

In 2008, the World Health Organization (WHO) recognized the BCR-ABL1 negative myeloproliferative neoplasm (MPN) chronic neutrophilic leukemia (CNL) as a distinct entity based on the features of an increased white blood cell count (WBC) with a preponderance of neutrophils, no increase in blasts, no dysplasia, and the exclusion of other clonal or non-clonal etiologies.1 More recently, it was observed that CSF3R is recurrently mutated in patients with CNL,2 which has led to the addition of this finding to the diagnostic criteria for CNL in the most recent 2016 WHO classification of myeloid neoplasms.3 CSF3R mutations have therapuetic implications; membrane proximal mutations result in activated signaling through the JAK-STAT pathway, and anectodal reports suggest the JAK1/2 inhibitor ruxolitinib may have efficacy in this setting.2,4,5
Here, we report the case of a 69-year-old CNL patient treated with ruxolitinib. He had no significant past medical history and, four months prior to presentation, developed increasing fatigue, abdominal distention, and easy bruising. He was noted to have a WBC of 100.1×109/L with an absolute neutrophil count (ANC) of 93.1×109/L, a hemoglobin of 10.0 g/dL, and a platelet count of 102×109/L (Figure 1). His peripheral blood smear showed marked leukocytosis, predominately neutrophils (Figure 2a). On physical examination, small areas of ecchymosis on the trunk and back, and significant splenomegaly, palpated 10 cm below the costal margin, were noted.
Figure 1.
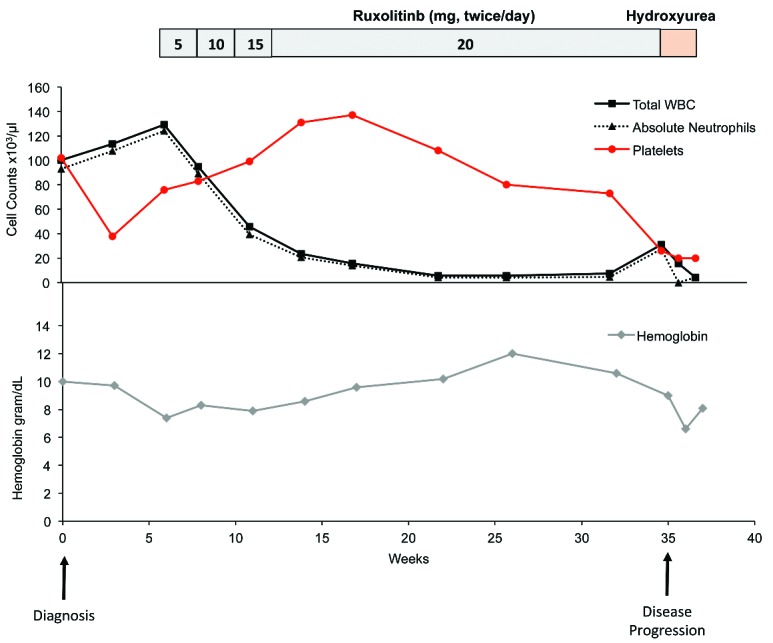
Graphical analysis of white blood cell count, absolute neutrophil count, platelets and hemoglobin at diagnosis, with escalating doses of ruxolitinib, and at the time of progression. Ruxolitinib resulted in a rapid and significant reduction in white blood cells (109/L), absolute neutrophil count (109/L) as well as improvement in platelets (109/L) and stabilization of hemoglobin (g/dL). At disease progression there was an increase in the white blood cells and neutrophils, mitigated by hydroxyurea as well as a decline in platelets and hemoglobin.
Figure 2.

Peripheral blood smear and bone marrow aspirate and biopsy for the patient at the time of diagnosis. The peripheral smear (A, 600×) is consistent with leukocytosis with a left shift, characterized by a prominent increase in mature neutrophils. This was in accordance with the aspirate smear (B, 600×), which revelated no overt dysplasia or increase in blasts, with mild reticulin fibrosis. The core biopsy specimen (C, 200×) is markedly hypercellular (>95%) for age, and is packed with sheets of left shifted myeloid forms.
