

Abstract
Repopulation of immunodeficient mice remains the primary method for functional assessment of human acute myeloid leukemia. Published data report engraftment in ~40–66% of cases, mostly of intermediate- or poor-risk subtypes. Here we report that extending follow-up beyond the standard analysis endpoints of 10 to 16 weeks after transplantation permitted leukemic engraftment from nearly every case of xenotransplanted acute myeloid leukemia (18/19, ~95%). Xenogeneic leukemic cells showed conserved immune pheno-types and genetic signatures when compared to corresponding pre-transplant cells and, furthermore, were able to induce leukemia in re-transplantation assays. Importantly, bone marrow biopsies taken at standardized time points failed to detect leukemic cells in 11/18 of cases that later showed robust engraftment (61%, termed “long-latency engrafters”), indicating that leukemic cells can persist over months at undetectable levels without losing disease-initiating properties. Cells from favorable-risk leukemia subtypes required longer to become detectable in NOD/SCID/IL2Rγnull mice (27.5±9.4 weeks) than did cells from intermediate-risk (21.9±9.4 weeks, P<0.01) or adverse-risk (17±7.6 weeks; P<0.0001) subtypes, explaining why the engraftment of the first was missed with previous protocols. Mechanistically, leukemic cells engrafting after a prolonged latency showed inferior homing to the bone marrow. Finally, we applied our model to favorable-risk acute myeloid leukemia with inv(16); here, we showed that CD34+ (but not CD34−) blasts induced robust, long-latency engraftment and expressed enhanced levels of stem cell genes. In conclusion, we provide a model that allows in vivo mouse studies with a wide range of molecular subtypes of acute myeloid leukemia subtypes which were previously considered not able to engraft, thus enabling novel insights into leukemogenesis.
Introduction
The proliferation and survival of acute myeloid leukemia (AML) cells depend largely on environmental cues that are yet to be deciphered, making in vivo models mandatory for functional studies on AML.1 In contrast to genetically modified mice, human AML xenografts better depict the disease heterogeneity observed in patients.
A variety of different strains of immunosuppressed mice are available for xenograft studies.2,3 Overall, a higher degree of immune suppression appears to facilitate human cell engraftment. As such, robust engraftment was reported from ~40% of human AML samples transplanted via intrafemoral injection into non-obese diabetes/severe combined immunodeficiency (NOD/SCID) mice that were given pre-transplant irradiation conditioning.4 The more severely immunosuppressed NOD/SCID/IL2Rγnull (NSG) mice, which lack T, B and functional natural killer cells,3,5 enabled engraftment of 66% of transplanted AML samples, but mice received 10-fold higher cell numbers (107 cells/mouse) and a particularly low threshold of >0.1% human among murine bone marrow (BM) cells was set to define engraftment.6 With both protocols, engraftment was preferentially observed from FLT3-mutated high and intermediate molecular-risk AML (defined according to European LeukemiaNet criteria)7,8 suggesting that especially favorable-risk AML cannot be studied using these models.9 Insufficient cross-reactivity between murine and human proteins may lead to inadequate cytokine activity in human cells growing in the murine environment, thereby specifically hampering engraftment of certain subtypes of AML;10,11 consistently, human cytokine knock-in mice were recently shown to improve engraftment.12
Time to AML development is not assessable in patients, since diagnosis is typically made at the time of overt disease. However, a longer time to relapse after apparent remission is observed in patients with favorable-risk as compared to adverse-risk AML, suggesting differences in in vivo leukemia kinetics.13,14 We, therefore, hypothesized that subsets of AML (e.g. favorable-risk types) may require longer time to produce detectable engraftment and induce leukemia in mice. We transplanted a mixed cohort of 19 human AML of various genetic backgrounds, including four favorable-risk AML and two cases of acute promyelocytic leukemia (APL), and extended the post-transplant observation period to 1 year, instead of the 10 to 16 weeks used in previous studies.4,6 Indeed, only 7/19 transplanted AML (~37%, termed “standard engrafters”) showed detectable engraftment by week 16 after transplantation, while 11/19 (~58%, termed “long-latency engrafters”) repopulated mice later. Consistent with our hypothesis, all favorable-risk AML were long-latency engrafters. Importantly, longitudinal assessment of murine BM at 8 to 16 weeks after transplantation showed no evidence of leukemic cells in long-latency engrafters, indicating that they would have been missed with standard protocols. Next, we used this model to investigate the mechanisms underlying the observed differences in engraftment latency and the leukemia-initiating cell (LIC) compartment of favorable risk AML with inv(16).
Methods
Primary acute myeloid leukemia cells
Peripheral blood (PB) samples from patients with AML (Table 1 and Online Supplementary Table S1) were collected following approval by the Ethics Review Board of the University Hospitals of Basel and Tuebingen, enriched for mononuclear cells using a Ficoll (Biocoll, Merck Millipore, Darmstadt, Germany) gradient and viably frozen in 10% dimethyl sulfoxide solution (AppliChem, Darmstadt, Germany).
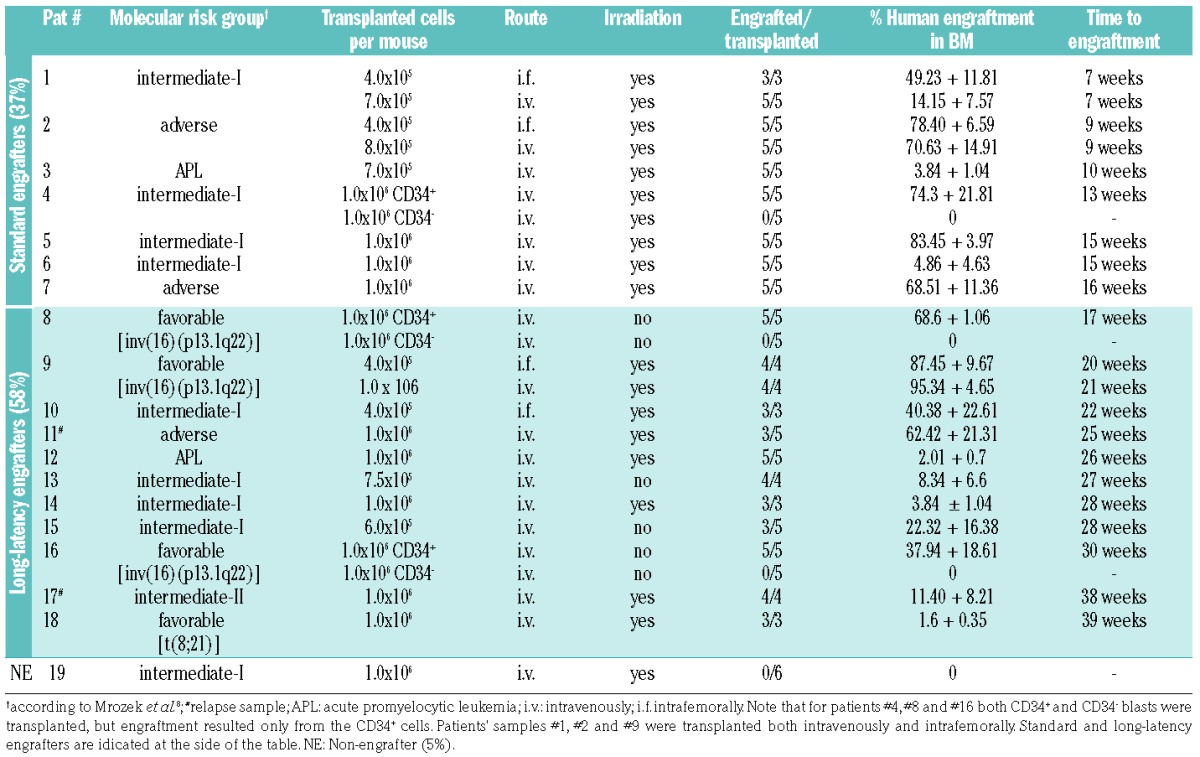
Table 1.
