

Abstract
It is becoming clear that the manner by which the immune response resolves or contains infection by a pathogen varies according to the tissue that is affected. Unlike many peripheral cell types, CNS neurons are generally non-renewable. Thus, the cytolytic and inflammatory strategies that are effective in controlling infections in the periphery could be damaging if deployed in the CNS. Perhaps for this reason, the immune response to some CNS viral infections favours maintenance of neuronal integrity and non-neurolytic viral control. This modified immune response — when combined with the unique anatomy and physiology of the CNS — provides an ideal environment for the maintenance of viral genomes, including those of RNA viruses. Therefore, it is possible that such viruses can reactivate long after initial viral exposure, contributing to CNS disease.
An oversimplification that is promoted in much of the scientific literature is that extracellular, receptor-binding ligands — including viruses, cytokines and interferons (IFNs) — transduce invariant signalling pathways, independent of cell type. Such generalizations limit our ability to fully appreciate the complexity and diversity of the cellular response to pathogens and potent pathogen-fighting proteins. There are also clinical ramifications of this myopic view: for example, ignoring the possibility that a particular cell population may behave uniquely upon cytokine encounter could limit drug efficacy or hinder the development of therapeutics. In this Review, we discuss some recently defined neuron-specific immune responses that broaden our view of how CNS infections, especially those caused by RNA viruses, are controlled.
Intuitively, the notion that neurons differ immunologically from other cell types makes sense: we cannot tolerate the loss of these generally non-renewable cells, as we can the lysis of more-easily replaceable epithelial cells. For example, herpes simplex virus (HSV) infection of epithelial cells results in massive immune- and virus-mediated cell death1–3; however, the lost cells are readily replaced, as observed in the healing that follows a cold sore. If lysis of irreplaceable neurons occurred in the same manner, neural circuits could become compromised, and, depending on the magnitude of damage, the host could be permanently impaired. Thus, the immune response to a viral challenge must be tailored to promote survival of infected neurons but to destroy infected epithelial or endothelial cells. However, such neuronal sparing might result in long-term consequences that are spatially or temporally separated from acute infection.
In this Review, we integrate insights from the fields of virology, neurobiology and immunology to provide an overview of the mechanisms by which the restricted environment of the CNS is accessed by both RNA viruses and DNA viruses, and to explain how the host response contends with such infections. We particularly focus on a developing literature that elucidates cell-specific immunity and the consequences of non-lytic viral clearance within the brain. We conclude with a forward-looking hypothesis: non-lytic clearance of neuronal infections may allow for persistence of RNA viruses that induce pathogenesis long after primary exposure.
Viral entry and spread into the CNS
Viral entry
The brain is shielded from external threats at both macro-and microscopic levels: it is encased in bone, to prevent physical injury, and separated from peripheral tissues and blood via highly specialized barriers. Although such characteristics may limit infections of CNS-resident cells, these barriers can be breached. Three major routes of viral entry into the brain have been identified: direct infection of the cells that comprise the blood–brain barrier (BBB) and blood–cerebrospinal fluid (BCSF) barrier (with consequent release of viral particles into the parenchyma), infection of cells that are able to cross these barriers, and transneuronal migration across synapses from the peripheral nervous system (PNS) into the CNS (FIG. 1).
Figure 1. Viral entry into the CNS.
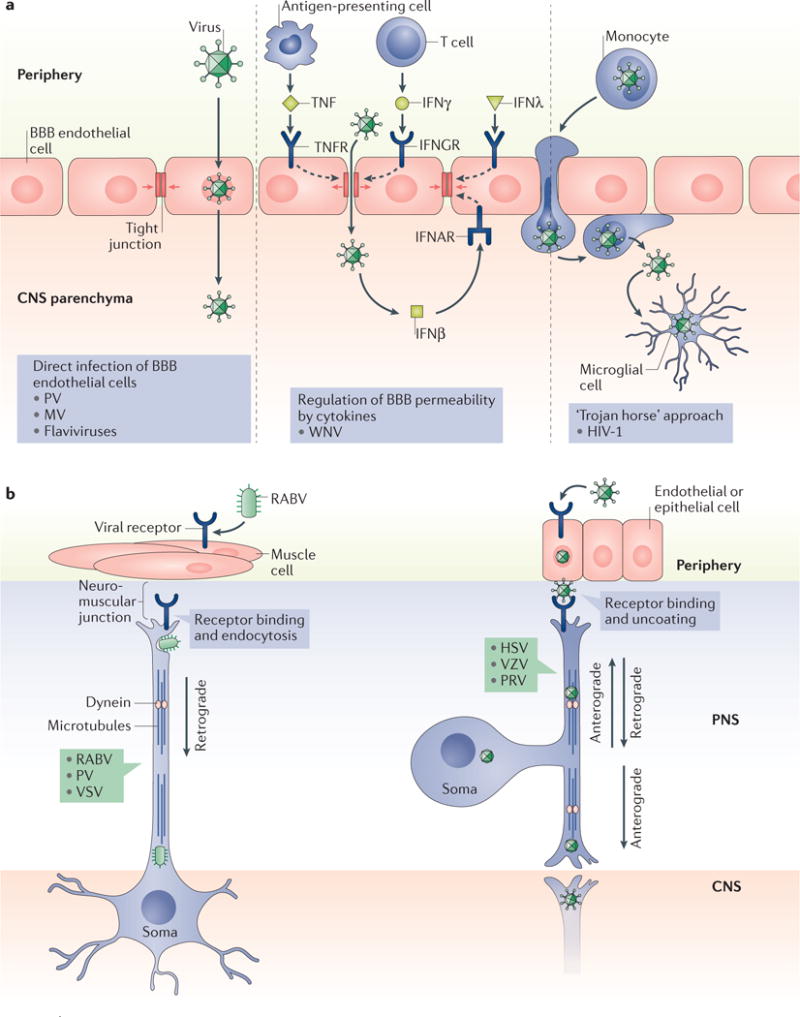
Three modes of viral entry into the brain are shown. a | Viruses may directly infect the cells comprising the blood–brain barrier (BBB), followed by release into the parenchymal space (left panel). Alternatively, viruses may diffuse across permeable regions of the BBB (middle panel). Of note, BBB permeability can be influenced by cytokines, such as tumour necrosis factor (TNF) and various interferons (IFNβ, IFNγ and IFNλ), which can loosen or reinforce the barrier integrity. In the ‘Trojan horse’ approach (right panel), infected lymphocytes or monocytes (including macrophages) traffic across the BBB or blood–cerebrospinal fluid barrier, releasing the virus once in the brain parenchyma. b | Trans-synaptic spread of viral particles involves the transport of viral genomes and associated proteins via microtubules and molecular motors. The left panel shows the movement of rabies virus (RABV) from the muscle, across the neuromuscular junction, and the dynein-mediated retrograde transport of this virus into the CNS. In the right panel, the transport of viruses (including herpes simplex virus (HSV), varicella zoster virus (VZV) and pseudorabies virus (PRV)) occurs across the epithelial or endothelial–neuron junction. In these neurons, retrograde transport brings the virus to the neuronal soma, and anterograde transport delivers the virus to the peripheral nervous system (PNS)–CNS synaptic junction. IFNAR, IFN α/β receptor; IFNGR, IFNγ receptor; HIV-1, human immunodeficiency virus type 1; MV, measles virus; PV, poliovirus; TNFR, TNF receptor; WNV, West Nile virus. Part a is adapted with permission from REF.9, PLoS. Part b is adapted with permission from REF.4, Elsevier.
Within the CNS, the BBB and BCSF barrier restrict the migration and diffusion of cells, pathogens, antibodies and macromolecules into the brain parenchyma. Neurotropic RNA viruses, including poliovirus (PV), measles virus (MV) and some flaviviruses, can circumvent these barriers by directly infecting the tightly associated endothelial or epithelial cells that comprise them4. Viral particles can then be released from the basolateral membrane into the parenchyma. For example, after MV infection of human brain microvascular endothelial cells, release of viral particles occurs from both the apical and basolateral membranes, without disrupting cell polarity or barrier integrity, allowing MV to spread into the parenchyma5. Alternatively, barrier integrity may be compromised when the tight junctions between cells loosen as a result of inflammation and cytokine exposure, allowing free viral particles to diffuse directly from the blood or CSF into the brain. For example, peripheral West Nile virus (WNV) infection acts through the engagement of Toll-like receptor 3 (TLR3) to induce the synthesis of cytokines — including tumour necrosis factor (TNF) — by circulating antigen-presenting cells6. In turn, TNF reduces BBB integrity by loosening tight junctions7, allowing for WNV migration through the less-restrictive BBB. In reality, however, modulating the barrier integrity is not as simple as this description implies. The balance of different cytokines can determine the extent to which the BBB is perturbed or stabilized. For example, IFNs, which are also produced in infected hosts, help to keep the barrier intact8; thus, the relative type and ratios of cytokines that are synthesized in response to various infections will differentially affect barrier integrity9.
