

Abstract
During insulin-resistant states such as type 2 diabetes mellitus (T2DM), insulin fails to suppress hepatic glucose production yet promotes lipid synthesis leading to hyperglycemia and hypertriglyceridemia. Defining the downstream signaling pathways underlying the control of hepatic metabolism by insulin is necessary for understanding both normal physiology and the pathogenesis of metabolic disease. Here, we summarize recent literature highlighting the importance of both hepatic and extra-hepatic mechanisms in insulin’s regulation of liver glucose and lipid metabolism. We posit that a failure of insulin to inappropriately regulate liver metabolism during T2DM is not exclusively from an inherent defect in canonical liver insulin signaling but, rather, due to a combination of hyperinsulinemia, altered substrate supply, and the input of several extra-hepatic signals.
Keywords: Liver metabolism, insulin signaling, de novo lipogenesis, hepatic glucose production
The pathological diabetic liver
Excess fat accumulation in metabolic organs, altered glucose homeostasis, and the development of insulin resistance are hallmarks type II diabetes mellitus (T2DM). The liver plays a fundamental role in coordinating systemic metabolic homeostasis and the adaptation to nutrient availability and deprivation. During the fasting state, the combination of high glucagon and low insulin levels increases hepatic glucose production (HGP) to meet the metabolic demands of peripheral tissues. This increase in hepatic glucose production (HGP) initially originates from the breakdown of stored glycogen, termed glycogenolysis. As glycogen stores are reduced, decreased glycolytic flux, increased substrate supply and the augmented expression and activity of genes involved in the gluconeogenic pathway enable the liver to generate glucose from 3-carbon precursors [1]. This process is opposed in response to a meal, as levels of glucagon decline and insulin concentrations rise, which signal to increase liver glucose uptake, synthesize glycogen, suppress HGP, and induce the synthesis of fatty acids for storage and subsequent utilization [2].
During insulin-resistant disorders such as T2DM, insulin fails to stimulate liver glucose uptake and to adequately suppress HGP in both the fasted and postprandial state [3]. The inability of insulin to inhibit HGP results from an initial failure to inhibit glycogenolysis followed by an inability of insulin to appropriately suppress gluconeogenesis [3]. Excess HGP coupled with the well-document defect in muscle glucose uptake leads to hyperglycemia [3]. However, despite insulin’s inability to regulate glucose homeostasis, insulin-resistant individuals exhibit increased de novo lipogenesis and re-esterification, inducing fat accumulation in the liver. In addition, insulin-resistant individuals have increased secretion and decreased clearance of triglyceride leading to elevations in blood lipid levels [4–7]. Therefore, during the progression of insulin resistance, the lipid promoting effects of insulin are maintained yet the control of HGP by insulin is defective, leading to hyperglycemia and hypertriglyceridemia [8]. In this review we focus on the recent findings detailing the molecular mechanisms governing lipid synthesis in liver and HGP, and highlight the essential role of both direct and indirect effects of insulin in coordinating hepatic metabolism. We suggest a unified model to reconcile the selective defects in insulin action in liver, a model that explains increased HGP and lipid synthesis independent of defects in canonical insulin signaling due to a combination of hyperinsulinemia, altered substrate supply, and the input of several extra-hepatic signals including free fatty acids.