Molecular risk groups of the transplanted cases of acute myeloid leukemia, transplantation procedure and outcome details.
Mice and xenotransplantation assays
NOD.Cg-Prkdcscid IL2rgtmWjl/Sz (also termed NOD/SCID/IL2Rγnull, NSG) mice purchased from Jackson Laboratory (Bar Harbor, ME, USA) were maintained under pathogen-free conditions according to German and Swiss federal and state regulations. Freshly thawed AML cells were used for primary transplants. Details on sample preparation are provided in the Online Supplementary Material. Gender-matched, 7- to 10-week old animals with or without prior sublethal irradiation were injected intrafemorally15 or via the tail vein with AML cells resuspended in 25 or 200 μL phosphate-buffered saline, respectively (Table 2). Engraftment, (defined as ≥1% leukemic cells in murine PB or BM),1,4 was assessed in PB and BM at signs of distress (e.g. decreased food and water consumption, rapid breathing, altered movement)16 or routinely every 4 to 5 weeks in one mouse per group for each AML case. Mice were euthanized at sickness (weight loss, ruffled coat, weakness, reduced motility, other severe pathology) or detection of engraftment.6,17 Kaplan-Meier survival analysis and final assessment were performed on all animals. For secondary transplants, BM cells freshly isolated from mice showing >40% BM infiltration and belonging to one experimental group were pooled, subjected to MACS purification for human CD33 to eliminate contamination by murine cells and then used for transplantation in equal numbers as in the corresponding primary transplants. Limiting dilution and homing assays were performed according to standard protocols (see Online Supplementary Material).
Flow cytometry and histopathology
For flow cytometry analyses, fluorescent antibodies against human CD33, CD34, CD133, CD117, CD45, (BD Biosciences), CD14, CD13 (eBiosciences, San Diego, CA, USA), CD3 and CD19 (Biolegend, San Diego, CA, USA) were used. The analyses were performed on either a FACS CantoII or LSR II Fortessa (both BD Biosciences). SytoxBlue or 7-aminoactinomycin D was used to discriminate living and dead cells. Histological studies (with hematoxylin & eosin) and immunohistochemical analyses (CD33, CD34; Ventana, Tucson, Arizona, USA; CD117, DAKO, Glostrup, Denmark) were performed on formalin-fixed paraffin-embedded organs as described elsewhere.18 Samples were evaluated for Ki67 protein expression using the monoclonal anti-Ki67 antibody (clone Mib 1, DAKO IR626, ready-to-use). The percentage of stained leukemic cells was counted and recorded in 5% increments.
Colony-forming unit assays
For colony-forming assays, 2.5×104 CD34+ or respectively CD34− cells, sorted by FACS, were plated in methylcellulose (Methocult H4434, StemCell Technologies, Vancouver, Canada) in triplicate, incubated at 37°C in 5% CO2 and scored for colony-forming units at day 14.
Next-generation sequencing and microarray gene expression analyses
For next-generation sequencing, the AML community panel from Thermo Fisher containing 19 genes frequently mutated in AML (see ampliseq.com), the Ion PGM platform and the Ion Reporter AML pipeline (version 5.0) were employed. Agilent SurePrint G3 Human Gene Expression 8×60K v2 Microarrays analyses were performed on RNA extracts. For a detailed description of the next-generation sequencing and microarrays see the Online Supplementary Data.
Statistical analysis
Data are presented as the mean ± standard error of the mean. P values are derived from the application of the Mann-Whitney U test or two-tailed Fisher exact test.
Results
Extended follow-up time improved the detection rate of human acute myeloid leukemia engraftment in NSG mice
Nineteen AML cases of various genetic backgrounds, including four favorable-risk AML [three with inv(16) and one with t(8;21)] and two APL, were investigated regarding engraftment in NSG mice (Table 1 and Online Supplementary Table S1 for details of the patients and AML characteristics). Xenotransplants were performed following standard procedures via the tail vein (18/19 AML cases, 7×105 to 1×106 cells per mouse) or intrafemoral injection (4/19 AML cases, 4×105 cells per mouse) (Table 1).
In previous studies, mice transplanted with human AML cells were assessed at defined time-points (10, 12 or 16 weeks) and considered engrafted if >0.1–1% of human among total BM cells were detected.4,6 Using this method, a considerable proportion of AML, mostly of intermediate-and favorable-risk subtypes, were scored non-engraftable (35–60%, depending on the transplantation procedure and detection threshold). We hypothesized that these AML would also engraft, if longer observation times were applied. We prolonged follow-up to 1 year after transplantation, during which we closely monitored animals by inspection (every other day) and by BM puncture and multi-parameter flow cytometry analysis (performed at signs of distress or routinely every 4 to 5 weeks in one mouse per group for every AML case). This longitudinal assessment in the BM revealed that engraftment detection did indeed progress over time (Figure 1A and Online Supplementary Table S2). Compared to analyses at 8–10 or 12–14 weeks after transplantation, the standard analysis at 16 weeks already improved engraftment, but late analysis at 1 year revealed better engraftment than at any of these early time-points (Figure 1A, P<0.0001, P<0.0001 and P<0.001, respectively). In fact, only 7/19 transplanted AML cases (37%) were standard engrafters, showing ≥1% of human leukemic cells in the BM within the first 16 weeks after transplantation, while the majority (11/19, 58%) of cases were long-latency engrafters and repopulated mice at detectable levels only later. Importantly, 11/11 and respectively 10/11 of long-latency engrafters were screened negative for human leukemic cells in routine BM biopsies performed at 8–10, 12–14 and 16 weeks after transplantation (Figure 1A and Online Supplementary Table S2), indicating that their engraftment would have been missed with protocols stopping experimental follow-up at these time-points. Consistently, the majority (63%) of transplanted mice were alive until 16 weeks after transplantation, but developed robust leukemic engraftment and were removed from the experiments progressively within the next months (Figure 1B). Of note, in the three AML cases transplanted using both techniques (patients #1, #2, and #9; Table 1), intrafemoral transplantation was more efficient and allowed comparable engraftment starting from 2-fold reduced cell numbers (Online Supplementary Figure S1).