Viruses may also passively access resident CNS cells by infecting lymphocytes or monocytes that can be transported across a cellular barrier. This strategy is often referred to as the ‘Trojan horse’ approach, because viral particles are released once the blood cell gains access to the parenchyma. A classic example of this mode of invasion is provided by the human immunodeficiency virus type 1 (HIV-1): CD16+ monocytes, permissive for HIV-1, traffic across the BBB and release virions that can then infect CNS microglia10,11.
A third mode of CNS entry is transneuronal migration, a strategy adopted by rabies virus (RABV) and many herpesviruses, including pseudorabies virus (PRV). Intracellular trafficking in PNS neurons, which is necessary to shuttle cellular components to and from the synapse, can be commandeered to facilitate viral travel within and among synaptically connected neurons. The best-characterized examples of this type of spread are provided by herpesvirus members such as HSV type 1 (HSV-1) and the closely related PRV4,12. After infection of epithelial cells in the oral mucosa, HSV-1 spreads to sensory and autonomic ganglia, establishing lifelong latency. Reactivation of the virus from latency — in response to decreases in immune monitoring, other infections or stress — leads to an active infection in PNS neurons, in which viral membrane proteins (including US9, glycoprotein E and glycoprotein I) can direct movement of newly replicated viral particles in an anterograde manner13. During transport, viral components are shuttled along axons via microtubule tracks and in association with their dynein and kinesin motors14,15. Sensory neurons have a pseudo-unipolar morphology in which one axon is in contact with epithelial cells and the other synapses are in contact with CNS neurons12. Beyond the value of these studies to understand how neurotropic viruses are propagated, viruses that spread across synapses (including RABV and MV) have provided a valuable method to trace neural circuits in vivo16,17; that is, the use of recombinant viruses encoding fluorescent proteins. These unique virological tools may also allow the development of strategies to deliver therapeutic payloads from the periphery to the CNS.
Viral spread
Once a virus has infected a neuron, there are two primary modes of subsequent spread to other cells: the release of infectious viral particles that can infect distant permissive cells or the transfer of viral nucleic acid, subviral particles or infectious virions between infected and uninfected cells that are in direct contact. The former mechanism requires the release of viral particles through the neuronal membrane (chiefly, via budding out of the infected cell), whereas the latter mechanism is primarily dependent on viral proteins that mimic or co-opt cellular processes to direct the insertion of viral fusion proteins into a host cell membrane or to direct the spread of viral capsids, as seen with HSV12,18. Both modes of viral spread occur in neurons; however, in most instances, viral transfer to adjacent neurons happens in the absence of syncytia formation (BOX 1), and little or no amount of extracellular infectious virus is detected, suggesting that neurons facilitate a distinct mode of spread for many viruses4. Interestingly, trans-synaptic spread of MV within primary mouse hippocampal neurons occurs independently of known MV receptors, which are crucial for syncytia formation in non-neuronal cells19,20. The paucity of viral particles in the extracellular space may protect the neuron from plasma membrane damage via budding and facilitate evasion of antibody detection. Although many neurotropic infections spread by direct contact at the presynaptic–postsynaptic junction, alternative modes of transport may also be used. For example, although RABV primarily spreads trans-synaptically in a retrograde manner, an electron microscopy study showed the presence of viral particles in the extracellular neuronal space, accompanied by direct neuronal budding21.
Box 1. Syncytia formation and trans-synaptic spread.
Viruses gain entry into permissive cells through an interaction between virally encoded glycoproteins, which are expressed on the outer surface of the virus particle, and cellular receptors. Entry can be achieved through endocytosis into vesicles or via membrane fusion114. For fusogenic viruses, exit from the cell occurs either via budding of virus particles through the plasma membrane or via fusion of an infected cell with an adjacent, uninfected cell114. The latter process results in the formation of multinucleated cells, or syncytia. The formation of syncytia may support further viral production but irrevocably leads to the death of the fused cells. Similarly, release of infectious particles by budding often leads to the death of the infected cell115.
However, viruses that are considered cytopathic in renewable cell types — including measles virus (MV), rabies virus and pseudorabies virus — can switch to a non-productive, non-syncytia-forming mode of spread when infecting neurons, promoting neuronal survival12,19,20,116,117. Often, this is correlated with absence of detectable extracellular viral particles. The spread of these viruses within neurons is primarily trans-synaptic, although the neuronal processes that enable a switch from viral budding and syncytia formation to non-cytolytic, trans-synaptic spread are not yet defined.
At least two possibilities might explain the viral movement across the synapse. In the first scenario, the spread of viral particles between neurons requires ligand–receptor interactions, similar to infection in non-neuronal cells. Directed transport to the synapse and focal fusion at the synaptic cleft might be required for a virus to migrate across the synapse: thus, the process that occurs in non-neuronal cells might also be operative in neurons. Trans-synaptic spread might require the same cellular and viral proteins that allow for fusion of non-neuronal cells or may be unique to the presynaptic–postsynaptic interface. For example, in MV neuronal infection, expression of the primary receptors that are used in non-neuronal cell infection is not required; however, a fusion event is still necessary for the spread to occur, perhaps, by forming a ‘pore’ through which the viral ribonucleic acid is transported118.
In the second scenario, the close approximation between the presynaptic and postsynaptic membrane, coupled with the unique attributes of the synaptic junction, may allow for the passive transport of viruses that have trafficked or assembled there. The release of neurotransmitters and uptake of their receptors make the synaptic interface particularly fluid, which might make it uniquely able to support receptor-independent trafficking.
Defining long-lasting infections
An outcome of viral neuroinvasion is that the viral genome, viral proteins and/or complete virus particles may remain in the brain long after initial exposure. To describe the myriad ways by which viruses establish enduring interactions with host neurons, numerous descriptors have been used, including ‘prolonged’, ‘persistent’, ‘latent’, ‘smouldering’, ‘quiescent’ and ‘chronic’ (REFS 4,22,23); however, their use is not consistent. Variables — including detection threshold, target organs and cell-specific influences on the viral life cycle — collectively contribute to the challenge of establishing an agreed-upon nomenclature. Moreover, some viruses can reactivate to cause the same disease as the acute infection (such as HSV), whereas others manifest differently upon reactivation (such as varicella zoster virus (VZV), which causes chicken pox as a primary infection but typically causes shingles upon reactivation). Other viruses result in pathogenesis only after protracted infection (such as tumour-causing viruses).
For this Review, we use three classifications. Latent infections are defined as those in which the virus establishes a non-lytic state during which host-to-host transmission is not possible, unless the virus reactivates to produce infectious virions. Chronic transmissible infections are characterized by the continuous production of infectious viral progeny and their ability to be transferred to new hosts. Chronic non-transmissible infections are those in which consistent detection of viral nucleic acid over extended periods of time is observed but in which transmission to new hosts does not occur.
Latency is most frequently attributed to herpesvirus infections, such as HSV-1, HSV-2 and VZV. After initial infection of epithelial cells, these viruses become non-lytic within PNS neurons, and viral nucleic acid is maintained in a heterochromatin episomal state with negligible transcription12. A small number of viral transcripts are synthesized during latency and are termed latency-associated transcripts. These RNA species do not encode functional proteins but are thought to prevent neuronal apoptosis and to disrupt both innate and adaptive immune signalling through mechanisms that include inhibition of caspase activity and of granzyme B-mediated killing24,25. VZV also produces various proteins, including ORF63, that prevent neuronal apoptosis24. The term ‘latent’ accurately conveys the status of these viruses: hidden, incapable of transmission, but able to fully reactivate, spread, and be transmitted to a new host. Another type of latency, which is not typically seen in neurons, occurs after viral nucleic acid is reverse transcribed from RNA to DNA and then integrated into the host genome. This process is unique to retroviruses, such as HIV-1 (REF.26). In this type of latency, integrated viral genomic DNA becomes indistinguishable from host DNA, and viral genes can be epigenetically silenced or activated throughout the cell lifetime and passed on to daughter cells.
In a chronic transmissible infection, the infectious virus can be continuously recovered from the host and can be disseminated to new hosts, as in hepatitis B and hepatitis C. Mice infected with the lymphocytic choriomeningitis virus (LCMV) offer a well-characterized model of a chronic transmissible CNS infection. LCMV infection of newborn mice leads to a non-cytopathic chronic infection in almost every tissue. Infectious LCMV particles can be recovered from multiple organs throughout life and can be shed in the faeces or transmitted vertically to offspring23,27,28. Although most strains of mice survive LCMV infection with no overt pathogenic consequences, some studies reported learning and memory deficits in these chronically infected animals29, underscoring the potentially subtle effects of long-term infection on CNS function.