A bone marrow examination revealed a markedly hypercellular marrow (>95%) with prominent left shifted granulopoiesis. Dysplasia was not noted, and blasts accounted for 1.5% of the bone marrow aspirate; mild reticulin fibrosis was observed (Figure 2b). Cytogenetic studies revealed a 46,Y,t(X;7)(p22.1;q36) in all 20 metaphases examined, believed to be a constitutional finding in this patient. Fluorescence in situ hybridization for the BCR/ABL1 rearrangement was negative. Next generation sequencing of DNA from the bone marrow aspirate using the TruSight myeloid sequencing panel with 568 amplicons interrogating 54 genes was performed with genomic DNA isolated using the Promega Maxwell® RSC Blood DNA Kit (Promega, Madison, WI) according to the manufacturer’s instructions. DNA quantitation was performed using NanoDrop (Thermal Fisher Scientific, Waltham, MA). 100 micrograms of DNA were processed using the TruSight Myeloid Sequencing Panel (Illumina, San Diego, CA), and the normalized libraries were pooled and loaded on a MiSeq for sequencing using the 600-cycleMiSeq Reagent Kit v3 to generate 2×250 read lengths. Mutations in SETBP1 (c.G2608A; p.G870S) and CSF3R (c.C1853T; p.T618I) were observed, and a diagnosis of CNL was made.
The patient initiated ruxolitinib 5 mg twice daily, and after two weeks experienced decreased fatigue and abdominal fullness; the ruxolitinib was rapidly titrated to 20 mg twice daily over a six-week period. After four months of treatment, the patient had stabilization of the blood counts, with a WBC of 5.5×109/L, hemoglobin 10.2 g/dL and platelets of 137×109/L (Figure 1), he no longer had a palpable spleen and, clinically, had returned to his usual baseline with respect to energy and activity tolerance. He declined a follow up bone marrow examination to formally assess his response. The ruxolitinib was continued.
Nine months after initiating ruxolitinib, he developed a rapid increase in fatigue and abdominal distention. His WBC increased to 31.2 ×109/L, and the hemoglobin and platelets dropped to 8.3 g/dL and 26 ×109/L, respectively, with an ANC of 27.4 ×109/L and 2% blasts in the peripheral blood. (Figure 1). The spleen was again palpable, now 8 cm below the costal margin, and a computed tomography scan confirmed splenomegaly (24.5 cm in length). A bone marrow examination revealed a hypercellular (>95%) marrow, with prominent neutrophils that still met diagnostic criteria for CNL; the only new finding was a small increase in the blast percentage, from 1.5% to about 5% of marrow cells. Cytogenetic findings were unchanged. The molecular panel was repeated, and three additional mutations were detected: KIT (c.2447A>T; p.D816V), GATA2 (c.1176_1177insA; p.K392fs), and RUNX1 (c.1009_1022del; p.337_341del); the prior mutations in CSF3R and SETBP1 were no longer observed at the clinically reportable level of detection (~7% allele frequency). Given the newly detected KIT mutation, the biopsy was reviewed carefully for the presence of an increased population of mast cells and no evidence of increased or atypical mast cells was observed.
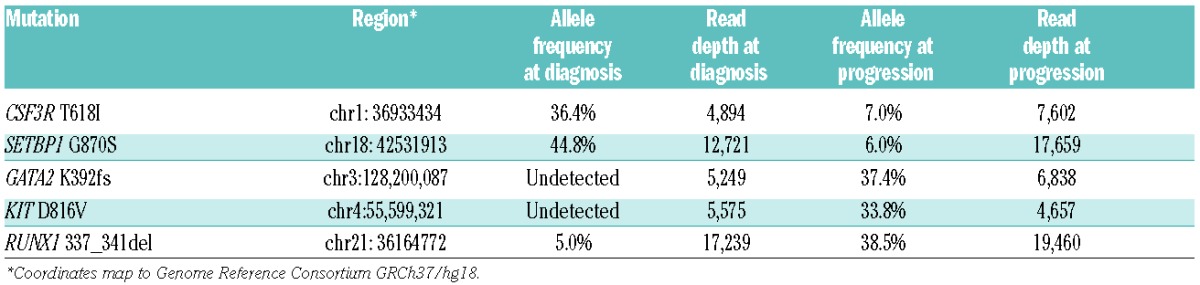
Re-analysis of the sequencing data to investigate mutations at or below the 7% clinically reportable cutoff did reveal evidence of the CSF3R and SETBP1 mutations, at 7% and 6%, respectively (Table 1). In addition, re-analysis of the sequencing data was also performed on the original bone marrow aspirate, revealing the sub-clinical presence of the RUNX1 mutation that was not originally detected, but no evidence for the presence of the GATA2 or KIT mutations that were subsequently noted (Table 1).
