T, Kretzschmar H, Dexter DT, Reynolds R. 2015. Common mechanisms in neurodegeneration and neuroinflammation: a BrainNet Europe gene expression microarray study. J Neural Transm (Vienna) 122:1055–1068. doi: 10.1007/s00702-014-1293-0. [DOI] [PubMed] [Google Scholar]
- 71.Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL III, Araoz C. 1989. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A 86:7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moynagh PN, Williams DC, O'Neill LA. 1994. Activation of NF-kappa B and induction of vascular cell adhesion molecule-1 and intracellular adhesion molecule-1 expression in human glial cells by IL-1. Modulation by antioxidants. J Immunol 153:2681–2690. [PubMed] [Google Scholar]
- 73.Meyuhas O, Kahan T. 2015. The race to decipher the top secrets of TOP mRNAs. Biochim Biophys Acta 1849:801–811. doi: 10.1016/j.bbagrm.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 74.Yoshihama M, Uechi T, Asakawa S, Kawasaki K, Kato S, Higa S, Maeda N, Minoshima S, Tanaka T, Shimizu N, Kenmochi N. 2002. The human ribosomal protein genes: sequencing and comparative analysis of 73 genes. Genome Res 12:379–390. doi: 10.1101/gr.214202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pellizzoni L, Cardinali B, Lin-Marq N, Mercanti D, Pierandrei-Amaldi P. 1996. A Xenopus homologue of La autoantigen binds the pyrimidine tract of 5′ UTR of ribosomal protein mRNAs in vitro: implication of a protein factor in complex formation. J Mol Biol 259:904–915. doi: 10.1006/jmbi.1996.0368. [DOI] [PubMed] [Google Scholar]
- 76.Zhu J, Hayakawa A, Kakegawa T, Kaspar RL. 2001. Binding of the La autoantigen to the 5′ untranslated region of a chimeric human translation elongation factor 1A reporter mRNA inhibits translation in vitro. Biochim Biophys Acta 1521:19–29. doi: 10.1016/S0167-4781(01)00277-9. [DOI] [PubMed] [Google Scholar]
- 77.Gunapala KM, Chang D, Hsu CT, Manaye K, Drenan RM, Switzer RC, Steele AD. 2010. Striatal pathology underlies prion infection-mediated hyperactivity in mice. Prion 4:302–315. doi: 10.4161/pri.4.4.13721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ziebell JM, Taylor SE, Cao T, Harrison JL, Lifshitz J. 2012. Rod microglia: elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J Neuroinflammation 9:247. doi: 10.1186/1742-2094-9-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Diedrich J, Wietgrefe S, Zupancic M, Staskus K, Retzel E, Haase AT, Race R. 1987. The molecular pathogenesis of astrogliosis in scrapie and Alzheimer's disease. Microb Pathog 2:435–442. doi: 10.1016/0882-4010(87)90050-7. [DOI] [PubMed] [Google Scholar]
- 80.Janeczko K. 1988. The proliferative response of astrocytes to injury in neonatal rat brain. A combined immunocytochemical and autoradiographic study. Brain Res 456:280–285. [DOI] [PubMed] [Google Scholar]
- 81.Le Prince G, Delaere P, Fages C, Duyckaerts C, Hauw JJ, Tardy M. 1993. Alterations of glial fibrillary acidic protein mRNA level in the aging brain and in senile dementia of the Alzheimer type. Neurosci Lett 151:71–73. doi: 10.1016/0304-3940(93)90048-P. [DOI] [PubMed] [Google Scholar]
- 82.Ling EA, Wong WC. 1993. The origin and nature of ramified and amoeboid microglia: a historical review and current concepts. Glia 7:9–18. doi: 10.1002/glia.440070105. [DOI] [PubMed] [Google Scholar]