Chronic non-transmissible infections are also characterized by sustained viral replication or consistent detection of viral nucleic acid over extended periods of time, but further host dissemination is absent. One example may be the rare cases of MV CNS infection. Acute infection can, in some instances, lead to the development of neuropathogenic diseases, including subacute sclerosing panencephalitis (SSPE) and measles inclusion-body encephalitis. These uncommon neurological diseases often present months or years after viral exposure and are characterized either by negligible replication or by persistence of replication-competent nucleic acid in the CNS30–32. In both diseases, no viral dissemination to uninfected hosts has been reported. Determining whether the state of the virus that causes these sequelae is ‘latent’ or ‘chronic non-transmissible’ is difficult, owing to both the small number of clinical specimens available and the lack of small animal models that mimic SSPE disease31,33,34. In humans, it may be that neurological symptoms appear only once viral replication reaches a crucial threshold or when the virus infects a key site within the brain, exceeding the host’s capacity to control the infection. Alternatively, non-replicating MV genomes may be maintained for prolonged periods and reactivated later. Either way, the MV genome remains intact, in some form, long after control of the acute infection is achieved, in the absence of further viral dissemination.
A final point of clarification: not all neurotropic viruses are associated with long-lasting infections. Some viruses, such as the reovirus, can induce neuronal apoptosis through the induction of pro-apoptotic proteins such as BAX35,36. The reason why some infections lead to neuronal suicide, whereas others lead to a long (potentially unhappy) ‘marriage’ between the host and the virus, is a major focus in the field of neurovirology, and answering this question may lead to the discovery of virus-specific therapies to prevent or minimize infection-triggered neuropathology.
Immune clearance of neuronal infection
The various permutations of neurotropic viral infections pose unique challenges for the host, including detecting antigens within the CNS, enabling T lymphocytes to engage with neurons that express negligible levels of proteins that are typically present on target cells, and mitigating the risks of neuroinflammation and widespread loss of generally non-renewable neurons.
Type I interferon signalling
The early response to an infection typically begins with the engagement of pathogenic motifs by pattern recognition receptors, which are expressed on (or in) virtually all cells. The binding of these receptors to conserved motifs, such as double-stranded RNA, lipopolysaccharides or glycoproteins, propagates signals that culminate in the production of type I IFNs, chiefly IFNα and IFNβ. These IFNs are secreted from the infected cell and act in both a paracrine and an autocrine manner by binding to IFN α/β receptor (IFNAR), a heterotetramer with phosphorylatable cytoplasmic domains. This engagement leads to the phosphorylation of tyrosine kinases (including Janus kinases) and the receptor itself, and is followed by tyrosine phosphorylation of cytoplasmic signal transducer and activator of transcription 1 (STAT1) and STAT2, which are usually abundant but inactive within the cytoplasm. Activated STAT1–STAT2 heterodimers couple with IFN regulatory factor 9 (IRF9) to form the complex termed ISGF3, which translocates into the nucleus and binds to IFN-stimulated response elements within the promoters of IFN-stimulated genes (ISGs). These genes encode proteins that eliminate infected cells or aid in viral clearance. Type I IFNs also bind to adjacent, uninfected cells to shield them from infection. Although this pathway is operative in many cells, alternative IFN-triggered pathways that limit viral spread but do not depend on induction of the ‘usual suspects’, the ISGs, can be induced in some cell types, including neurons37.
Neurons also secrete type I IFNs, which can act in an autocrine or paracrine manner on neurons or other parenchymal cell types38. RABV, which infects muscle cells and peripheral neurons after a bite from an infected animal, induces copious IFN secretion early after infection, in vivo and in vitro39. By contrast, IFN-induced STAT phosphorylation in primary hippocampal neurons is delayed, with maximal activation occurring only after ~24 hours40,41. Delayed STAT activation coincides with delayed expression of traditional ISGs41. The protracted interval between receptor binding and STAT activation may be due to a greatly reduced basal expression of STATs in these hippocampal neurons, as compared with other cell types40–42. Interestingly, lower homeostatic STAT expression is not unique to neurons but has also been observed in another non-renewable cell type, cardiac myocytes43. Like neurons, cardiac myocytes have high basal IFNβ expression, which may protect them from infection41,43. Perhaps, the disparity between expression of IFNs and the signal transduction molecules that they induce may skew towards protection from infection rather than towards induction of a potentially cytotoxic response. Surprisingly, synthesis of ISGs can differ within a single neuron: IFNβ induces a non-canonical, local antiviral response in axons that is not observed in the neuronal soma44,45. The startling implication of this finding is that neurons, especially those with long processes (as in the PNS), may ‘compartmentalize’ the response to extracellular immune mediators.
Although much of this Review focuses on neuronal responses to infections and antiviral cytokines, it is important to underscore that differential responses to, and production of, type I IFNs have been demonstrated in other parenchymal cell populations and may influence the neuronal response. For example, when comparing microglia and oligodendroglia collected from mice that were infected with a neurotropic strain of mouse hepatitis virus (MHV), it was shown that microglia are the main producers of type I IFN and downstream ISG products46. Overall, the fact that different cell types show distinct homeostatic expression of key signal transducers and of their downstream gene targets underscores the cellular diversity that can follow cytokine engagement.
Perhaps predictably, for many neurotropic RNA viruses, including MV, Theiler’s murine encephalomyelitis virus, Murray Valley encephalitis virus, WNV and others, experimentally induced loss of type I IFN signalling results in pathogenesis, altered viral tropism (generally accompanied by increased neurovirulence) and an inability to control viral spread both in vivo and in vitro41,47–54. Although most of these studies were performed using IFNAR-knockout mice lacking receptor expression on all cells, selective disruption of neuronal IFN signalling (using neuron-specific knockouts of IFNAR) also results in death after vesicular stomatitis virus infection55. Moreover, infection of olfactory neurons and mucosa with either a neurotropic RNA virus (vesicular stomatitis virus) or a neurotropic DNA virus (cytomegalovirus) leads to a robust type I IFN response deep within the brain, preventing viral spread and attendant disease56. Thus, infection of cells in direct contact with the environment (including sensory olfactory neurons) can trigger a long-distance warning (production of type I IFNs) that ultimately limits or precludes viral spread to remote regions of the brain.
Antigen presentation and CNS immunity
For some time, it was known that the primary cell populations of the adaptive immune system, T cells and B cells, contributed to viral control within the brain; however, the apparent absence of a CNS lymphatic drainage system made it complicated to understand how antigens could exit the parenchyma to promote the activation and proliferation of naive antigen-specific T cells7. Recent findings have begun to resolve this mystery: these include the identification of lymphatic drainage portals from the CNS into deep cervical lymph nodes and the presence of a fluid gradient that flushes the brain of extracellular proteins (which are termed ‘glymphatics’ because of the crucial role of glia in this process)57–59. CSF moves towards the perivascular space, where it is transported into the dense brain parenchyma via aquaporin 4 water channels that are expressed on cortical astrocytes. The CSF movement drives the interstitial fluid towards perivenous spaces, where it then drains towards the newly identified meningeal or dura mater lymphatic vessels, and ultimately to the deep cervical lymph nodes, where T cell activation and proliferation can occur57,59,60. These studies indicated how antigens and professional antigen-presenting cells can exit the CNS to alert immature T cells in the lymph nodes.
T cell-mediated pathogen clearance
After T cells mature in lymphoid tissues, they enter the bloodstream, where they can interact with adhesion molecules that are expressed on the surface of blood vessel endothelia within infected tissues. Mature T cells chiefly engage with selectins (and later, integrins) on the surface of the BBB or BCSF barrier. The expression of these adhesion molecules is induced by chemokines that are produced within the parenchyma by infected neurons and adjacent glia. This results in migration of T cells across the barrier (diapedesis). Although it was previously believed that neurons do not express major histocompatibility complex (MHC) class I molecules (and thus could not be recognized, at least in the canonical manner, by CD8+ T cells), we now know that some neuronal populations constitutively synthesize these cell surface proteins and that others can induce their expression after injury or infection61,62. Even so, most neurons do not express typical levels of MHC class I antigens under non-inflammatory conditions63, and thus T cell effector functions, including cytokine production, may not be triggered by the infected cell (the neuron) directly but rather by adjacent MHC class I-expressing cells (usually glia) that can display antigenic peptides via cross-presentation64. Although cross-presenting glia may not be directly infected, this strategy allows for elaboration of antiviral processes. Resident CNS cells may not only be invisible to immune cells as a result of reduced expression of MHC recognition molecules but may also express immunomodulatory molecules, such as programmed cell death ligand 1 (PDL1)65, that down-modulate T effector functions. Remarkably, the MHC class I expression system that is key to T cell recognition is likely to have also other functions in neurons, including those involved in neurodevelopment and neuronal plasticity66,67.
One of the major strategies that is used by activated T cells to combat neuronal viral infections is the production of IFNγ. Similar to type I IFNs, IFNγ transduces a signal via receptor binding, leading to STAT1 activation and homodimerization. Activated STAT1 homodimers translocate into the nucleus and bind to gamma-activated sequences (GASs) in the promoters of approximately 100 genes (which overlap with, but are generally distinct from, the ISGs that are induced by type I IFNs), promoting their transcription and translation (FIG. 2a). The products of these genes, similar to ISG proteins, combat viral infection or induce apoptosis of the infected cell37.
Figure 2. The receptor-occupancy hypothesis.