Liver insulin signaling and hepatic lipid metabolism
In the postprandial state, insulin controls the synthesis and storage of lipids in liver in part by increasing de novo lipogenesis, suppressing fatty acid oxidation, and promoting triglyceride esterification and secretion [9]. Studies in rodent models and humans have defined the significance of hepatic insulin action to the regulation of hepatic lipid metabolism and the development of steatosis during insulin resistance. To address directly the role of hepatic insulin signaling in the regulation of lipid homeostasis, Kahn and colleagues generated mice lacking the insulin receptor in hepatocytes (LIRKO). Despite significant alterations in glucose homeostasis and pronounced insulin resistance, LIRKO mice are protected from the lipid abnormalities commonly associated with T2DM. When challenged with a high-fat diet or genetic forms of obesity, mice lacking the hepatic insulin receptor neither accumulate excess hepatic triglyceride nor develop fatty liver disease [10, 11]. Mechanistically, LIRKO mice have reduced serum triglycerides, increased fatty acid oxidation, and fail to induce de novo lipogenesis and the lipogenic gene program in response to a meal [10, 11]. These rodent studies are supported by human studies that demonstrate that patients with insulin receptor loss of function mutations display extreme insulin resistance and hyperglycemia, yet are protected from the development of fatty liver [12]. These clinical studies support the genetic loss-of-function mouse experiments highlighting the essential role of direct insulin action in the liver in the initiation and progression of fatty liver disease during insulin resistance and T2DM. To summarize, studies in both humans and animal models indicate that hepatic insulin signaling required for both de novo lipogenesis and hepatic lipid synthesis. As a result, there has been considerable interest in the metabolism field in unraveling the specific signaling pathways mediating liver insulin action to explain the paradoxical increase in lipid synthesis simultaneous to enhanced HGP.
During the last decade, many studies have pointed to one signaling cascade in particular as being integral to the metabolic actions of insulin, the phosphoinositide 3-kinase (PI3K)-Akt pathway. Upon insulin engagement of its cognate receptor, PI3K is recruited to phosphorylate insulin receptor substrates (IRS) and generate 3′ phosphoinositides. Phosphatidylinositol (3,4,5)-trisphosphate (PIP3) generation promotes the recruitment of Pyruvate Dehydrogenase Kinase 1 (PDK1) and Akt (also known as protein kinase B) leading to the subsequent phosphorylation of Akt by PDK1 on Thr308 and by mammalian target of rapamycin (mTORC) 2 on Ser473 [13]. Akt then signals to multiple downstream pathways to control cell growth, survival and glucose and lipid metabolism including mTORC1, glycogen synthase kinase, and the FoxO transcription factors [14, 15] (Figure 1). In support of the Akt pathway as mediator of insulin’s control of hepatic lipid metabolism, mice with hepatic deficiency of Phosphatase And Tensin Homolog (PTEN, a negative regulator of Akt) or overexpression of a constitutively active, myristoylated Akt display hepatomegaly and a dramatic increase in liver fat [16, 17]. Consistent with this, germline Akt2 knockout out mice (Akt2KO) as well as liver-specific Akt2KO mice are protected from the development of fatty liver in response to genetic or diet-induced obesity and insulin resistance, despite marked hyperinsulinemia [18]. Moreover, in mice lacking Pten in liver, enhanced Akt2 signaling is required for the induction of the lipogenic gene program and hepatic steatosis. This direct requirement for Akt2 signaling acting downstream of Pten was nicely demonstrated using combined genetic knockout of both Akt2 and Pten specifically in hepatocytes [19]. Genetic loss-of-function experiments have uncovered the signaling pathways coordinating insulin’s effects on hepatic lipid metabolism. These data strongly support the notion that Akt signaling downstream of the insulin receptor is essential to increase hepatic lipid synthesis and accumulation of liver fat during insulin resistance.
Figure 1. Insulin-dependent signaling mechanisms in liver in the regulation of hepatic lipid and glucose metabolism.