Figure 1.

Extended post-transplant follow-up enhances the detection efficiency of human acute myeloid leukemia cell engraftment in NSG mice. Longitudinal engraftment analysis by routine bone marrow punctures performed every 4 weeks revealed dependency of engraftment efficiency on post-transplant follow-up time. Lower percentages of engrafted mice were detected at standard post-transplantation analysis time-points used in previous protocols as shown by (A) quantification of engrafted samples at 8–10 weeks, 12–14 weeks and 16 weeks versus 1 year and by (B) Kaplan-Meier survival analysis of 109 mice xenotransplanted with human AML cells, indicating that with extended follow-up time, ~95% of all transplanted mice develop leukemia. A Fisher exact test was used to calculate statistical significance between engrafters and non-engrafters at each analyzed time to our endpoint of 1 year of observation in (A).
Conserved immunophenotypic and molecular features in xenogeneic versus corresponding pre-transplant acute myeloid leukemia cells
Mice engrafted with human AML cells were analyzed by multicolor flow cytometry, next-generation sequencing and whole-body histopathology. Various degrees of leukemic infiltration were detected in murine PB, BM and organs (Figure 2A,B). Engraftment of healthy human CD3+ and CD19+ cells was not observed at any time-point (presumably because CD33 or CD34 pre-selection was applied to cells from AML samples showing <95% blasts). Xenogeneic leukemic cells derived from murine BM mostly showed conserved immunophenotypes when compared to corresponding pre-transplant samples (Figure 2C and Online Supplementary Figure S2), with the exception of a down-regulation of CD117 (cKIT), which was observed in 15/18 analyzed liver and 17/18 PB but not in corresponding spleen or BM-derived samples (Online Supplementary Figure S2B). Of note, 10/18 (56%) of AML samples that showed robust engraftment in BM and/or organs did not show leukemic cells in the PB (Figure 2A). These data suggest that there are differences in leukemic cells residing in different sites in the mouse, and show that screening of PB alone cannot be used to monitor engraftment reliably.
Figure 2.
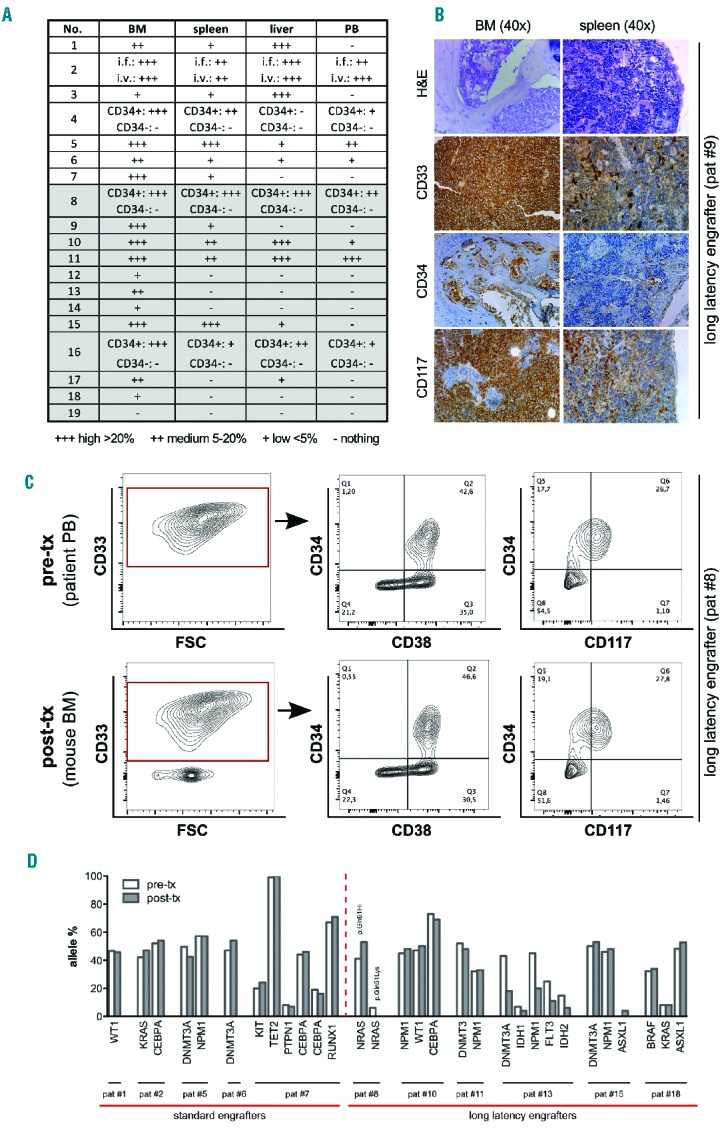
Conserved immunophenotypic and genetic features in patient-derived and corresponding xenogeneic acute myeloid leukemia cells. (A) Whole-body flow cytometry and (B) histopathological analysis of peripheral blood (PB), bone marrow (BM), spleen and liver of transplanted mice revealed heterogeneous infiltration with human leukemic blasts. (A) Semi-quantitative analysis summarizing high (>20%, +++), medium (5–20%, ++) and low (<5%, +) degrees of human leukemic cells among murine cells. Note that cells could be undetectable in PB despite robust BM/organ infiltration. (B) A representative histopathological analysis of BM and spleen from one engrafted sample [patient #9, AML with inv(16)] using hematoxylin & eosin and antibody staining against human CD33, CD34 and CD117 indicating the leukemic origin. (C) A representative multicolor flow cytometric analysis of immunophenotypic profiles of pre- (patient PB-derived) versus post-transplant (mouse BM-derived) AML cells showing no phenotypic changes upon engraftment [patient #8, AML with inv(16)]. (D) Highly conserved genetic patterns in pre- (patient-derived) and post-transplant (mouse BM-derived) samples. Allele frequencies for each mutation as detected by next-generation sequencing are shown. Note the loss of one out of two NRAS mutations (*p.Gln61Hi, # p.Gln61Lys; patient #8) and the gain of a low level ASXL1 mutation (patient #15).