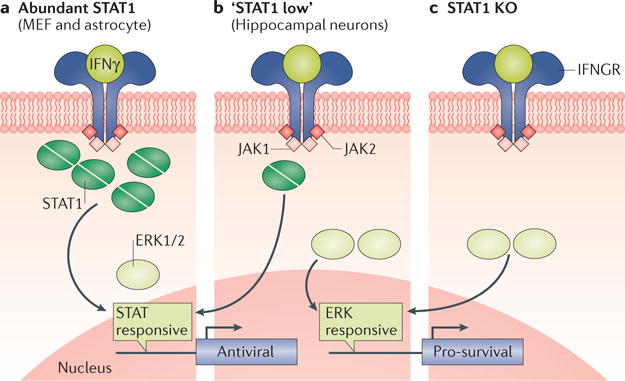
In cells with abundant levels of signal transducer and activator of transcription 1 (STAT1) signalling proteins, engagement of the interferon-γ (IFNγ) receptor (IFNGR) by its ligand transduces a primarily STAT1-driven cellular response, leading to activation of gene products that are chiefly antiviral (part a). By contrast, when a particular cell population (such as hippocampal neurons) expresses reduced homeostatic levels of STAT1 (part b) or when STAT1 is removed by genetic deletion (part c), alternative signalling molecules with an affinity to the IFNGR may bind to this receptor, transducing unique cellular responses. In the case of neurons, this includes activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2), which then can result in the induction of genes encoding pro-survival proteins. JAK1, Janus kinase 1; KO, knockout; MEF, mouse embryonic fibroblast.
STAT1 can be activated in neurons after IFNγ exposure, but the kinetics of induction are markedly slower than those observed in treated mouse embryonic fibroblasts, similar to the delayed response that is seen after type I IFN exposure40. In addition, IFNγ induces transcription of both traditional genes (that is, those that are typically expressed in response to IFNγ in other cellular populations) and non-traditional genes in primary hippocampal neurons after exposure68. This diverse profile of genes that are induced may affect the cellular outcome: although IFNγ can induce necroptosis, in neurons the virus is controlled in a non-cytolytic manner (presumably owing to the paucity of STAT1 and non-traditional GAS gene induction)69,70 (FIG. 2b). This feature is not unique to neurons: IFNγ is also essential for controlling MHV infection of oligodendrocytes via non-cytolytic pathways71,72. How are genes activated when basal levels of available STAT1 are low in resting neurons? Interestingly, when challenged with a neuron-restricted MV infection, most STAT1-knockout mice survive. By contrast, all IFNγ-knockout mice show severe signs of chronic disease, with approximately 50% succumbing to infection68,70, suggesting that the requirement for IFNγ is decoupled from the main transducer through which it signals. This observation led to the identification of an IFNγ-dependent, STAT1-independent activation of antiviral and pro-survival genes68,73, which might be facilitated by the access of other signalling factors — including extracellular signal-regulated kinases 1 and 2 (ERK1/2) and AKT — to the activated IFNγ receptor when STAT1 is absent or not abundant (FIG. 2b,c).
IFNγ is crucial for the control of multiple neurotropic viral infections in mice and primary neuronal cultures. Recently, IFNγ was identified as a key suppressor of HSV and VZV reactivation in the trigeminal ganglion of both humans and mice24,74–77. What makes these studies particularly intriguing is the type of T cell that is shown to be constitutively secreting IFNγ: T resident memory cells (Trm)74–79. Trm (defined by CD103 and CD69 expression) are in direct proximity to latently infected PNS neurons and do not re-enter circulation. Furthermore, these brain-resident lymphocytes have a unique molecular signature that distinguishes them from other types of cytotoxic T cells or from memory T cells80. Trm populations expand and contract in their resident tissue, acting as a first line of defence against reinfection81. Moreover, as we suggest below, these cells may be crucial sentinels that keep chronic neuronal infections at bay; therefore, their loss may contribute to viral reactivation.
In addition to cytokine secretion, some T lymphocytes kill infected cells through perforin- and/or granzyme-mediated mechanisms. Perforins, which are found in the lytic granules of CD8+ cytotoxic T cells, punch holes in the membrane of infected target cells, allowing for the delivery of granzymes that lead to lysis of the infected cell. Granzymes are serine proteases that induce caspase cleavage and activation of pro-apoptotic cellular proteins, such as BH3-interacting domain death agonist (BID). This mode of T cell-mediated killing, which efficiently eliminates ‘viral factories’, has been primarily studied in rapidly dividing cells. Interestingly, in some neuronal infections, the secretion of granzymes does not lead to lysis but rather aids in preventing viral reactivation and replication while sparing the infected neuron82. In addition to their ability to kill cells, granzymes can directly cleave eukaryotic translation initiation factor 4G3 (eIF4G3) (a cellular protein that is important for host and viral translation) and ICP4 (a herpesvirus-specific protein needed for the transcription of early and late viral genes)82,83. By cleaving eIF4G3, granzymes block viral translation but fail to induce neuronal apoptosis, further preventing viral dissemination within the host and sparing the infected neuron. Cleavage of ICP4 by granzymes directly prevents reactivation of latent HSV from infected neurons. In these instances, granzymes are acting on proteins other than their traditional protein targets to induce an alternative neuronal response.
It has also been speculated that viral RNAs and proteins can contribute to non-lytic outcomes. For instance, HSV latency-associated transcripts inhibit the action and expression of various caspase proteins, which are key mediators of the cell death process84. Nevertheless, in some cases, bystander immune-mediated neuronal death may occur. For example, Theiler’s murine encephalomyelitis virus infection of mice results in hippocampal neuron death through a mechanism that is dependent on inflammatory monocyte infiltration and activation85.
Humoral responses within the CNS
The notable absence of B cells in the brain of virus-infected mice led to the misperception that B cells and the antibodies that they secrete play a minor part in viral control. In fact, numerous human CNS infections, including those caused by MV, PV, VZV, HSV and flavi-viruses, are characterized by the presence of intrathecal antibodies in the CSF86–88. Humoral responses seem to be associated with protective rather than pathogenic functions, as observed for Japanese Encephalitis virus and some neurotropic retroviruses86. Antibodies may be particularly beneficial for those infections that result in extracellular infectious virus production.
Neuronal subtypes and infection
A central theme of this Review has been the notion that infected cells, such as neurons, respond to immune effectors in cell-specific ways. However, the existence of many subpopulations of neurons, which are segregated by location and function, raises the issue of whether responses may differ within these neuronal subsets. Recent studies showed that cerebellar granule neurons and cortical neurons pretreated with type I IFNs vary in their ability to control a WNV infection89. Type I IFN treatment had a much greater impact on the spread of infection in cerebellar granule neurons than it did in cortical neurons (100-fold versus 15-fold reduction), and this difference correlated with discrete patterns of ISG induction89. Animal model studies have also shown differences in the propensity for a virus to infect individual neuronal subpopulations and regions of the brain (FIG. 3); for example, the hippocampus is heavily infected by RABV, whereas MV is more often found in the midbrain90–95. Whether these distinctions can be attributed to differences in viral tropism or intrinsic variations in the neuronal response to soluble immune effector proteins (or, perhaps, to the way a virus gains access to the brain) is not known. Answering this question will require further studies that must necessarily integrate virology, immunology and neurobiology.
Figure 3. Tropism of neurotropic RNA viruses for distinct brain regions and neuronal subpopulations.
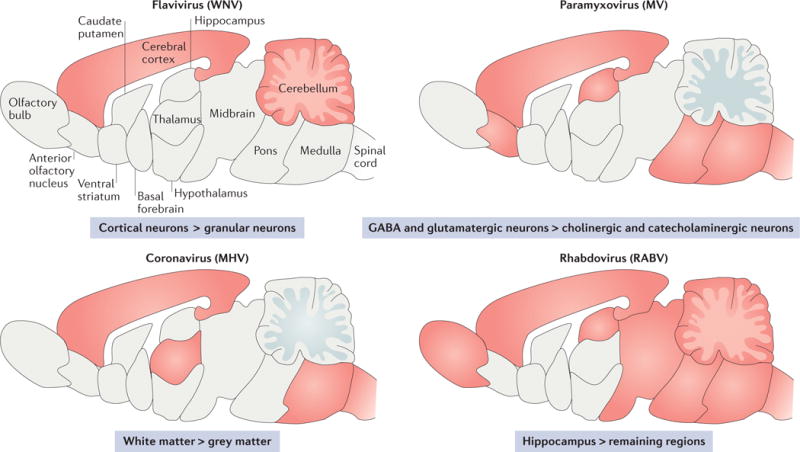
The schematics show a simplified sagittal view of the mouse brain with the regions that are known to be infected by various viruses indicated in red. The symbol ‘>‘ indicates higher propensity for a virus to infect a certain cell type or region of the brain than another cell type or region. MHV, mouse hepatitis virus; MV, measles virus; RABV, rabies virus; WNV, West Nile virus.