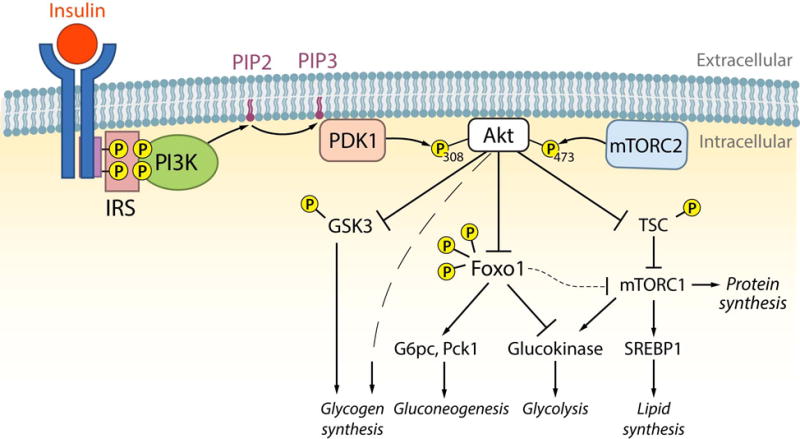
In response to a meal, insulin concentrations rise and signal in liver to regulate hepatic lipid and glucose metabolism. Upon engagement of its receptor (insulin receptor) resulting in autophosphorylation, insulin receptor substrates (IRS) are recruited and phosphorylated. IRS proteins then recruit and activate PI3K, which phosphorylates PIP2 to generate PIP3. PDK1 is then activated by PIP3, which phosphorylates Akt at Thr308. Additionally, Akt is activated by phosphorylation by mTORC2 at Ser473. Once fully activated, Akt signals via phosphorylation to control multiple metabolic processes in liver including glycogen synthesis, gluconeogenesis, glycolysis, and lipid synthesis. Active Akt induces glycogen synthesis by both multiple mechanisms including GSK3-dependent and independent pathways. Akt inhibits gluconeogeneic gene expression by phosphorylating and inhibiting FoxO1, which is a transcription factor involved in the activation of G6pc and Pck1. In addition, inhibition of FoxO1 in part leads to the induction of glucokinase gene expression. Lastly, Akt activates the mTORC1 and protein synthesis pathways by phosphorylating and inhibiting the TSC proteins. By activating mTORC1 and inhibiting FoxO1, Akt promotes the activation of SREBP1c and the lipogenic gene program increasing lipid synthesis.
With the role of Akt in hepatic lipid metabolism so clearly defined, attention has turned to elucidating the more distal signaling events downstream of Akt in the regulation of lipid metabolism. Recent studies have focused on the transcriptional control of de novo lipogenesis by insulin, highlighting the importance of several lipogenic transcription factors including the sterol regulatory element binding protein 1c (SREBP1c) and the carbohydrate response element binding-protein (ChREBP). SREBPs are ubiquitous transcription factors that play a critical role in the synthesis of fatty acids, triglycerides and cholesterol [20], and hepatic SREBP1c activity is increased in mouse models of insulin resistance such as ob/ob mice and lipodystrophic diabetes [21]. In hepatocytes, insulin activation of Akt enhances both the synthesis and processing of the predominant SREBP isoform, SREBP1c [22, 23]. The requirement for SREBP1c in mediating the hepatic steatosis and fatty liver during insulin resistance and carbohydrate-induced hypertriglyceridemia has been demonstrated in vivo by Moon et al, who generated mice with liver-specific knockout of Scap, an escort protein essential for cleavage of all SREBPS. [24]. These mice were protected from hepatic steatosis when fed a high fat diet or crossed with mice deficient for leptin, consistent with an essential role for SREBPs in mediating hepatic lipid accumulation downstream of insulin.
The role of ChREBP downstream of hepatic insulin action is less well defined. ChREBP is a glucose-responsive transcription factor and activates similar lipogenic genes as SREBP1c, including Fasn, Scd1, and Elovl6 [25]. Dietary fructose administration induces hepatic steatosis, an effect dependent on ChREBP [26]. Hepatic overexpression of ChREBP is sufficient to induce fatty liver in mice and increased ChREBP has been correlated with NAFLD in humans [27]. Germline deletion of ChREBP reduces basal amounts of fatty acid synthesis independent of changes in SREBP [28]. Moreover, liver-specific knockdown of ChREBP reduces de novo lipogenesis and hepatic triglyceride in ob/ob mice [29]. Although it is now well-established that de novo lipogenesis and fat accumulation are increased in the livers of insulin-resistant people [4–7], the activation status of SREPB1c and ChREBP is not known. Future studies will be needed to better understand the specific effects of these key lipogenic transcription factors and how these molecules interact in the regulation of lipogenic gene expression and de novo lipogenesis in humans.