- 83.Vidovic M, Sparacio SM, Elovitz M, Benveniste EN. 1990. Induction and regulation of class II major histocompatibility complex mRNA expression in astrocytes by interferon-gamma and tumor necrosis factor-alpha. J Neuroimmunol 30:189–200. doi: 10.1016/0165-5728(90)90103-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mumaw CL, Levesque S, McGraw C, Robertson S, Lucas S, Stafflinger JE, Campen MJ, Hall P, Norenberg JP, Anderson T, Lund AK, McDonald JD, Ottens AK, Block ML. 2016. Microglial priming through the lung-brain axis: the role of air pollution-induced circulating factors. FASEB J 30:1880–1891. doi: 10.1096/fj.201500047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Taetzsch T, Levesque S, McGraw C, Brookins S, Luqa R, Bonini MG, Mason RP, Oh U, Block ML. 2015. Redox regulation of NF-kappaB p50 and M1 polarization in microglia. Glia 63:423–440. doi: 10.1002/glia.22762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. 1998. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 152:307–317. [PMC free article] [PubMed] [Google Scholar]
- 87.Matsumoto Y, Ohmori K, Fujiwara M. 1992. Microglial and astroglial reactions to inflammatory lesions of experimental autoimmune encephalomyelitis in the rat central nervous system. J Neuroimmunol 37:23–33. doi: 10.1016/0165-5728(92)90152-B. [DOI] [PubMed] [Google Scholar]
- 88.Kreutzberg GW. 1996. Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 89.Block ML, Zecca L, Hong JS. 2007. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 90.Pais TF, Figueiredo C, Peixoto R, Braz MH, Chatterjee S. 2008. Necrotic neurons enhance microglial neurotoxicity through induction of glutaminase by a MyD88-dependent pathway. J Neuroinflammation 5:43. doi: 10.1186/1742-2094-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rudy B, Fishell G, Lee S, Hjerling-Leffler J. 2011. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol 71:45–61. doi: 10.1002/dneu.20853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sik A, Hajos N, Gulacsi A, Mody I, Freund TF. 1998. The absence of a major Ca2+ signaling pathway in GABAergic neurons of the hippocampus. Proc Natl Acad Sci U S A 95:3245–3250. doi: 10.1073/pnas.95.6.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ayukawa K, Taniguchi S, Masumoto J, Hashimoto S, Sarvotham H, Hara A, Aoyama T, Sagara J. 2000. La autoantigen is cleaved in the COOH terminus and loses the nuclear localization signal during apoptosis. J Biol Chem 275:34465–34470. doi: 10.1074/jbc.M003673200. [DOI] [PubMed] [Google Scholar]
- 94.Huang M, Ida H, Kamachi M, Iwanaga N, Izumi Y, Tanaka F, Aratake K, Arima K, Tamai M, Hida A, Nakamura H, Origuchi T, Kawakami A, Ogawa N, Sugai S, Utz PJ, Eguchi K. 2005. Detection of apoptosis-specific autoantibodies directed against granzyme B-induced cleavage fragments of the SS-B (La) autoantigen in sera from patients with primary Sjogren's syndrome. Clin Exp Immunol 142:148–154. doi: 10.1111/j.1365-2249.2005.02888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rutjes SA, Utz PJ, der Heijden A, Broekhuis C, van Venrooij WJ, Pruijn GJ. 1999. The La (SS-B) autoantigen, a key protein in RNA biogenesis, is dephosphorylated and cleaved early during apoptosis. Cell Death Differ 6:976–986. doi: 10.1038/sj.cdd.4400571. [DOI] [PubMed] [Google Scholar]
- 96.Tran HB, Ohlsson M, Beroukas D, Hiscock J, Bradley J, Buyon JP, Gordon TP. 2002. Subcellular redistribution of La/SS-B autoantigen during physiologic apoptosis in the fetal mouse heart and conduction system: a clue to the pathogenesis of congenital heart block. Arthritis Rheum 46:202–208. doi:. [DOI] [PubMed] [Google Scholar]
- 97.Stennicke HR, Jurgensmeier JM, Shin H, Deveraux Q, Wolf BB, Yang X, Zhou Q, Ellerby HM, Ellerby LM, Bredesen D, Green DR, Reed JC, Froelich CJ, Salvesen GS. 1998. Pro-caspase-3 is a major physiologic target of caspase-8. J Biol Chem 273:27084–27090. doi: 10.1074/jbc.273.42.27084. [DOI] [PubMed] [Google Scholar]