Emerging principles in neurovirology
Preservation of virus-challenged neurons from immune-mediated lysis seems to be advantageous to the host, but this leaves open the possibility of long-term viral maintenance in surviving neurons (TABLE 1). Previously, many researchers believed that neurotropic RNA viruses were sterilely cleared from the CNS. Indeed, unlike DNA viruses or retroviruses that can establish latent infections through episome formation or integration, RNA viruses do not have known means to ‘survive’ within a host cell. This is especially relevant given the lability of RNA within the cytoplasm, which arises owing to the inherently unstable ribose subunit and the susceptibility of the 2′ hydroxyl group to deprotonation. On the other hand, RNA viral genomes are unlikely to persist in the cytoplasm as naked RNA. Ribonucleoprotein complexes would provide some protection, and viral RNAs (like other cellular RNAs) may also be sequestered in stress granules. Thus, mechanisms must exist to protect RNA viral genomes, allowing for their long-term stability in the cytoplasm.
Table 1.
Evidence of long-term persistence of RNA viruses in CNS tissue
| Virus | Model | Species detected | Infectious? | Time after infection | Tissue | Method | Refs | ||
|---|---|---|---|---|---|---|---|---|---|
| RNA | mRNA | Protein | |||||||
| Measles | Human | Yes | Yes | Unknown | Unknown | Decades (lifespan) | Brain | PCR | 96,100 |
| Mouse hepatitis | Mouse | Yes | Yes | NT | NT | 10 months | Spinal cord and liver | In situ hybridization | 104 |
| Sendai | Mouse | Yes | Unknown | NT | Unknown | 423 days (lifespan) | Brain | Dot blot | 105 |
| Sindbis | Mouse | Yes | Unknown | NT | Yes | 17 months | Brain | PCR | 106 |
| Rabies | Mouse | Yes (genomic) | NT | Unknown | Unknown | 6 months | Brain | qRT-PCR | 95 |
NT, not tested; qRT-PCR, quantitative reverse transcription PCR.
Do these long-term infections have pathogenic potential? A set of studies from the late 1980s showed that MV RNA can persist in human brains for decades after resolution of the peripheral infection without causing neurological symptoms96–100; in these studies, organs from individuals who had died of non-viral, non-CNS-related causes were screened, and a high proportion of brain tissues were found to be MV RNA positive. In addition, some scientists have argued that MV entry into the human CNS may occur at a higher rate than previously thought101, although only a small fraction of acutely infected people will manifest neurological consequences. Accordingly, viral RNAs were generally considered ‘fossils’ that were unlikely to contribute to human disease. Surprisingly, autopsy studies performed on the brain of patients that succumbed to SSPE have shown regions of the brain with no detectable MV proteins, despite the presence of MV RNA, suggesting that RNA, even with its inherent instability, can be maintained in a translationally silent state102.
The long-term persistence of viral RNA in the CNS is not unique to MV. For example, infection of mice with the MHV strain A59, which is used to model the demyelinating disease multiple sclerosis, leads to encephalitis and hepatitis. The infectious virus is cleared from the liver and CNS in 20 days; however, the mice develop a progressive, immune-mediated demyelinating disease103, and viral nucleic acid persists104. The potential importance of viral nucleic acid persistence in demyelination has been subordinated by the prevailing view that long-term disease is caused by an overactivation of the host response towards myelin proteins. Other neurotropic RNA viruses that are known to persist within the mouse brain (sometimes for periods longer than 1 year post exposure) in the absence of detectable antigen or infectious viral progeny include Sindbis virus, Sendai virus and RABV95,105,106. However, the lack of recoverable infectious virus does not preclude the possibility that these viruses are actively suppressed in the CNS, similar to the control of neuronal herpesvirus infections by Trm. Could decreases in the magnitude or quality of the host response (for example, with ageing or after immunosuppressive therapy) lead to loss of resident memory cells and reactivation of viral replication that are temporally separated from the initial infection?
The short answer is that we do not yet know. However, it was recently shown that an endogenous retrovirus, which was integrated into the host genome millions of years ago, could contribute to human neurological disease. Amyotrophic lateral sclerosis is a progressive neurological disease of poorly understood aetiology that is characterized by consistent inflammatory response and immune-mediated pathogenesis. The expression of this human endogenous retrovirus, specifically the expression of the envelope protein, was proposed as a possible cause for the neuropathology that is seen in amyotrophic lateral sclerosis107.
Perspectives
Limits of detection, reproducibility, consistency in the brain regions that are analysed and patient-to-patient variability all contribute to the challenges and dangers of ascribing neurotropic infections to be the aetiologic causes of poorly understood CNS diseases. Moreover, the association of ‘new’ viruses with CNS disease (including Zika virus, which is linked to microcephaly108,109) or the emergence of more neurovirulent influenza strains110 are reminders that our understanding of the pathogenic consequences of CNS infections remain quite primitive. Translational studies have provided insights into the links between infections and disease but are not without controversy. For example, the prevalence of human cytomegalovirus in patients with glioblastoma has been hotly debated111, although anti-cytomegalovirus treatments lead to reduction in tumour burden in some patients111. Furthermore, losses in host immune status due to age or chemotherapy are well known to provoke disease, as observed with JC (John Cunningham) virus infection and progressive multifocal leukoencephalopathy112.
Whether CNS virus infections have a larger role in human diseases of unknown aetiology remains controversial. In support of this notion, CNS neurons may be an ideal harbour for long-term infections: non-lytic immune mechanisms spare neuronal loss while providing an avenue for a non-cytopathic virus to persist. Moreover, trans-synaptic spread is likely to enable viral escape from antibody recognition or phagocytosis by antigen-presenting cells. From an evolutionary perspective, neuronal survival is paramount; thus, sparing infected neurons a lytic fate may promote survival early on but could potentially open the door for viral reactivation later in life. We do not know if there are viruses that are typically found in CNS tissues of overtly healthy individuals; with the advent of the RNA sequencing technology, new RNA sequencing studies might shed light on the potential ‘virome’ within the brain of both asymptomatic individuals and people with neurological conditions.
One final point that is worth noting concerns the utility of mouse models (on which many of the studies cited in this Review were based) to study human CNS diseases. Scientists often make the mistake of assuming that mouse survival is equivalent to an absence of disease. This may mean that the long-term ramifications of acute virus infections, especially those of RNA viruses that are not generally considered to be lifelong, may be overlooked. However, we are increasingly becoming aware that the presence of viral fragments or latent viruses that can reactivate might evoke non-lethal pathogenic consequences resulting from either viral replication and cell damage or immune responses directed against viral antigens. Such pathogenic consequences, as seen with the learning defects in LCMV-infected mice, may be subtle. Consequently, the parallel development of more-precise approaches to assess CNS disease in mice, including tools to evaluate the impacts on learning, behaviour and memory, should refine how we describe neuropathogenesis in the many valuable mouse models that are currently in use. Finally, determining whether or not persistent viral nucleic acids detected within the brain are replication competent and how these viruses evade complete clearance could promote the development of novel antiviral therapies to treat or prevent devastating and prevalent human neurological and neurodegenerative diseases95,113.
Acknowledgments
The authors acknowledge L. Enquist, O. Koyuncu and C. Matullo for their input and contributions to this manuscript. They also gratefully acknowledge support from the F. M. Kirby Foundation.