In addition to SREBPs/ChREBP, the FoxO transcription factors have been implicated as an important downstream transcriptional regulator of hepatic lipid metabolism [14]. Akt controls the activity of the FoxO proteins through a well-characterized phosphorylation mechanism that plays a central role in multiple metabolic processes [30]. Many of the studies of FoxO transcription factors have focused on the predominant FoxO transcription factor in liver, FoxO1. Transgenic mice that express in liver a mutated version of FoxO1 (FoxoAAA) that cannot be phosphorylated by Akt and therefore is constitutively active [31, 32], display reduced de novo lipogenesis, and triglyceride secretion, and fail to induce of the lipogenic gene program in response to a meal [33–35]. Consistent with this, hepatic deletion of all three FoxO transcription factors induce de novo lipogenesis, hepatic steatosis, and increases in lipogenic gene expression [36]. The transcriptional mechanisms responsible for Foxo1’s direct control of hepatic lipid metabolism are not completely defined. Studies by Deng et al suggest that Foxo1 can bind directly to the SREBP1c promoter and affect the transcriptional activity of Sp1 and SREBP1c [33]. Foxo1 also controls glucokinase gene expression, which likely contributes to the lipogenic response by increasing glucose-6-phosphate levels and subsequently activating ChREBP [37]. Collectively, the data support the hypothesis that FoxO transcription factors, in particular FoxO1, play an important role in mediating insulin’s control of hepatic lipid synthesis.
Several studies have reported that mTORC1 is downstream of Akt in the activation of SREBP in cell culture models [23, 38]. Experiments in mice confirm that mTORC1 is required although not sufficient to induce de novo lipogenesis and the lipogenic gene program [39, 40]. Moreover in the absence of Akt, genetic activation of mTORC1 is not sufficient to induce de novo lipogenesis nor the SREBP1-dependent lipogenic gene program without concomitant inactivation of FoxO1 [37]. In addition to SREBP1, activation of mTORC1 and inhibition of FoxO1 are both essential to induce glucokinase expression, which contributes to increased glycolytic flux and activation of ChREBP. These changes, along with the activation of SREBP1 and other genes involved in lipogenesis such as Fasn, are required to induce de novo lipogenesis downstream of Akt [37, 41]. Despite this complexity downstream of Akt, the most critical point is that the regulation of lipid metabolism and the development of fatty liver require insulin signaling via Akt. We now turn our attention to defining the mechanisms governing the control of HGP by insulin.
Direct regulation of HGP by insulin
In response to a meal, increased insulin concentrations in the blood signal the liver to regulate glucose metabolism by stimulating liver glucose uptake and glycogen storage and suppressing HGP (Figure 1). The precise coordination of glucose fluxes by insulin is essential to adequately control postprandial glucose concentrations. In response to an oral glucose challenge, the liver is responsible for approximately 30% of the disposal of glucose. However, this does not take into account the acute suppression of hepatic production, which further contributes to liver’s important role in postprandial glycemic control [42, 43]. During the progression of insulin resistance, defects in both hepatic glucose uptake and production contribute to postprandial hyperglycemia [42]. Given the well-document defect in insulin’s ability to suppress HGP in T2DM, in the remaining sections, we will discuss recent data defining the mechanisms of regulation of HGP by insulin in vivo.
Similar to its control of hepatic lipid metabolism, insulin acts directly on hepatocytes to suppress HGP via multiple mechanisms, including acute changes in metabolic pathways and more long-term alterations in gene expression [2]. Elegant physiological studies have demonstrated that insulin directly suppresses HGP [43], by acutely inhibiting glycogenolysis [44]. Importantly, insulin’s direct control of HGP can occur independently of changes in glucagon and circulating free fatty acids, further suggesting that the direct action of insulin on hepatocytes is essential for proper regulation of HGP and hepatic glucose homeostasis [45, 46]. Mechanistically, studies both in vivo and in the perfused livers of mice lacking hepatic Akt2 demonstrate that Akt is required for insulin’s direct effect on glycogenolysis [47]. Insulin alters liver gluconeogenic gene expression through multiple mechanisms including the allosteric regulation of pyruvate carboxylase and potentially by inhibiting the transcription of gluconeogenesis including glucose-6-phosphatase (G6pc) and phosphoenolpyurvate carboxy kinase (Pck1) [2, 48]. Both genetic and pharmacological approaches have clearly demonstrated the requirement for hepatic Akt signaling in mediating insulin’s direct control of hepatic gluconeogenic gene expression [23, 49]. These data implicate a direct action of insulin on hepatocytes in the physiological response to nutrient intake, and in the regulation of blood glucose.