Outgrowth of human leukemic cells in a xenograft animal model – especially if observed after periods of long latency – might indicate selection of specific clones or de novo acquisition of permissive mutations. To investigate this aspect, we analyzed the mutational status of pre-transplant and mouse-derived (post-transplant) leukemic cells using next-generation sequencing. Post-transplant samples were generated from pooled BM of all mice engrafted with one AML case. Paired samples of pre-transplant and corresponding xenogeneic cells were available for analysis from 12 of 18 engrafted AML cases. As shown in Figure 2D, high conservation of genetic signatures was observed in both standard and long-latency engrafters (see also Online Supplementary Table S3 for a detailed annotation of individual mutations). In one mouse-derived sample, a de novo-occurring ASLX1 mutation was detected at a low allelic burden (4%), while in another a low allelic burden NRAS mutation was lost, suggesting that genetic evolution can occur, but may not be a frequent event (Figure 2D, Online Supplementary Table S3). Furthermore, reduced frequencies of all five detected mutations were observed in post- versus pre-transplant samples of another case, suggesting possible co-engraftment with non-leukemic human cells (patient #13, Figure 2D). Finally, xenogeneic leukemic cells were shown to induce leukemia efficiently upon retransplantation in secondary recipients (Figure 3). Interestingly, faster repopulation was observed with xenogeneic secondary AML cells compared with the primary AML cells from the same donor for both standard and long-latency engrafting AML (Figure 3).
Figure 3.
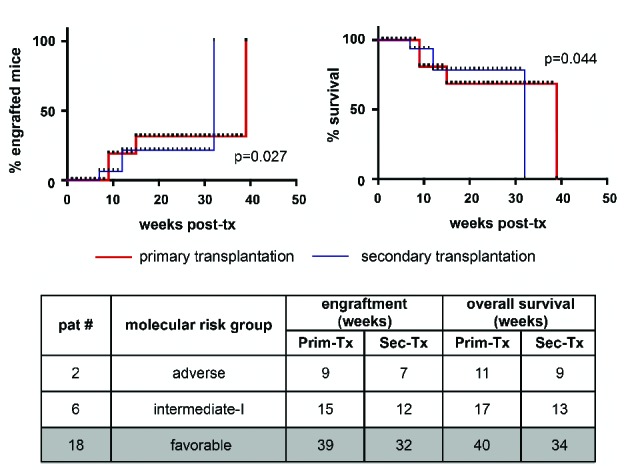
Xenogeneic human cells robustly induce leukemia in secondary transplantation assays. Fresh xenogeneic cells derived from murine BM purified by MACS recognizing leukemic antigens were retransplanted at equal numbers as in the corresponding primary transplantation assay. Note that robust engraftment occurred from all retransplanted cases, including the long-latency engrafting AML. Of note, engraftment occurred faster (left) and mice survived for a shorter time (right) after secondary transplantation (blue line) than after the primary transplantations (red line). This is also summarized in the table below. A log-rank (Mantel-Cox) test was applied to calculate statistical significance.
Taken together, leukemic engraftment in mice was confirmed by phenotype, histopathology, molecularly by next-generation sequencing and functionally in retransplantation assays. Mouse xenografts appear to mimic human disease faithfully, even though engraftment occurs after a long latency.
Favorable-risk acute myeloid leukemia required a longer time to become detectable in mice
Favorable-risk AML cells were previously scored non-engraftable, because they were not detectable in transplanted NSG mice analyzed 10 to 16 weeks after transplantation. Our study confirms these results: at these time-points, leukemic cells could not be detected in BM punctures performed on mice transplanted with favorable-risk AML cells (Online Supplementary Table S2), even though, when followed-up for longer periods, these mice eventually showed robust engraftment (Figure 1, Table 1 and Online Supplementary Table S2).
To analyze the relationship between molecular risk classification and engraftment capacity in NSG mice, we divided our cohort according to the European LeukemiaNet molecular risk score into favorable-, intermediate (I or II)- or adverse-risk AML. The time to detectable engraftment was indeed longer in mice transplanted with favorable-risk versus adverse- or intermediate-risk AML cells (favorable versus adverse P<0.0001, favorable versus intermediate P=0.004, Figure 4A, see also Table 1 for detailed numbers of engrafted among transplanted mice). Consistently, the longest survival was observed in mice transplanted with favorable-risk AML (27.5±9.4 weeks), followed by those transplanted with intermediate-risk AML (21.9±9.4 weeks, P=0.008) and adverse-risk cells (17±7.6 weeks, P<0.0001) (Figure 4B). The two analyzed APL samples showed heterogeneous behavior, with one sample belonging to the standard engraftment group and the other to the long-latency group (Online Supplementary S3A).
Figure 4.
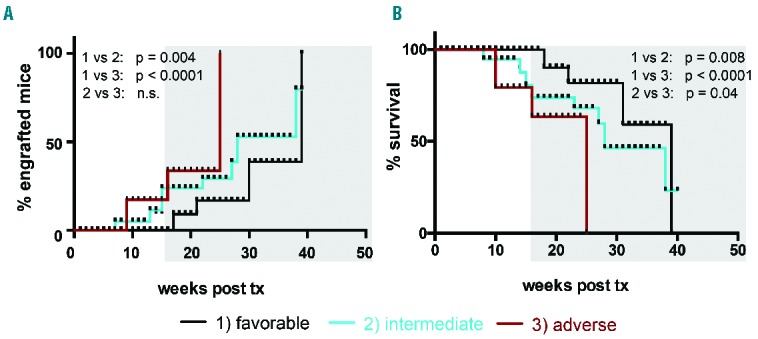
Time to in vivo engraftment and mouse survival correlate with molecular risk group stratification of the transplanted acute myeloid leukemia. (A) Favorable-risk AML showed longer time to engraftment when compared to intermediate- or adverse-risk AML. (B) Kaplan-Meier survival analysis of 99 mice transplanted with human AML indicates that mouse survival depends on the AML molecular risk group. Note that animals transplanted with favorable-risk AML cases showed the longest survival. (black: favorable, n=4; blue: intermediate (I+II), n=8; red: adverse risk AML, n=3). See Table 1 for transplanted/engrafted mouse numbers per individual AML case. A log-rank (Mantel-Cox) test was applied to calculate statistical significance.
Consistent with previous data reporting a high frequency of FLT3-ITD mutations among engrafting AML, FLT3-ITD mutations were observed in 4/7 (57%) of standard versus only 3/11 (27%) long-latency engrafters (Online Supplementary Table S1). However, when all mice were analyzed together, FLT3-ITD mutated AML did not show significantly faster engraftment or shorter survival when compared to samples without a mutated FLT3 (Online Supplementary Figure 3B).
Investigation of factors regulating engraftment latency
Leukemia has been proposed to initiate from rare subpopulations of blasts termed leukemia-initiating cells (LIC).1,4,19 The observed differences in leukemia latency could derive from: (i) different numbers of LIC (resulting from different LIC frequency and/or homing capacity to BM niches); (ii) differences in survival and/or proliferation capacity of human AML cells in the murine microenvironment; or (iii) a combination of these factors.