Glossary
- Cytokines
Small proteins released by cells that affect cell signalling and act to regulate cell growth, maturation and effector functions
- Interferons
(IFNs). Signalling proteins that are released by cells in response to infection to promote an antiviral state
- RNA viruses
Viruses with genomic material that is composed of RNA rather than DNA. Genomic viral RNA can be double stranded, single stranded, positive sense or negative sense
- DNA viruses
Viruses with genomic material that is composed of DNA
- Virions
Complete forms of an infectious viral particle
- Permissive cells
Cells that actively express viral receptor proteins, thereby facilitating viral entry and infection
- Budding
The final step of viral release during which a virion gains its outer membrane by bursting through the host cell membrane
- Viral fusion proteins
Viral glycoproteins that are essential in mediating the virus–host interaction in which the viral membrane fuses with the host membrane releasing a virion into the host cell
- Syncytia
The result of infected cells fusing with adjacent uninfected cells, producing large, multinucleated clusters
- Cytotoxic T cells
A subset of T cells that are primed to kill target cells
- Memory T cells
A subset of T cells that have previously interacted with their cognate antigen
- Perforin
A protein that is stored by cytotoxic T cells and that creates holes in target cell membranes, allowing for the delivery of cytotoxic granzymes
- Stress granules
Dense aggregates of protein and RNA that are present in the cytoplasm and are typically associated with the endoplasmic reticulum
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. Recognition of herpesviruses by the innate immune system. Nat Rev Immunol. 2011;11:143–154. doi: 10.1038/nri2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones CA, et al. Herpes simplex virus type 2 induces rapid cell death and functional impairment of murine dendritic cells in vitro. J Virol. 2003;77:11139–11149. doi: 10.1128/JVI.77.20.11139-11149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braaten DC, Sparks-Thissen RL, Kreher S, Speck SH, Virgin HW. An optimized CD8+ T-cell response controls productive and latent gammaherpesvirus infection. J Virol. 2005;79:2573–2583. doi: 10.1128/JVI.79.4.2573-2583.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koyuncu OO, Hogue IB, Enquist LW. Virus infections in the nervous system. Cell Host Microbe. 2013;13:379–393. doi: 10.1016/j.chom.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dittmar S, et al. Measles virus-induced block of transendothelial migration of T lymphocytes and infection-mediated virus spread across endothelial cell barriers. J Virol. 2008;82:11273–11282. doi: 10.1128/JVI.00775-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang T, et al. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–1373. doi: 10.1038/nm1140. This paper shows that WNV-induced, TLR3-dependent cytokine production mediates loss of BBB permeability, facilitating viral entry into the CNS. [DOI] [PubMed] [Google Scholar]
- 7.Libbey J, Fujinami RS. Adaptive immune response to viral infections in the central nervous system. Hanb Clin Neurol. 2014;123:225–247. doi: 10.1016/B978-0-444-53488-0.00010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daniels BP, et al. Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. mBio. 2014;5:e01476–14. doi: 10.1128/mBio.01476-14. The authors demonstrate a unique duality between type I IFN signalling and activation in the brain, and suggest a role for cytokines that are produced peripherally by T cells in the regulation of BBB integrity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daniels BP, Klein RS. Knocking on closed doors: host interferons dynamically regulate blood-brain barrier function during viral infections of the central nervous system. PLoS Pathog. 2015;11:e1005096. doi: 10.1371/journal.ppat.1005096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGavern DB, Kang SS. Illuminating viral infections in the nervous system. Nature. 2011;11:318–329. doi: 10.1038/nri2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kramer-Hämmerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005;111:194–213. doi: 10.1016/j.virusres.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 12.Kramer T, Enquist L. Directional spread of alphaherpesviruses in the nervous system. Viruses. 2013;5:678–707. doi: 10.3390/v5020678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howard PW, Howard TL, Johnson DC. Herpes simplex virus membrane proteins gE/gI and US9 act cooperatively to promote transport of capsids and glycoproteins from neuron cell bodies into initial axon segments. J Virol. 2012;87:403–414. doi: 10.1128/JVI.02465-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kramer T, et al. Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe. 2012;12:806–814. doi: 10.1016/j.chom.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaichick SV, et al. The herpesvirus VP1/2 Protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe. 2013;13:193–203. doi: 10.1016/j.chom.2013.01.009. This paper shows that tegument proteins of the herpesvirus PRV tether viral capsids to dynein and dynactin to enhance microtubule transport, neuroinvasion and pathogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Granstedt AE, Brunton BW, Enquist LW. Imaging the transport dynamics of single alphaherpesvirus particles in intact peripheral nervous system explants from infected mice. mBio. 2013;4:e00358–13. doi: 10.1128/mBio.00358-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hagendorf N, Conzelmann K-K. Recombinant fluorescent rabies virus vectors for tracing neurons and synaptic connections. Cold Spring Harb Protoc. 2015 doi: 10.1101/pdb.top089391. http://dx.doi.org/10.1101/pdb.top089391. [DOI] [PubMed]
- 18.Flint J, Racaniello V, Rall GF, Skalka AM, Enquist LW. Principles of Virology. 4th. American Society for Microbiology; 2015. [Google Scholar]
- 19.Lawrence DMP, et al. Measles virus spread between neurons requires cell contact but not CD46 expression, syncytium formation, or extracellular virus production. J Virol. 2000;74:1908–1918. doi: 10.1128/jvi.74.4.1908-1918.2000. This article demonstrates that the spread of MV in neurons occurs via a receptor-independent mechanism and in the absence of the syncytia formation that is typically observed upon MV infection of non-neuronal cell types. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makhortova NR, et al. Neurokinin-1 enables measles virus trans-synaptic spread in neurons. Virology. 2007;362:235–244. doi: 10.1016/j.virol.2007.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwasaki Y, Ohtani S, Clark HF. Maturation of rabies virus by budding from neuronal cell membrane in suckling mouse brain. J Virol. 1975;15:1020–1023. doi: 10.1128/jvi.15.4.1020-1023.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78:5535–5545. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oldstone MBA. Anatomy of viral persistence. PLoS Pathog. 2009;5:e1000523. doi: 10.1371/journal.ppat.1000523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kinchington PR, Leger AJS, Guedon JMG, Hendricks RL. Herpes simplex virus and varicella zoster virus, the house guests who never leave. Herpesviridae. 2012;3:5. doi: 10.1186/2042-4280-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang X, et al. The herpes simplex virus type 1 latency-associated transcript can protect neuron-derived C1300 and Neuro2A cells from granzyme B-induced apoptosis and CD8 T-cell killing. J Virol. 2011;85:2325–2332. doi: 10.1128/JVI.01791-10. The authors demonstrate that the expression of latency-associated transcripts alone (which is observed during HSV latency) can potently reduce the lytic effects of granzyme B in neuronal cell lines. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohammadi P, Ciuffi A, Beerenwinkel N. Dynamic models of viral replication and latency. Curr Opin HIV AIDS. 2015;10:90–95. doi: 10.1097/COH.0000000000000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oldstone MBA. Viral persistence: parameters, mechanisms and future predictions. Virology. 2006;344:111–118. doi: 10.1016/j.virol.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 28.Traub E. Persistence of lymphocytic choriomeningitis virus in immune animals and its relation to immunity. J Exp Med. 1936;63:847–861. doi: 10.1084/jem.63.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brot MD, Rall GF, Oldstone MBA, Koob GF, Gold LH. Deficits in discriminated learning remain despite clearance of long-term persistent viral infection in mice. J Neurovirol. 1997;3:265–273. doi: 10.3109/13550289709029467. [DOI] [PubMed] [Google Scholar]
- 30.Norrby E, Kristensson K. Measles virus in the brain. Brain Res Bull. 1997;44:213–220. doi: 10.1016/s0361-9230(97)00139-1. [DOI] [PubMed] [Google Scholar]
- 31.Gutierrez J, Issacson RS, Koppel BS. Subacute sclerosing panencephalitis: an update. Dev Med Child Neurol. 2010;52:901–907. doi: 10.1111/j.1469-8749.2010.03717.x. [DOI] [PubMed] [Google Scholar]
- 32.Griffin DE. Measles virus and the nervous system. Handb Clin Neurol. 2014;123:577–590. doi: 10.1016/B978-0-444-53488-0.00027-4. [DOI] [PubMed] [Google Scholar]
- 33.Griffin DE, Lin WH, Pan CH. Measles virus, immune control, and persistence. FEMS Microbiol Rev. 2012;36:649–662. doi: 10.1111/j.1574-6976.2012.00330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayashi M, et al. Neurodegenerative mechanisms in subacute sclerosing panencephalitis. J Child Neurol. 2002;17:725–730. doi: 10.1177/08830738020170101101. [DOI] [PubMed] [Google Scholar]
- 35.Clarke P, Beckham JD, Leser JS, Hoyt CC, Tyler KL. Fas-mediated apoptotic signaling in the mouse brain following reovirus infection. J Virol. 2009;83:6161–6170. doi: 10.1128/JVI.02488-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berens HM, Tyler KL. The proapoptotic Bcl-2 protein Bax plays an important role in the pathogenesis of reovirus encephalitis. J Virol. 2011;85:3858–3871. doi: 10.1128/JVI.01958-10. This paper shows that the pro-apoptotic protein BAX is important for reovirus growth and pathogenesis in neurons and that it mediates its effects via release of mitochondrial cytochrome c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goodbourn S, Didcock L, Randall RE. Interferons: cell signalling, immune modulation, antiviral responses and virus countermeasures. J Gen Virol. 2000;81:2341–2364. doi: 10.1099/0022-1317-81-10-2341. [DOI] [PubMed] [Google Scholar]