This conventional view of insulin regulation of HGP in the liver suggests that Akt phosphorylates and inactivates glycogen synthase kinase 3beta (GSK3) and FoxO1 to control glycogenolysis and gluconeogenic gene expression, respectively [2]. However, recent experiments have questioned this canonical view. For example, insulin still suppresses HGP and glycogenolysis in a mouse with mutant GSK3 that cannot be phosphorylated by Akt, suggesting that Akt mediates the control of glycogenolysis by insulin through GSK3-dependent as well as GSK3-independent mechanisms [47]. Moreover, and quite surprisingly, although regulation of gluconeogenic gene expression and HGP are the most well characterized metabolic roles of FoxO1 in liver [50], hepatic deletion of FoxO1 is sufficient to normalize glucose tolerance and peripheral insulin sensitivity in mice lacking the insulin receptor, insulin receptor substrates, or Akt isoforms in liver [49, 51–53]. Yet deletion of FoxO1 in liver alleviates the requirement for direct insulin action to suppress HGP suggesting direct liver insulin action is not essential for in vivo control of HGP [37, 49, 52]. Lastly, transgenic expression of the insulin receptor in the livers of mice lacking insulin receptor in all other tissues failed to correct systemic insulin resistance and glucose intolerance despite restoring insulin signaling in hepatocytes [54]. These data contradict the canonical model of hepatic insulin action and provide genetic evidence that suggest insulin may regulate HGP by additional extra-hepatic mechanisms.
Extra-hepatic control of HGP
Central Nervous System
Several studies indicate that the central nervous system plays a critical role in the regulation of peripheral metabolism and energy balance by insulin [55]. Direct intra-cerebral infusion of insulin suppresses HGP and gluconeogenic gene expression in rodents without activating hepatic insulin signaling [56]. Moreover, deletion of the insulin receptor in agouti related peptide (Agrp) expressing neurons in the arcuate nucleus of the hypothalamus prevents insulin-mediated suppression of HGP during hyperinsulinemic-euglycemic clamps with peripherally infused insulin [57]. However, HGP was found to be suppressed by insulin in mice devoid of hepatic insulin signaling that were subjected to selective hepatic vagotomy, suggesting that non-autonomous regulation of HGP can occur independent of neural signaling [37]. These results are consistent with the observation that hepatic vagotomy does not alter HGP during a peripheral infusion of insulin [58], although hepatic vagotomy does not completely rule out central regulation by vagus-independent neuronal pathways or indirect mechanisms. Moreover, the effect of insulin action in the CNS to acutely suppress HGP is not observed in canine models, where insulin action in the CNS induced a slow and modest reduction in hepatic gluconeogenic genes without altering HGP or gluconeogenesis [59, 60]. These data indicate that acute effects of insulin to suppress HGP occur independent of a direct brain-liver mechanism.