Limiting dilution assays did in fact reveal a strong association between engraftment latency (Figure 5A, left), mouse survival (Figure 5A, right) and, respectively, the number of transplanted cells, and lower LIC frequency in long-latency versus standard engrafters (1:197.599 versus 1:102.237). Interestingly, reduced homing rates to the BM20 were noted in long-latency versus standardly engrafting AML (Figure 5B) and the lowest percentage of homing cells was actually observed in the one AML sample that did not engraft (patient #19, data not shown).
Figure 5.
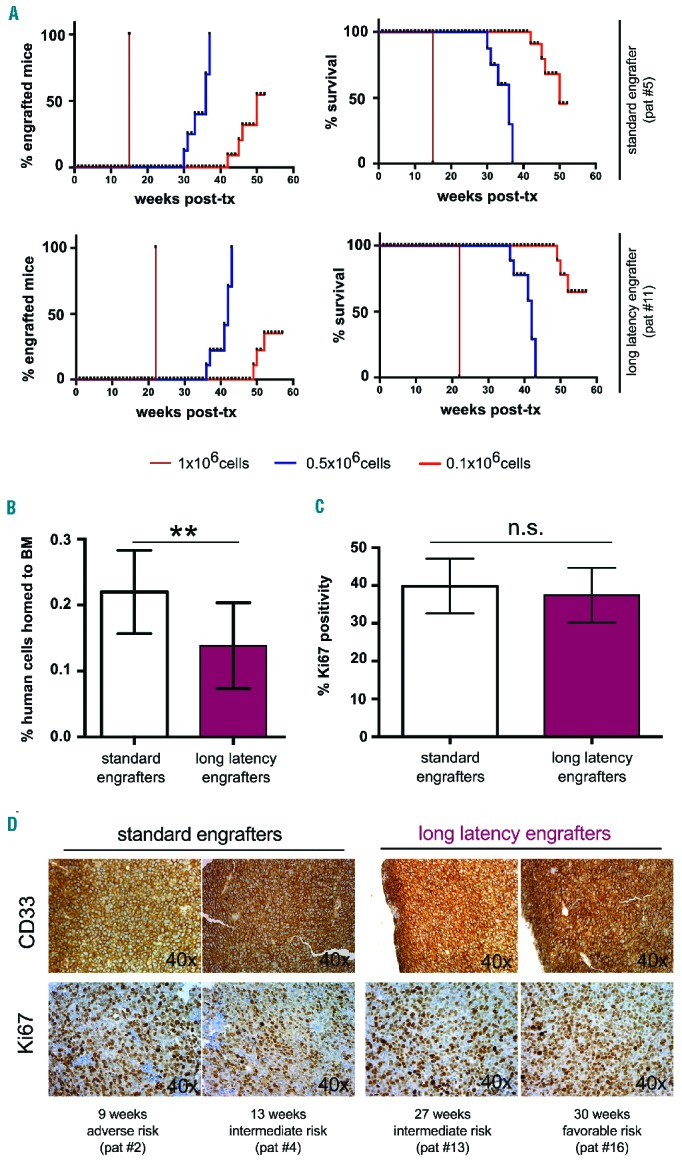
Investigation of factors regulating leukemia latency. (A) Limiting dilution assays showed that lowering the number of transplanted cells under certain thresholds resulted in engraftment failure (left) and better survival (right) in both standard (upper) and long-latency (lower) engrafters. (B) The frequency of leukemic cells homing to the BM was higher in standard engrafters than in long-latency engrafters. The data shown are from three standard (patients #2, #5, #6) and three long-latency engrafting AML cases (patients #11, #17, #18). (C, D) Comparable Ki67 positivity in standard vs. long-latency engrafting AML cells infiltrating murine BM (C). Representative pictures for both CD33 and Ki67 staining of two standard engrafters (patients #2, #4) and two long-latency engrafters (patients #13, #16) (D). For reasons of consistency only BM of mice showing >20% infiltration with human cells were included in the analysis.
To investigate in vivo proliferation, we performed Ki67 staining on the BM of mice infiltrated with human AML cells; for reasons of consistency, only mice showing >20% leukemic BM infiltration were included in the analysis. Interestingly, similar high Ki67 positivity was detected in standardly versus long-latency engrafting AML cells, indicating comparable in vivo proliferation at this advanced disease stage (Figure 5C,D). Supporting the notion that AML cells of both groups can rapidly expand in the BM microenvironment of NSG mice, the time from the last negative BM puncture to detectable engraftment was comparably short in standard versus long-latency engrafters (3.36±1.03 weeks versus 3.17±1.17 weeks, P=0.72) (Online Supplementary Table S2).
Favorable-risk acute myeloid leukemia with inv(16) engraft from CD34-expressing leukemia-initiating cells
Next, we used this model to investigate leukemia initiation from understudied favorable-risk AML with inv(16). All three inv(16) samples available to us were transplanted and showed engraftment, although only after a long latency (patients #8, #9 and #16; see Table 1 and Online Supplementary Table S1). Engraftment was confirmed as described above by multicolor flow cytometry and histopathology analyses detecting human leukemic cells of conserved phenotype in murine PB, BM and organs (Figure 2A,B and Online Supplementary Figure 2A). As shown by next-generation sequencing analysis, two NRAS mutations were found in the patient who engrafted first (patient #8), of which one was lost upon engraftment, while no further AML-specific mutations were detected with our panel in patient #9 in either pre- or post-transplant cells (Figure 2D and Online Supplementary Table S3). AML cells from patient #18, which required the longest time to engraft, showed well-conserved mutations in BRAF, KRAS (at low allele frequency) and ASXL1 (Figure 2D and Online Supplementary Table S3).
To investigate in more detail the relevance of CD34 expression as a marker for LIC in this genetic subtype, we sorted CD34+ and CD34− AML blasts with inv(16) and analyzed them functionally in colony-forming unit and xenotransplantation assays, as well as molecularly by gene expression arrays (Figure 6A). Interestingly, colony-formation capacity was higher in CD34+ blasts than in CD34− ones (Figure 6B). Even with prolonged follow-up, in vivo leukemia initiation was only observed from transplanted CD34+ but not CD34− leukemic inv(16) cells (Figure 6C), indicating that LIC are comprised in the CD34+ subpopulation. When observation times were longer, CD34+ inv(16) AML blasts induced engraftment even in the absence of pre-transplant irradiation conditioning. In mice, CD34+ blasts recapitulated the original AML phenotype generating both CD34+ and CD34− subpopulations (Online Supplementary Figure S2). Supporting these functional results, microarray gene expression analyses performed on CD34+ and CD34− inv(16) AML blasts revealed enhanced stem cell gene expression in CD34+ cells, as shown by gene set enrichment analysis, comparing our results with common datasets (Figure 6D and Online Supplementary Figure S4).4 Collectively these data indicate that, albeit with longer latency, favorable-risk AML with inv(16) can robustly engraft NSG mice from CD34+ LIC (Figure 6 and Table 1).
Figure 6.