- 98.Filippi-Chiela EC, Oliveira MM, Jurkovski B, Callegari-Jacques SM, da Silva VD, Lenz G. 2012. Nuclear morphometric analysis (NMA): screening of senescence, apoptosis and nuclear irregularities. PLoS One 7:e42522. doi: 10.1371/journal.pone.0042522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Heise T, Kota V, Brock A, Morris AB, Rodriguez RM, Zierk AW, Howe PH, Sommer G. 2016. The La protein counteracts cisplatin-induced cell death by stimulating protein synthesis of anti-apoptotic factor Bcl2. Oncotarget 7:29664–29676. doi: 10.18632/oncotarget.8819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Holcik M, Korneluk RG. 2000. Functional characterization of the X-linked inhibitor of apoptosis (XIAP) internal ribosome entry site element: role of La autoantigen in XIAP translation. Mol Cell Biol 20:4648–4657. doi: 10.1128/MCB.20.13.4648-4657.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nakatake M, Monte-Mor B, Debili N, Casadevall N, Ribrag V, Solary E, Vainchenker W, Plo I. 2012. JAK2(V617F) negatively regulates p53 stabilization by enhancing MDM2 via La expression in myeloproliferative neoplasms. Oncogene 31:1323–1333. doi: 10.1038/onc.2011.313. [DOI] [PubMed] [Google Scholar]
- 102.Trotta R, Vignudelli T, Pecorari L, Intine RV, Guerzoni C, Santilli G, Candini O, Byrom MW, Goldoni S, Ford LP, Caligiuri MA, Maraia R, Perrotti D, Calabretta B. 2003. BCR/ABL activates mdm2 mRNA translation via the La antigen. Cancer Cell 13:145–160. doi: 10.1016/S1535-6108(03)00020-5. [DOI] [PubMed] [Google Scholar]
- 103.Kole AJ, Annis RP, Deshmukh M. 2013. Mature neurons: equipped for survival. Cell Death Dis 4:e689. doi: 10.1038/cddis.2013.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. 1996. Subregion- and cell type-restricted gene knockout in mouse brain. Cell 87:1317–1326. doi: 10.1016/S0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- 105.Lafontaine DL. 2015. Noncoding RNAs in eukaryotic ribosome biogenesis and function. Nat Struct Mol Biol 22:11–19. doi: 10.1038/nsmb.2939. [DOI] [PubMed] [Google Scholar]
- 106.Maraia RJ, Intine RV. 2002. La protein and its associated small nuclear and nucleolar precursor RNAs. Gene Expr 10:41–57. [PMC free article] [PubMed] [Google Scholar]
- 107.Maraia RJ, Intine RV. 2001. Recognition of nascent RNA by the human La antigen: Conserved and diverged features of structure and function. Mol Cell Biol 21:367–379. doi: 10.1128/MCB.21.2.367-379.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Scheper GC, Voorma HO, Thomas AA. 1994. Basepairing with 18S ribosomal RNA in internal initiation of translation. FEBS Lett 352:271–275. doi: 10.1016/0014-5793(94)00975-9. [DOI] [PubMed] [Google Scholar]
- 109.Boria I, Garelli E, Gazda HT, Aspesi A, Quarello P, Pavesi E, Ferrante D, Meerpohl JJ, Kartal M, Da Costa L, Proust A, Leblanc T, Simansour M, Dahl N, Frojmark AS, Pospisilova D, Cmejla R, Beggs AH, Sheen MR, Landowski M, Buros CM, Clinton CM, Dobson LJ, Vlachos A, Atsidaftos E, Lipton JM, Ellis SR, Ramenghi U, Dianzani I. 2010. The ribosomal basis of Diamond-Blackfan Anemia: mutation and database update. Hum Mutat 31:1269–1279. doi: 10.1002/humu.21383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Phipps KR, Charette J, Baserga SJ. 2011. The small subunit processome in ribosome biogenesis—progress and prospects. Wiley Interdiscip Rev RNA 2:1–21. doi: 10.1002/wrna.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Briggs MW, Burkard KT, Butler JS. 1998. Rrp6p, the yeast homologue of the human PM-Scl 100-kDa autoantigen, is essential for efficient 5.8 S rRNA 3′ end formation. J Biol Chem 273:13255–13263. doi: 10.1074/jbc.273.21.13255. [DOI] [PubMed] [Google Scholar]