- 38.Delhaye S, et al. Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci USA. 2006;103:7835–7840. doi: 10.1073/pnas.0602460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Faul EJ, et al. Rabies virus infection induces type I interferon production in an IPS-1 dependent manner while dendritic cell activation relies on IFNAR signaling. PLoS Pathog. 2010;6:e1001016. doi: 10.1371/journal.ppat.1001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rose RW, Vorobyeva AG, Skipworth JD, Nicolas E, Rall GF. Altered levels of STAT1 and STAT3 influence the neuronal response to interferon gamma. J Neuroimmunol. 2007;192:145–156. doi: 10.1016/j.jneuroim.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cavanaugh SE, Holmgren AM, Rall GF. Homeostatic interferon expression in neurons is sufficient for early control of viral infection. J Neuroimmunol. 2015;279:11–19. doi: 10.1016/j.jneuroim.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Podolsky MA, et al. Extended JAK activation and delayed STAT1 dephosphorylation contribute to the distinct signaling profile of CNS neurons exposed to interferon-gamma. J Neuroimmunol. 2012;251:33–38. doi: 10.1016/j.jneuroim.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zurney J, Howard KE, Sherry B. Basal expression levels of IFNAR and Jak-STAT components are determinants of cell-type-specific differences in cardiac antiviral responses. J Virol. 2007;81:13668–13680. doi: 10.1128/JVI.01172-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosato PC, Leib DA. Neuronal interferon signaling is required for protection against herpes simplex virus replication and pathogenesis. PLoS Pathog. 2015;11:e1005028. doi: 10.1371/journal.ppat.1005028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song R, et al. Two modes of the axonal interferon response limit alphaherpesvirus neuroinvasion. mBio. 2016;7:e02145–115. doi: 10.1128/mBio.02145-15. This paper shows that axons respond differentially to type I and type II IFNs: the response to type I IFNs is rapid and restricted to the axon, whereas the response to type II IFNs involves long-distance signalling to the PNS cell body. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kapil P, Butchi NB, Stohlman SA, Bergmann CC. Oligodendroglia are limited in type I interferon induction and responsiveness in vivo. Glia. 2012;60:1555–1566. doi: 10.1002/glia.22375. The authors show that oligodendrocytes are poor sensors of viral infection and require exogenous IFNα and IFNβ to establish an antiviral state. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paul S, Ricour C, Sommereyns C, Sorgeloos F, Michiels T. Type I interferon response in the central nervous system. Biochimie. 2007;89:770–778. doi: 10.1016/j.biochi.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 48.Holmgren AM, Miller KD, Cavanaugh SE, Rall GF. Bst2/tetherin is induced in neurons by type I interferon and viral infection but is dispensable for protection against neurotropic viral challenge. J Virol. 2015;89:11011–11018. doi: 10.1128/JVI.01745-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ireland DDC, Stohlman SA, Hinton DR, Atkinson R, Bergmann CC. Type I interferons are essential in controlling neurotropic coronavirus infection irrespective of functional CD8 T cells. J Virol. 2007;82:300–310. doi: 10.1128/JVI.01794-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weber E, et al. Type I Interferon protects mice from fatal neurotropic infection with langat virus by systemic and local antiviral responses. J Virol. 2014;88:12202–12212. doi: 10.1128/JVI.01215-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fensterl V, et al. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog. 2012;8:e1002712. doi: 10.1371/journal.ppat.1002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samuel MA, Diamond MS. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol. 2005;79:13350–13361. doi: 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lobigs M, Mullbacher A, Wang Y, Pavy M, Lee E. Role of type I and type II interferon responses in recovery from infection with an encephalitic flavivirus. J Gen Virol. 2003;84:567–572. doi: 10.1099/vir.0.18654-0. [DOI] [PubMed] [Google Scholar]
- 54.Nayak D, et al. Type I interferon programs innate myeloid dynamics and gene expression in the virally infected nervous system. PLoS Pathog. 2013;9:e1003395. doi: 10.1371/journal.ppat.1003395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Detje CN, et al. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J Immunol. 2009;182:2297–2304. doi: 10.4049/jimmunol.0800596. The authors illustrate the crucial role of neuron specific IFN signalling in controlling vesicular stomatitis virus infection within the brain. [DOI] [PubMed] [Google Scholar]
- 56.van den Pol AN, Ding S, Robek MD. Long-distance interferon signaling within the brain blocks virus spread. J Virol. 2014;88:3695–3704. doi: 10.1128/JVI.03509-13. This paper shows that IFNβ, which is released by infected neurons in the olfactory bulb, can induce ISGs in the posterior regions of the brain, activating an antiviral state and preventing further virus invasion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aspelund A, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212:991–999. doi: 10.1084/jem.20142290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jessen NA, Munk ASF, Lundgaard I, Nedergaard M. The glymphatic system: a beginner’s guide. Neurochem Res. 2015;40:2583–2599. doi: 10.1007/s11064-015-1581-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Louveau A, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–341. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iliff JJ, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neumann H, Cavalie A, Jenne DE, Wekerle H. Induction of MHC class I genes in neurons. Science. 2016;269:549–552. doi: 10.1126/science.7624779. This article demonstrates that transcription of MHC class I genes is rare in healthy neurons but detected in electrically silent neurons, suggesting that immunosurveillance by cytotoxic T cells may be focused on functionally impaired neurons. [DOI] [PubMed] [Google Scholar]
- 62.Neumann H, Schmidt H, Cavalie A, Jenne D, Wekerle H. Major histocompatibility complex (MHC) class I gene expression in single neurons of the central nervous system: differential regulation by interferon (IFN) J Exp Med. 1997;185:305–316. doi: 10.1084/jem.185.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Joly E, Mucke L, Oldstone MBA. Viral persistence in neurons explained by lack of major histocompatibility class I expression. Science. 2016;253:1283–1285. doi: 10.1126/science.1891717. [DOI] [PubMed] [Google Scholar]
- 64.Calzascia T, et al. Cutting edge: cross-presentation as a mechanism for efficient recruitment of tumor-specific CTL to the brain. J Immunol. 2003;171:2187–2191. doi: 10.4049/jimmunol.171.5.2187. [DOI] [PubMed] [Google Scholar]
- 65.Jeon S, St Leger AJ, Cherpes TL, Sheridan BS, Hendricks RL. PD-L1/B7-H1 regulates the survival but not the function of CD8+ T cells in herpes simplex virus type 1 latently infected trigeminal ganglia. J Immunol. 2013;190:6277–6286. doi: 10.4049/jimmunol.1300582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huh GS, et al. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290:2155–2159. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goddard CA, Butts DA, Shatz CJ. Regulation of CNS synapses by neuronal MHC class I. Proc Natl Acad Sci USA. 2007;104:6828–6833. doi: 10.1073/pnas.0702023104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O’Donnell LA, et al. STAT1-independent control of a neurotropic measles virus challenge in primary neurons and infected mice. J Immunol. 2012;188:1915–1923. doi: 10.4049/jimmunol.1101356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burdeinick-Kerr R, Govindarajan D, Griffin DE. Noncytolytic clearance of sindbis virus infection from neurons by gamma interferon is dependent on Jak/Stat Signaling. J Virol. 2009;83:3429–3435. doi: 10.1128/JVI.02381-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patterson CE, Lawrence DMP, Echols LA, Rall GF. Immune-mediated protection from measles virus-induced central nervous system disease is noncytolytic and gamma interferon dependent. J Virol. 2002;76:4497–4506. doi: 10.1128/JVI.76.9.4497-4506.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parra B, et al. IFN-γ is required for viral clearance from central nervous system oligodendroglia. J Immunol. 1999;162:1641–1647. [PubMed] [Google Scholar]
- 72.González JM, et al. Inhibition of interferon-γ signaling in oligodendroglia delays coronavirus clearance without altering demyelination. Am J Pathol. 2006;168:796–804. doi: 10.2353/ajpath.2006.050496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Donnell LA, et al. Interferon gamma induces protective non-canonical signaling pathways in primary neurons. J Neurochem. 2015;135:309–322. doi: 10.1111/jnc.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.St Leger AJ, Hendricks RL. CD8+ T cells patrol HSV-1-infected trigeminal ganglia and prevent viral reactivation. J Neurovirol. 2011;17:528–534. doi: 10.1007/s13365-011-0062-1. [DOI] [PubMed] [Google Scholar]