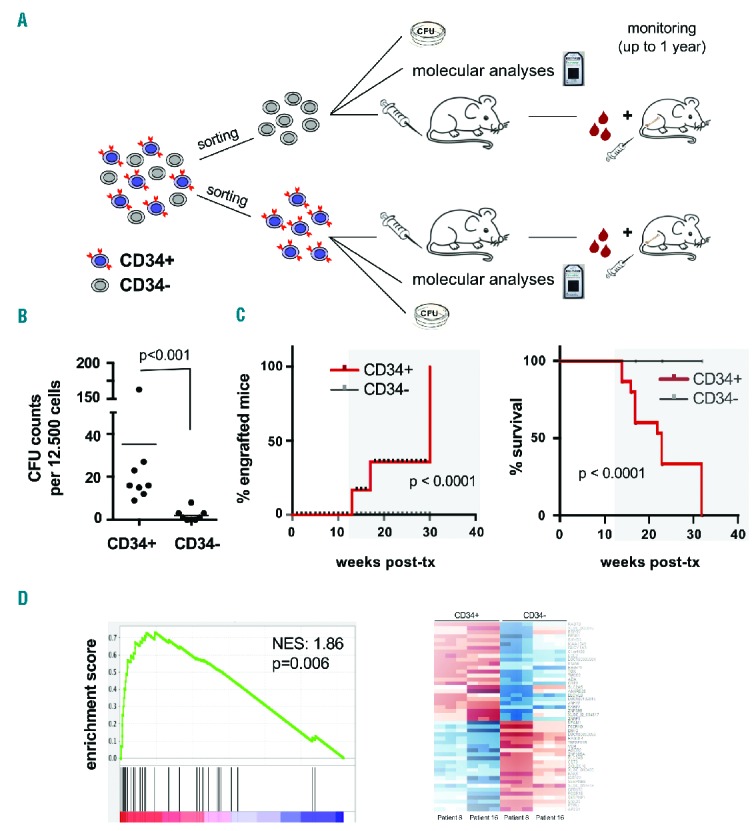
Leukemia initiating cells are within the CD34+ compartment of acute myeloid leukemia with inv(16). (A) Experimental setup to analyze CD34+ versus CD34− blasts in AML with inv(16). (B, C) Functional analyses of sorted CD34+ versus CD34− AML with inv(16) blasts revealed that CD34+ subsets are enriched for cells with in vitro colony-forming capacity (B) and contain in vivo engrafting LIC. Kaplan-Meier survival analysis indicating survival differences in mice transplanted with non-leukemogenic CD34− blasts versus leukemia-inducing CD34+ blasts (C); (red: CD34+ blasts (engraftable); gray: CD34− blasts (non-engraftable) of the same patient in each panel; patients #8 and #16; five transplanted mice per condition, 1×106 transplanted cells/mouse). For colony-forming unit (CFU) counts, three biological replicates performed on samples from patients #8 and #16 are shown. (D) Enrichment plot of a common LIC-gene signature4 in gene expression profiles of CD34+ versus CD34− blasts from two inv(16) AML cases (patients #8 and #16) indicating a common gene signature in both comparisons (left) and a heatmap of the CD34+ versus CD34− gene signatures in AML with inv(16) (right). NES denotes normalized enrichment score.
Discussion
Human healthy and malignant hematopoietic cells engraft immunosuppressed mice, thereby providing a valuable tool for studies on human hematopoiesis and leukemogenesis. Higher degrees of murine immune suppression facilitate repopulation with human cells,3,5 yet a substantial proportion of AML, particularly of favorable-risk and APL subtypes, remained non-engraftable in antecedent studies.21–24
The current study indicates that some AML require more time to become detectable in mice, and thus may have been falsely scored non-engraftable in previous reports. By performing longitudinal BM analyses of transplanted mice at 10, 12 and 16 weeks, and afterwards prolonging follow-up time to 1 year, we demonstrate that AML that score negative for human leukemic cells at conventional time-points eventually do engraft and induce symptomatic leukemia. Besides providing a tool for in vivo investigation of additional AML subtypes, these data demonstrate that leukemic cells can survive long periods of time at undetectable levels in mice before they eventually spread out and induce leukemia. This evolution in NSG mice mimics well the clinical course of AML in patients.
As hypothesized, the molecular risk group of the transplanted AML significantly influenced engraftment and leukemia kinetics in mice, with animals transplanted with favorable-risk AML showing the slowest engraftment and longest survival. FLT3-mutated AML, in previous studies overrepresented within engrafted AML,7 did not show significantly shorter time to engraftment nor was the overall survival of transplanted mice shorter compared to outcomes of FLT3-non-mutated samples.6,7 Higher sample numbers or exclusion of specific genetic backgrounds (e.g. aggressive AML without FLT3 mutation but with EVI1 overexpression, patient #2) might be required to detect significant differences.9 These findings are consistent with previously published data that correlate the capacity of AML cells to engraft NOD-SCID mice with patients’ outcome,22 and the side-by-side analysis of NOD-SCID and NSG mice indicating that AML considered non-engraftable show equally impaired repopulation.25
There are several possible explanations for the engraftment deficiency observed with some AML subtypes. Delayed engraftment, as assessed during 1 year in our study, might reflect clonal adaptation or slower growth rate of some AML subtypes in the mouse environment. In our study, both standard and long-latency engrafters showed conserved phenotypic and genetic features in mouse-derived cells in comparison with the corresponding pre-transplant cells, findings which stand in contrast to those of previous studies in which engraftment of individual subclones was detected.26,27 We hypothesize that the more homogeneous results consistently observed in the xenogeneic samples analyzed in our study reflect the fact that they were induced by higher numbers of LIC, which enhances the chance that several subclones are depicted in the eventually analyzed sample. In contrast to the previous studies, we used pre-transplant irradiation conditioning26 and comparatively younger mice,27 which might augment LIC homing to BM niches and thus enable long-term engraftment and disease initiation from higher numbers of cells. In support of this hypothesis, previous studies also showed engraftment of different subclones from one AML case in individual mice, suggesting that the ability to repopulate mice is not in fact restricted to certain genetic features. Further investigations (e.g. using exome sequencing comparison of patient-derived versus corresponding xenogeneic AML cells) are required to investigate whether atypical mutations (not captured by our next-generation sequencing platform) may however be acquired to facilitate growth in the murine environment. This latter could actually also explain the faster outgrowth of leukemic cells observed in retransplant assays.