- 112.Thomson E, Tollervey D. 2010. The final step in 5.8S rRNA processing is cytoplasmic in Saccharomyces cerevisiae. Mol Cell Biol 30:976–984. doi: 10.1128/MCB.01359-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Waldron C, Lacroute F. 1975. Effect of growth rate on the amounts of ribosomal and transfer ribonucleic acids in yeast. J Bacteriol 122:855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ozanick SG, Wang X, Costanzo M, Brost RL, Boone C, Anderson JT. 2009. Rex1p deficiency leads to accumulation of precursor initiator tRNAMet and polyadenylation of substrate RNAs in Saccharomyces cerevisiae. Nucleic Acids Res 37:298–308. doi: 10.1093/nar/gkn925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Turowski TW, Tollervey D. 2015. Cotranscriptional events in eukaryotic ribosome synthesis. Wiley Interdiscip Rev RNA 6:129–139. doi: 10.1002/wrna.1263. [DOI] [PubMed] [Google Scholar]
- 116.Daneshvar DH, Goldstein LE, Kiernan PT, Stein TD, McKee AC. 2015. Post-traumatic neurodegeneration and chronic traumatic encephalopathy. Mol Cell Neurosci 66:81–90. doi: 10.1016/j.mcn.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 117.De Chiara G, Marcocci ME, Sgarbanti R, Civitelli L, Ripoli C, Piacentini R, Garaci E, Grassi C, Palamara AT. 2012. Infectious agents and neurodegeneration. Mol Neurobiol 46:614–638. doi: 10.1007/s12035-012-8320-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang X, Huang T, Bu G, Xu H. 2014. Dysregulation of protein trafficking in neurodegeneration. Mol Neurodegener 9:31. doi: 10.1186/1750-1326-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nalavade R, Griesche N, Ryan DP, Hildebrand S, Krauss S. 2013. Mechanisms of RNA-induced toxicity in CAG repeat disorders. Cell Death Dis 4:e752. doi: 10.1038/cddis.2013.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Heppner FL, Ransohoff RM, Becher B. 2015. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16:358–372. doi: 10.1038/nrn3880. [DOI] [PubMed] [Google Scholar]
- 121.Lull ME, Block ML. 2010. Microglial activation and chronic neurodegeneration. Neurotherapeutics 7:354–365. doi: 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Maragakis NJ, Rothstein JD. 2006. Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol 2:679–689. [DOI] [PubMed] [Google Scholar]
- 123.Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. 2007. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Block ML, Hong JS. 2005. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 125.Navarrete M, Araque A. 2014. The Cajal school and the physiological role of astrocytes: a way of thinking. Front Neuroanat 8:33. doi: 10.3389/fnana.2014.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Davies JE, Huang C, Proschel C, Noble M, Mayer-Proschel M, Davies SJ. 2006. Astrocytes derived from glial-restricted precursors promote spinal cord repair. J Biol 5:7. doi: 10.1186/jbiol35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Baldwin KT, Carbajal KS, Segal BM, Giger RJ. 2015. Neuroinflammation triggered by beta-glucan/dectin-1 signaling enables CNS axon regeneration. Proc Natl Acad Sci U S A 112:2581–2586. doi: 10.1073/pnas.1423221112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chan PP, Lowe TM. 2016. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res 44:D184–D189. doi: 10.1093/nar/gkv1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, Depledge DP, Fischer S, Gajria B, Gao X, Gardner MJ, Gingle A, Grant G, Harb OS, Heiges M, Hertz-Fowler C, Houston R, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Logan FJ, Miller JA, Mitra S, Myler PJ, Nayak V, Pennington C, Phan I, Pinney DF, Ramasamy G, Rogers MB, Roos DS, Ross C, Sivam D, Smith DF, Srinivasamoorthy G, Stoeckert CJ Jr, Subramanian S, Thibodeau R, Tivey A, Treatman C, Velarde G, Wang H. 2010. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res 38:D457–D462. doi: 10.1093/nar/gkp851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, Heiges M, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Miller JA, Nayak V, Pennington C, Pinney DF, Roos DS, Ross C, Stoeckert CJ Jr, Treatman C, Wang H. 2009. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res 37:D539–D543. doi: 10.1093/nar/gkn814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Basu S, Fey P, Pandit Y, Dodson R, Kibbe WA, Chisholm RL. 2013. DictyBase 2013: integrating multiple Dictyostelid species. Nucleic Acids Res 41:D676–D683. doi: 10.1093/nar/gks1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Eichinger L, Pachebat JA, Glockner G, Rajandream MA, Sucgang R, Berriman M, Song J, Olsen R, Szafranski K, Xu Q, Tunggal B, Kummerfeld S, Madera M, Konfortov BA, Rivero F, Bankier AT, Lehmann R, Hamlin N, Davies R, Gaudet P, Fey P, Pilcher K, Chen G, Saunders D, Sodergren E, Davis P, Kerhornou A, Nie X, Hall N, Anjard C, Hemphill L, Bason N, Farbrother P, Desany B, Just E, Morio T, Rost R, Churcher C, Cooper J, Haydock S, van Driessche N, Cronin A, Goodhead I, Muzny D, Mourier T, Pain A, Lu M, Harper D, Lindsay R, Hauser H, James K, Quiles M, Madan Babu M, Saito T, Buchrieser C, Wardroper A, Felder M, Simmonds M, Spiegler S, Tivey A, Sugano S, White B, Walker D, Woodward J, Weinstock G, Rosenthal A, Cox EC, Chisholm RL, Gibbs R, Loomis WF, Platzer M, Kay RR, Williams J, Dear PH, Noegel AA, Barrell B, Kuspa A. 2005. The genome of the social amoeba Dictyostelium discoideum. Nature 435:43–57. doi: 10.1038/nature03481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.De Robertis EM, Olson MV. 1979. Transcription and processing of cloned yeast tyrosine tRNA genes microinjected into frog oocytes. Nature 278:137–143. doi: 10.1038/278137a0. [DOI] [PubMed] [Google Scholar]
- 134.Melton DA, De Robertis EM, Cortese R. 1980. Order and intracellular location of the events involved in the maturation of a spliced tRNA. Nature 284:143–148. doi: 10.1038/284143a0. [DOI] [PubMed] [Google Scholar]
- 135.Foretek D, Wu J, Hopper AK, Boguta M. 2016. Control of Saccharomyces cerevisiae pre-tRNA processing by environmental conditions. RNA 22:339–349. doi: 10.1261/rna.054973.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Huang Y, Intine RV, Mozlin A, Hasson S, Maraia RJ. 2005. Mutations in the RNA polymerase III subunit Rpc11p that decrease RNA 3′ cleavage activity increase 3′-terminal oligo(U) length and La-dependent tRNA processing. Mol Cell Biol 25:621–636. doi: 10.1128/MCB.25.2.621-636.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Van Horn DJ, Yoo CJ, Xue D, Shi H, Wolin SL. 1997. The La protein in Schizosaccharomyces pombe: a conserved yet dispensable phosphoprotein that functions in tRNA maturation. RNA 3:1434–1443. [PMC free article] [PubMed] [Google Scholar]
- 138.De Robertis EM, Black P, Nishikura K. 1981. Intranuclear location of the tRNA splicing enzymes. Cell 23:89–93. doi: 10.1016/0092-8674(81)90273-7. [DOI] [PubMed] [Google Scholar]
- 139.Hopper AK, Shaheen HH. 2008. A decade of surprises for tRNA nuclear-cytoplasmic dynamics. Trends Cell Biol 18:98–104. doi: 10.1016/j.tcb.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 140.Yoshihisa T, Ohshima C, Yunoki-Esaki K, Endo T. 2007. Cytoplasmic splicing of tRNA in Saccharomyces cerevisiae. Genes Cells 12:285–297. doi: 10.1111/j.1365-2443.2007.01056.x. [DOI] [PubMed] [Google Scholar]