Table 1.
Gene mutations and allele frequencies with read depths at diagnosis and progression.
The patient was admitted for respiratory failure from sepsis, developed multi-organ failure and expired.
The clinical experience with ruxolitinib in CSF3R-mutated myeloproliferative neoplasms is limited, but anectodal reports suggest it can be active,2,4,5 and this is the focus of an ongoing clinical trial (clinicaltrials.gov Identifier: 02092324). Ninety percent of CNL patients, and about 40% of atypical chronic myeloid leukemia (aCML) patients have mutations in CSF3R; membrane proximal mutations activate JAK/STAT signaling,2 suggesting the JAK1/2 inhibitor ruxolitinib may be active, supported by pre-clinical studies.6
At diagnosis, our patient had clinically detectable mutations in CSF3R and SETBP1, among the most commonly mutated genes in BCR-ABL1 negative myeloproliferative neoplasms.2,7 Retrospectively, the presence of the RUNX1 mutation, at low levels, was appreciated. His clinical and laboratory response with ruxolitinib ended abruptly; repeated genomic analysis at the time of disease progression revealed the development of new mutations in GATA2 and KIT, neither of which were detected at the time of diagnosis. The development of these mutations, and/or the increased allele burden of RUNX1, were possible contributors to ruxolitinib resistance and subsequent disease progression. KIT is involved in activated signaling,8 while GATA2 and RUNX1 are myeloid transcription factors; in addition, RUNX1 has been identified as a driver mutation in the evolution of a myeloproliferative neoplasm to acute myeloid leukemia.9 The proportional decreased allele frequenices for CSF3R and SETBP1 after treatment with ruxolitnib (Table 1) suggest that a single sub-clone harbored these mutations and was suscepitble to inhibition of the JAK/STAT signaling pathway, as was predicted. Although a previous case report did not show a reduction in CSF3R-T618I allele frequency in a patient who clinically responded to ruxolitinib,5 it is possible that depth of responses may vary, and that this patient experienced a deeper response.
In conclusion, we report here for the first time changes in the allele frequencies of CSF3R-T618I and SETBP1-G870S with response to ruxolitnib as well as insights into the clonal evolution of CNL under selective pressure from ruxolitinib. Ruxolitinib has activity in CSF3R-mutated myeloproliferative neoplasms, and can decrease the allele frequency of this mutation and others within a sub-clone. Although from this case we cannot state definitively whether disease progression was independent of treatment or due to selective pressure from ruxolitinib, we show that clonal evolution in CNL can occur and can be explained by the acqusition of driver gene mutations. Similar studies with more CNL patients undergoing treatment with targeted therapies are warranted to further understand this process.
Supplementary Material
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Swerdlow SH, Harris NL, Jaffe ES, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, 2008. [Google Scholar]
- 2.Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. [DOI] [PubMed] [Google Scholar]
- 4.Stahl M, Xu ML, Steensma DP, Rampal R, Much M, Zeidan AM. Clinical response to ruxolitinib in CSF3R T618-mutated chronic neutrophilic leukemia. Ann Hematol. 2016;95(7):1197–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dao KH, Solti MB, Maxson JE, et al. Significant clinical response to JAK1/2 inhibition in a patient with CSF3R-T618I-positive atypical chronic myeloid leukemia. Leuk Res Rep. 2014;3(2):67–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fleischman AG, Maxson JE, Luty SB, et al. The CSF3R T618I mutation causes a lethal neutrophilic neoplasia in mice that is responsive to therapeutic JAK inhibition. Blood. 2013;122(22):3628–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meggendorfer M, Bacher U, Alpermann T, et al. SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations. Leukemia. 2013;27(9):1852–1860. [DOI] [PubMed] [Google Scholar]
- 8.Chaix A, Arcangeli ML, Lopez S, et al. KIT-D816V oncogenic activity is controlled by the juxtamembrane docking site Y568-Y570. Oncogene. 2014;33(7):872–881. [DOI] [PubMed] [Google Scholar]
- 9.Engle EK, Fisher DA, Miller CA, et al. Clonal evolution revealed by whole genome sequencing in a case of primary myelofibrosis transformed to secondary acute myeloid leukemia. Leukemia. 2015; 29(4):869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.