AML with specific genetic backgrounds (e.g. favorable risk) may particularly rely on cytokine and/or niche support for optimal in vivo survival and growth. Their growth in NSG mice and capacity to engraft by outcompeting endogenous hematopoietic stem cells may thus be hampered by limited cross-reactivity of murine factors with human receptors. Immunosuppressed mice engineered to express human cytokines (“humanized mice”), or Kit mutations impairing murine host hematopoietic stem cells, respectively, were found to improve engraftment of healthy human hematopoietic and recently also leukemic cells.28–30 Further supporting the notion that engraftment is impaired by cross-species differences, implanted humanized BM ossicles – which provide a humanized niche environment within mice – were shown to facilitate repopulation with APL and myelofibrosis cells.31 In our study, similar (high) Ki67 positivity was detected in the BM of mice infiltrated with different AML at times of high leukemic burden. Furthermore, the time period from the last detected negative BM sample to engraftment and leukemia induction remained short in all transplanted AML irrespectively of engraftment latency or molecular risk subtype (data not shown). Thus, at times of advanced disease, the murine environment appears to support the proliferation of AML cells of various genetic backgrounds (including favorable-risk) comparably. However, AML cells are known to secrete cytokines and by themselves contribute to a pro-leukemogenic environment; if they are expanded to detectable numbers, inherent milieu disadvantages (e.g. lack of cross-reactive cytokines in NSG mice) might thereby be overruled. The situation might be different at the time of disease initiation, when only a few human AML cells are present in the mouse. Thus, the data presented here do not exclude the possibility that some AML engraft with long latency because they proliferate less in the murine BM at early (pre-detection) time-points, due to oncogene-specific differences in interactions with the healthy BM environment. Further studies are needed to investigate whether, specifically at stages of minimal disease burden, NSG mice provide suboptimal conditions for the outgrowth of certain genetic backgrounds, and to test whether further genetic modification of NSG mice via knock-in of human cytokines could accelerate disease induction thereby reducing more elevated costs raised by long-term mouse maintenance.
Previous studies in mouse xenograft models have shown that leukemia derives from rare LIC.1,4,19,25,32 Consistently, lowering transplanted cell numbers below a critical level (as part of limiting dilution assays) resulted in engraftment failure, which could not be compensated by extending follow-up. Reduced LIC frequency, as also measured by us in long-latency versus standardly engrafting AML, might thus be another explanation for the negative or delayed engraftment observed with certain AML subtypes (particularly intermediate- and favorable-risk). Supporting this notion, Griessinger and colleagues recently reported a surrogate ex vivo short-term co-culture system showing lower frequencies of long-term culture-initiating cells in good- versus intermediate- and poor-risk AML.33 Furthermore, indirect support of a lower LIC frequency in favorable-risk AML was provided by mRNA analyses, in which favorable clinical outcome was associated with reduced expression of stem cell genes.4,32,34 Alternatively, the reduced expression of stem cell genes might derive from qualitative alterations of LIC in these subtypes.4
LIC evaluation ultimately requires in vivo studies. LIC frequency and phenotype are thus largely understudied in AML previously considered non-engraftable, particularly in favorable-risk AML. In our study, we observed lower LIC frequencies in long-latency versus standard engrafters, suggesting that this factor does indeed contribute to the observed difference in leukemia induction latency. However, follow-up studies involving larger numbers of patients are necessary to confirm this conclusion. Furthermore, long-latency engrafting AML contained fewer numbers of leukemic cells able to home to the BM, when compared to standard engrafting AML. The low homing capacity and corresponding delay in leukemia induction observed with some AML may result from reduced expression of adhesion molecules, which in healthy hematopoietic stem and progenitor cells have been shown to essentially regulate BM homing.35,36,37
Homing is a core property of healthy hematopoietic stem cells.38 Although understudied in the setting of AML, homing appears as a pre-requisite for BM niche occupation and long-term leukemia induction, and LIC should be included within the subset of homing AML cells. Future studies are needed to evaluate homing as a surrogate assay to measure LIC content and perhaps even as a short-term functional assay that can predict disease aggressiveness.
Several attempts have been made to identify the phenotype of LIC. While they were reported to reside in the CD34+ or CD34+CD38− blast subpopulations, CD34+CD38+ and CD34− LIC have also been described (e.g. recently via expression of GPR56)32 and specifically linked to certain genetic backgrounds (e.g. AML with NPM1 mutation).1,4,19 We next used our model to investigate in vivo leukemogenesis and LIC from AML with inv(16). All three transplanted samples of this subtype robustly engrafted NSG mice, albeit with longer latency. Engraftment was achieved even in the absence of prior irradiation, indicating that conditioning and knock-in of human cytokines are not mandatory for leukemia induction, although they likely accelerate this process.12 However, in spite of extended follow-up, in vivo leukemogenesis was only observed from CD34+ (but not CD34−) inv(16) blasts. Furthermore, gene expression analyses demonstrated enhanced expression of stem cell genes in CD34+ cells, reinforcing CD34 surface expression as a LIC marker in this AML subtype.
In conclusion, we present a model that enables in vivo studies of AML subtypes previously considered non-engraftable, using widely accessible and well-characterized NSG mice. We show that in vivo xenotransplantation of human AML cells in NSG mice faithfully recapitulates human disease since xenogeneic leukemic cells: (i) retain the phenotypic, genetic and functional leukemia-initiating properties of the corresponding pre-transplant AML samples; (ii) follow disease kinetics and mortality induction in mice according to molecular risk groups established in humans; and (iii) importantly, can persist in animals over several months at undetectable levels without losing disease-initiating properties, thus mimicking the clinical course of AML in humans. Mechanistically, the longer time to detectable engraftment observed with some AML (e.g. favorable molecular risk) may be due to lower numbers of transplanted LIC or, alternatively, to qualitative alterations in LIC resulting in lower homing to the BM and/or reduced growth rates in the mouse microenvironment. When applied to favorable-risk AML with inv(16), our model indicates that in this subtype leukemia initiation occurs from CD34+ blasts with enhanced expression of stem cell genes.