- 75.Liu T, Khanna KM, Chen X, Fink DJ, Hendricks RL. CD8+ T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J Exp Med. 2000;191:1459–1466. doi: 10.1084/jem.191.9.1459. The authors demonstrate that CD8+ T cells residing alongside trigeminal ganglionic neurons infected with HSV are essential in preventing viral reactivation in the absence of neuronal death. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Theil D, et al. Latent herpesvirus infection in human trigeminal ganglia causes chronic immune response. Am J Pathol. 2014;163:2179–2184. doi: 10.1016/S0002-9440(10)63575-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu T, Khanna KM, Carriere BN, Hendricks RL. Gamma interferon can prevent herpes simplex virus type 1 reactivation from latency in sensory neurons. J Virol. 2001;75:11178–11184. doi: 10.1128/JVI.75.22.11178-11184.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu T, Tang Q, Hendricks RL. Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. J Virol. 1996;70:264–271. doi: 10.1128/jvi.70.1.264-271.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci USA. 2010;107:17872–17879. doi: 10.1073/pnas.1010201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wakim LM, et al. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J Immunol. 2012;189:3462–3471. doi: 10.4049/jimmunol.1201305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med. 2015;21:688–697. doi: 10.1038/nm.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Knickelbein JE, et al. Noncytototoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science. 2008;322:268–271. doi: 10.1126/science.1164164. This paper shows that non-cytotoxic CD8+ T cells mediate prevention of HSV reactivation from latency that is triggered by granzyme B-mediated cleavage of an essential early HSV protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marcet-Palacios M, et al. Granzyme B inhibits vaccinia virus production through proteolytic cleavage of eukaryotic initiation factor 4 gamma 3. PLoS Pathog. 2011;7:e1002447. doi: 10.1371/journal.ppat.1002447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Himmelein S, et al. Latent herpes simplex virus 1 infection does not induce apoptosis in human trigeminal ganglia. J Virol. 2011;89:5747–5750. doi: 10.1128/JVI.03481-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Howe CL, et al. Hippocampal protection in mice with an attenuated inflammatory monocyte response to acute CNS picornavirus infection. Sci Rep. 2012;2:545. doi: 10.1038/srep00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Phares T, Stohlman S, Bergmann C. Intrathecal humoral immunity to encephalitic RNA viruses. Viruses. 2013;5:732–752. doi: 10.3390/v5020732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Skoldenberg B, Kalimo K, Carlstrom A, Forgren M, Halonen P. Herpes simplex encephalitis: a serological follow-up study. Acta Neuropathol. 1981;63:273–285. [PubMed] [Google Scholar]
- 88.Burke DS, Nisalak A, Lorsomrudee W, Ussery MA, Laorpongse T. Virus-specific antibody producing cells in blood and cerebrospinal fluid in acute Japanese encephalitis. J Med Virol. 1985;17:283–292. doi: 10.1002/jmv.1890170310. [DOI] [PubMed] [Google Scholar]
- 89.Cho H, et al. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat Med. 2013;19:458–464. doi: 10.1038/nm.3108. The authors demonstrate that neurons vary in susceptibility to viral infection in a subtype- and brain region-specific manner, as a result of basal differences in ISG expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jehmlich U, Ritzer J, Grosche J, Härtig W, Liebert UG. Experimental measles encephalitis in Lewis rats: dissemination of infected neuronal cell subtypes. J Neurovirol. 2013;19:461–470. doi: 10.1007/s13365-013-0199-1. [DOI] [PubMed] [Google Scholar]
- 91.Liebert UG, Baczko K, Budka H, ter Meulen V. Restricted expression of measles virus proteins in brains from cases of subacute sclerosing panencephalitis. J Gen Virol. 1986;67:2435–2444. doi: 10.1099/0022-1317-67-11-2435. [DOI] [PubMed] [Google Scholar]
- 92.Backzo K, et al. Restriction of measles virus gene expression in measles inclusion body encephalitis. J Infect Dis. 1988;158:144–150. doi: 10.1093/infdis/158.1.144. [DOI] [PubMed] [Google Scholar]
- 93.Zerboni L, Arvin A. Neuronal subtype and satellite cell tropism are determinants of varicella-zoster virus virulence in human dorsal root ganglia xenografts in vivo. PLoS Pathog. 2015;11:e1004989. doi: 10.1371/journal.ppat.1004989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lucas TM, Richner JM, Diamond MS. The interferon-stimulated gene Ifi27l2a restricts West Nile virus infection and pathogenesis in a cell-type and region-specific manner. J Virol. 2015;90:2600–2615. doi: 10.1128/JVI.02463-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gomme EA, Wirblich C, Addya S, Rall GF, Schnell MJ. Immune clearance of attenuated rabies virus results in neuronal survival with altered gene expression. PLoS Pathog. 2012;8:e1002971. doi: 10.1371/journal.ppat.1002971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Katayama Y, et al. Detection of measles virus mRNA from autopsied human tissues. J Clin Microbiol. 1998;36:299–301. doi: 10.1128/jcm.36.1.299-301.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nakayama T, et al. Detection of measles virus genome directly from clinical samples by reverse transcriptase-polymerase chain reaction and genetic variability. Virus Res. 1995;35:1–16. doi: 10.1016/0168-1702(94)00074-m. [DOI] [PubMed] [Google Scholar]
- 98.Kawashima H, et al. A case of intractable epilepsy positive for the detection of measles virus genome in the cerebrospinal fluid and peripheral mononuclearcells using reverse transcriptase-polymerase chain reaction. Brain Dev. 1996;18:220–223. doi: 10.1016/0387-7604(95)00154-9. [DOI] [PubMed] [Google Scholar]
- 99.Haase AT, et al. Natural history of restricted synthesis and expression of measles virus genes in subacute sclerosing panencephalitis. Proc Natl Acad Sci USA. 1985;82:3020–3024. doi: 10.1073/pnas.82.9.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Katayama Y, Hotto H, Nishimura A, Tatsuno Y, Homma M. Detection of measles virus nucleoprotein mRNA in autopsied brain tissues. J Gen Virol. 1995;76:3201–3204. doi: 10.1099/0022-1317-76-12-3201. The authors show that MV mRNA is detectable in the brain of humans who died of natural causes in the absence of clinical CNS disease, presumably, decades after acute MV infection. [DOI] [PubMed] [Google Scholar]
- 101.Hanninen P, Arstila P, Lang H, Salmi A, Panelius M. Involvement of the central nervous system in acute, uncomplicated measles virus infection. J Clin Microbiol. 1980;11:610–613. doi: 10.1128/jcm.11.6.610-613.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Allen IV, McQuaid S, McMahon J, Kirk J, McConnell R. The significance of measles virus antigen and genome distribution in the CNS in SSPE for mechanims of viral spread and demyelination. J Neuropathol Exp Neurol. 1996;55:471–480. doi: 10.1097/00005072-199604000-00010. [DOI] [PubMed] [Google Scholar]
- 103.Matthews AE, et al. Antibody is required for clearance of infectious murine hepatitis virus A59 from the central nervous system, but not the liver. J Immunol. 2001;167:5254–5263. doi: 10.4049/jimmunol.167.9.5254. [DOI] [PubMed] [Google Scholar]
- 104.Lavi E, Gilden DH, Hihkin MK, Weiss S. Persistence of mouse hepatits virus A59 RNA in a slow virus demyelinating infection in mice as detected by in situ hybriization. J Virol. 1984;51:563–566. doi: 10.1128/jvi.51.2.563-566.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Koch EM, Neubert WJ, Hofschneider PH. Lifelong persistence of paramyxovirus Sendai-6/94 in C129 mice: detection of latent viral RNA by hybridization with a cloned genomic cDNA probe. Virology. 1984;136:78–88. doi: 10.1016/0042-6822(84)90249-6. [DOI] [PubMed] [Google Scholar]
- 106.Griffin DE, Levine B. Persistence of viral RNA in mouse brains after recovery from acute alphavirus encephalitis. J Virol. 1992;66:6429–6435. doi: 10.1128/jvi.66.11.6429-6435.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li W, et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med. 2015;7:307ra153. doi: 10.1126/scitranslmed.aac8201. The authors identify a human endogenous retrovirus as a causative agent in neurodegenerative disease, positing an aetiology of amyotrophic lateral sclerosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brasil P, et al. Zika virus infection in pregnant women in Rio de Janeiro — preliminary report. N Engl J Med. 2016 doi: 10.1056/NEJMoa1602412. http://dx.doi.org/10.1056/NEJMoa1602412. [DOI] [PMC free article] [PubMed]
- 109.Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. Zika virus and birth defects — reviewing the evidence for causality. N Engl J Med. 2016;374:1981–1987. doi: 10.1056/NEJMsr1604338. [DOI] [PubMed] [Google Scholar]
- 110.Wiley CA, Bhardwaj N, Ross TM, Bissel SJ. Emerging Infections of CNS: avian influenza A virus, Rift Valley fever virus and human parechovirus. Brain Pathol. 2015;25:634–650. doi: 10.1111/bpa.12281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Söderberg-Nauclér C, Johnsen JI. Cytomegalovirus in human brain tumors: role in pathogenesis and potential treatment options. WJEM. 2015;5:1–10. doi: 10.5493/wjem.v5.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sudhakar P, Bachman DM, Mark AS, Berger JR, Kedar S. Progressive multifocal leukoencephalopathy. J Neuroophthamol. 2015;35:296–305. doi: 10.1097/WNO.0000000000000271. [DOI] [PubMed] [Google Scholar]
- 113.Maehlen J, Wallen P, Love A, Norrby E, Kristensson K. Paramyxovirus infections alter certain functional properties in cultured sensory neurons. Brain Res. 1991;540:123–130. doi: 10.1016/0006-8993(91)90498-k. [DOI] [PubMed] [Google Scholar]
- 114.Zhong P, Agosto LM, Munro JB, Mothes W. Cell-to-cell transmission of viruses. Curr Opin Virol. 2013;3:44–50. doi: 10.1016/j.coviro.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Watanabe S, et al. Measles virus mutants possessing the fusion protein with enhanced fusion activity spread effectively in neuronal cells, but not in other cells, without causing strong cytopathology. J Virol. 2015;89:2710–2717. doi: 10.1128/JVI.03346-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Taylor MP, Kobiler O, Enquist LW. Alphaherpesvirus axon-to-cell spread involves limited virion transmission. Proc Natl Acad Sci USA. 2012;109:17046–17051. doi: 10.1073/pnas.1212926109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lancaster KZ, Pfeiffer JK. Limited trafficking of a neurotropic virus through inefficient retrograde axonal transport and the type I interferon response. PLoS Pathog. 2010;6:e1000791. doi: 10.1371/journal.ppat.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sattentau Q. Avoiding the void: cell-to-cell spread of human viruses. Nat Rev Microbiol. 2008;6:815–826. doi: 10.1038/nrmicro1972. [DOI] [PubMed] [Google Scholar]