- 141.Yoshihisa T, Yunoki-Esaki K, Tanaka N, Endo T. 2003. Possibility of cytoplasmic pre-tRNA splicing: the yeast tRNA splicing endonuclease mainly localizes on the mitochondria. Mol Biol Cell 14:3266–3279. doi: 10.1091/mbc.E02-11-0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kramer EB, Hopper AK. 2013. Retrograde transfer RNA nuclear import provides a new level of tRNA quality control in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 110:21042–21047. doi: 10.1073/pnas.1316579110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cook AG, Fukuhara N, Jinek M, Conti E. 2009. Structures of the tRNA export factor in the nuclear and cytosolic states. Nature 461:60–65. doi: 10.1038/nature08394. [DOI] [PubMed] [Google Scholar]
- 144.Trotta CR, Abelson J. 1999. tRNA splicing: an RNA world add-on or an ancient reaction?, p 561–584. In Gesteland RF, Cech TR, Atkins JF (ed), The RNA world, 2nd ed Cold Spring Harbor Press, Cold Spring Harbor, NY. [Google Scholar]
- 145.Trotta CR, Miao F, Arn EA, Stevens SW, Ho CK, Rauhut R, Abelson JN. 1997. The yeast tRNA splicing endonuclease: a tetrameric enzyme with two active site subunits homologous to the archaeal tRNA endonucleases. Cell 89:849–858. doi: 10.1016/S0092-8674(00)80270-6. [DOI] [PubMed] [Google Scholar]
- 146.Trotta CR, Paushkin SV, Patel M, Li H, Peltz SW. 2006. Cleavage of pre-tRNAs by the splicing endonuclease requires a composite active site. Nature 441:375–377. doi: 10.1038/nature04741. [DOI] [PubMed] [Google Scholar]
- 147.Karaca E, Weitzer S, Pehlivan D, Shiraishi H, Gogakos T, Hanada T, Jhangiani SN, Wiszniewski W, Withers M, Campbell IM, Erdin S, Isikay S, Franco LM, Gonzaga-Jauregui C, Gambin T, Gelowani V, Hunter JV, Yesil G, Koparir E, Bainbridge MN, Gezdirici A, Seven M, Muzny DM, Boerwinkle E, Ozen M, Baylor Hopkins Center for Mendelian Genetics, Clausen T, Tuschl T, Yuksel A, Hess A, Gibbs RA, Martinez J, Penninger JM, Lupski JR. 2014. Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell 157:636–650. doi: 10.1016/j.cell.2014.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schaffer AE, Eggens VR, Caglayan AO, Reuter MS, Scott E, Coufal NG, Silhavy JL, Xue Y, Kayserili H, Yasuno K, Rosti RO, Abdellateef M, Caglar C, Kasher PR, Cazemier JL, Weterman MA, Cantagrel V, Cai N, Zweier C, Altunoglu U, Satkin NB, Aktar F, Tuysuz B, Caksen H, Bilguvar K, Fu XD, Trotta CR, Gabriel S, Reis A, Gunel M, Baas F, Gleeson JG. 2014. CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell 157:651–663. doi: 10.1016/j.cell.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Battini R, D'Arrigo S, Cassandrini D, Guzzetta A, Fiorillo C, Pantaleoni C, Romano A, Alfei E, Cioni G, Santorelli FM. 2014. Novel mutations in TSEN54 in pontocerebellar hypoplasia type 2. J Child Neurol 29:520–525. doi: 10.1177/0883073812470002. [DOI] [PubMed] [Google Scholar]
- 150.Breuss MW, Sultan T, James KN, Rosti RO, Scott E, Musaev D, Furia B, Reis A, Sticht H, Al-Owain M, Alkuraya FS, Reuter MS, Abou Jamra R, Trotta CR, Gleeson JG. 2016. Autosomal-recessive mutations in the tRNA splicing endonuclease subunit TSEN15 cause pontocerebellar hypoplasia and progressive microcephaly. Am J Hum Genet 99:228–235. doi: 10.1016/j.ajhg.2016.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Huang da W, Sherman BT, Lempicki RA. 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mi H, Dong Q, Muruganujan A, Gaudet P, Lewis S, Thomas PD. 2010. PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res 38:D204–D210. doi: 10.1093/nar/gkp1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. 2003. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wang J, Huang Q, Liu ZP, Wang Y, Wu LY, Chen L, Zhang XS. 2011. NOA: a novel network ontology analysis method. Nucleic Acids Res 39:e87. doi: 10.1093/nar/gkr251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Preibisch S, Saalfeld S, Tomancak P. 2009. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 25:1463–1465. doi: 10.1093/bioinformatics/btp184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.