Supplementary Material
Acknowledgments
This study was supported by grants from the DFG (Deutsche Forschungsgemeinschaft, LE 2483/7-1) and SNF (Schweizerischer Nationalfonds, NMS1820) to CL. We thank Joëlle S. Müller for help with the isolation of mononuclear cells from patients’ samples and the animal and FACS core facilities of the Department of Biomedicine at the University Hospital Basel.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/5/854
References
- 1.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–648. [DOI] [PubMed] [Google Scholar]
- 2.Meyer LH, Debatin KM. Diversity of human leukemia xenograft mouse models: implications for disease biology. Cancer Res. 2011;71(23):7141–7144. [DOI] [PubMed] [Google Scholar]
- 3.Theocharides AP, Rongvaux A, Fritsch K, Flavell RA, Manz MG. Humanized hematolymphoid system mice. Haematologica. 2016;101(1):5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17(9):1086–1093. [DOI] [PubMed] [Google Scholar]
- 5.McDermott SP, Eppert K, Lechman ER, Doedens M, Dick JE. Comparison of human cord blood engraftment between immunocompromised mouse strains. Blood. 2010;116(2):193–200. [DOI] [PubMed] [Google Scholar]
- 6.Sanchez PV, Perry RL, Sarry JE, et al. A robust xenotransplantation model for acute myeloid leukemia. Leukemia. 2009;23(11): 2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rombouts WJ, Blokland I, Löwenberg B, Ploemacher RE. Biological characteristics and prognosis of adult acute myeloid leukemia with internal tandem duplications in the Flt3 gene. Leukemia. 2000;14(4):675–683. [DOI] [PubMed] [Google Scholar]
- 8.Mrózek K, Marcucci G, Nicolet D, et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol. 2012;30(36):4515–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rombouts WJ, Martens AC, Ploemacher RE. Identification of variables determining the engraftment potential of human acute myeloid leukemia in the immunodeficient NOD/SCID human chimera model. Leukemia. 2000;14(5):889–897. [DOI] [PubMed] [Google Scholar]
- 10.Rongvaux A, Takizawa H, Strowig T, et al. Human hematolymphoid system mice: current use and future potential for medicine. Annu Rev Immunol. 2013;31:635–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manz MG. Human-hematolymphoid-system mice: opportunities and challenges. Immunity. 2007;26(5):537–541. [DOI] [PubMed] [Google Scholar]
- 12.Ellegast JM, Rauch PJ, Kovtonyuk LV, et al. inv(16) and NPM1mut AMLs engraft human cytokine knock-in mice. Blood. 2016;128(17):2130–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Estey EH. Treatment of relapsed and refractory acute myelogenous leukemia. Leukemia. 2000;14(3):476–479. [DOI] [PubMed] [Google Scholar]
- 14.Breems DA, Van Putten WL, Huijgens PC, et al. Prognostic index for adult patients with acute myeloid leukemia in first relapse. J Clin Oncol. 2005;23(9):1969–1978. [DOI] [PubMed] [Google Scholar]
- 15.Mazurier F, Doedens M, Gan OI, Dick JE. Rapid myeloerythroid repopulation after intrafemoral transplantation of NOD-SCID mice reveals a new class of human stem cells. Nat Med. 2003;9(7):959–963. [DOI] [PubMed] [Google Scholar]
- 16.Jacobsen KR, Kalliokoski O, Teilmann AC, Hau J, Abelson KS. Postsurgical food and water consumption, fecal corticosterone metabolites, and behavior assessment as noninvasive measures of pain in vasectomized BALB/c mice. J Am Assoc Lab Anim Sci. 2012;51(1):69–75. [PMC free article] [PubMed] [Google Scholar]
- 17.Konantz M, André MC, Ebinger M, et al. EVI-1 modulates leukemogenic potential and apoptosis sensitivity in human acute lymphoblastic leukemia. Leukemia. 2013;27(1):56–65. [DOI] [PubMed] [Google Scholar]
- 18.Kunder S, Calzada-Wack J, Hölzlwimmer G, et al. A comprehensive antibody panel for immunohistochemical analysis of formalin-fixed, paraffin-embedded hematopoietic neoplasms of mice: analysis of mouse specific and human antibodies cross-reactive with murine tissue. Toxicol Pathol. 2007;35(3):366–375. [DOI] [PubMed] [Google Scholar]
- 19.Taussig DC, Vargaftig J, Miraki-Moud F, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(−) fraction. Blood. 2010;115(10): 1976–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C, Sashida G, Saraya A, et al. Depletion of Sf3b1 impairs proliferative capacity of hematopoietic stem cells but is not sufficient to induce myelodysplasia. Blood. 2014;123(21):3336–3343. [DOI] [PubMed] [Google Scholar]
- 21.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. [DOI] [PubMed] [Google Scholar]
- 22.Pearce DJ, Taussig D, Zibara K, et al. AML engraftment in the NOD/SCID assay reflects the outcome of AML: implications for our understanding of the heterogeneity of AML. Blood. 2006;107(3):1166–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ailles LE, Gerhard B, Kawagoe H, Hogge DE. Growth characteristics of acute myel-ogenous leukemia progenitors that initiate malignant hematopoiesis in nonobese diabetic/severe combined immunodeficient mice. Blood. 1999;94(5):1761–1772. [PubMed] [Google Scholar]
- 24.Grimwade D, Enver T. Acute promyelocytic leukemia: where does it stem from? Leukemia. 2004;18(3):375–384. [DOI] [PubMed] [Google Scholar]
- 25.Vargaftig J, Taussig DC, Griessinger E, et al. Frequency of leukemic initiating cells does not depend on the xenotransplantation model used. Leukemia. 2012;26(4):858–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klco JM, Spencer DH, Miller CA, et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell. 2014;25(3):379–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quek L, Otto GW, Garnett C, et al. Genetically distinct leukemic stem cells in human CD34− acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. J Exp Med. 2016;213(8):1513–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rongvaux A, Willinger T, Martinek J, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol. 2014;32(4):364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cosgun KN, Rahmig S, Mende N, et al. Kit regulates HSC engraftment across the human-mouse species barrier. Cell Stem Cell. 2014;15(2):227–238. [DOI] [PubMed] [Google Scholar]
- 30.Feuring-Buske M, Gerhard B, Cashman J, Humphries RK, Eaves CJ, Hogge DE. Improved engraftment of human acute myeloid leukemia progenitor cells in beta 2-microglobulin-deficient NOD/SCID mice and in NOD/SCID mice transgenic for human growth factors. Leukemia. 2003;17(4):760–763. [DOI] [PubMed] [Google Scholar]
- 31.Reinisch A, Thomas D, Corces MR, et al. A humanized bone marrow ossicle xenotransplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nat Med. 2016;22(7):812–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pabst C, Bergeron A, Lavallée VP, et al. GPR56 identifies primary human acute myeloid leukemia cells with high repopulating potential in vivo. Blood. 2016;127(16):2018–2027. [DOI] [PubMed] [Google Scholar]
- 33.Griessinger E, Anjos-Afonso F, Vargaftig J, et al. Frequency and dynamics of leukemia-initiating cells during short-term ex vivo culture informs outcomes in acute myeloid leukemia patients. Cancer Res. 2016;76(8): 2082–2086. [DOI] [PubMed] [Google Scholar]
- 34.Verhaak RG, Wouters BJ, Erpelinck CA, et al. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. 2009;94(1):131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cancelas JA, Williams DA. Stem cell mobilization by beta2-agonists. Nat Med. 2006;12(3):278–279. [DOI] [PubMed] [Google Scholar]
- 36.Lapidot T, Dar A, Kollet O. How do stem cells find their way home? Blood. 2005;106(6):1901–1910. [DOI] [PubMed] [Google Scholar]
- 37.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12(10):1167–1174. [DOI] [PubMed] [Google Scholar]
- 38.Suárez-Álvarez B, López-Vázquez A, López-Larrea C. Mobilization and homing of hematopoietic stem cells. Adv Exp Med Biol. 2012;741:152–170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.