Glucagon
Glucagon is the major hormone that opposes the effects of insulin on hepatic glucose metabolism, functioning as a positive regulator of gluconeogenesis and glycogenolysis [61]. Physiological studies in dogs demonstrate an antagonistic role of insulin on glucagon secretion during euglycemic clamps [1, 62]. Moreover, modern genetic studies support the importance of glucagon signaling in the control of HGP [63–65]. Deletion of the insulin receptor in pancreatic alpha cells enhances glucagon secretion and leads to mild glucose intolerance, hyperglycemia, and hyperglucagonemia in the fed state, supporting a role for insulin in intraislet communication [66]. Surprisingly, the metabolic abnormalities associated with streptozotocin-induced insulin deficiency are abrogated by loss of the glucagon receptor [67, 68]. However, antagonism of glucagon action in liver failed to prevent the ability of insulin to suppress HGP during hyperinsulinemic-euglycemic clamps in mice also lacking hepatic insulin signaling, indicating that peripheral actions of insulin to regulate HGP are independent of acute changes of liver glucagon signaling [37], consistent with older studies that show the extra-hepatic action of insulin suppresses HGP regardless of portal concentrations of glucagon [69]. Although insulin controls HGP independent of acute changes in glucagon, several clinical studies have demonstrated reductions in blood glucose levels with glucagon receptor antagonists [70]
Free fatty acids
Insulin markedly suppresses adipocyte lipolysis, reducing the levels of circulating free fatty acids (FFA) and glycerol. It has been known for decades that an increase in circulating FFAs impairs the ability of insulin to suppress endogenous glucose production and directly enhances HGP in humans [71]. The ability of FFAs to augment HGP is likely mediated multiple metabolic pathways that stimulate gluconeogenesis including increased ATP production, mitochondrial Acetyl-CoA, and changes in NAD+/NADH ratio [43]. The time course of insulin-mediated suppression of HGP correlates with the reduction of FFA [72] and prevention of the decline in FFA levels during hyperinsulinemic-euglycemic clamps in the presence of somatostatin partially blocks the reduction in HGP by peripheral insulin infusion in dogs [73]. Moreover, lowering serum FFA during a somatostatin infusion is sufficient to reduce HGP independent of exogenous insulin [74] while elevating FFAs fails to increase HGP [73]. These data support the notion that an increase in the delivery of free fatty acids from visceral adipose depots results in increased HGP and may explain the disproportionate association of abdominal compared to subcutaneous adiposity with metabolic disease [75].
Recent reports have reinforced the conclusion that free fatty acids act as an extra-hepatic signal and reductions in circulating free fatty acids contribute to insulin’s ability to suppress HGP in vivo. Infusion of acetate to mimic an elevation in FFA by increasing intra-hepatic acetyl-CoA effectively blocked the ability of insulin to suppress HGP in fasted mice as well as mice lacking hepatic insulin signaling [48]. Similarly, preventing the reduction in free fatty acids during a hyperinsulinemic-euglycemic clamp blocks the ability of insulin to suppress HGP in mice lacking hepatic insulin signaling [37].
While these studies agree on the main point that insulin suppression of lipolysis underlies its non-hepatocyte-autonomous effects on HGP, an important difference in these studies concerns the role of the extra-hepatic pathway when insulin signaling is intact in hepatocytes. Perry et al. suggest that the effect of insulin on lipolysis is the predominant mechanism of action by which insulin suppresses HGP, regardless of whether the hepatocyte remains capable of responding directly to insulin [48] whereas Titchenell et al found that an intact insulin-signaling pathway in liver was sufficient to mediate the suppression of HGP even when the decrease in FFA was abrogated [37]. One explanation for the discrepancy between the physiological role of FFAs in control HGP may relate to the use of acetate to mimic the reduction in free fatty acids and subsequent reduction in hepatic Acetyl-CoA levels [48]. In this regard, previous studies using FFAs rather than acetate demonstrated that the direct actions of insulin on the liver are dominant over peripheral effects [76–78]. These mouse experiments are also supported by experiments in dogs, which similarly found that the direct actions of insulin on the liver are dominant in the control of HGP [45]. In addition, the study by Perry et al used overnight fasted mice, which are glycogen depleted. In doing so, the direct inhibition of glycogenolysis by insulin may be underrepresented. In conclusion, this suggests that indirect control of the regulation of HGP becomes the major determinant only under conditions in which the direct pathway is defective. However, in the setting of obesity associated diabetes, where direct liver insulin regulation of HGP is defective, there is agreement that adipocyte-derived FFA drives HGP independent of the direct actions of insulin on hepatocytes. Future work will be required to address this issue in more depth.
Concluding remarks and future perspectives
Recent understanding of signaling mechanisms downstream of the hepatic insulin receptor have illuminated the critical role of activation of Akt and its downstream effectors in the direct control of lipid metabolism by insulin. In addition, multiple lines of evidence using distinct models have demonstrated the important contributions of hepatic and extra-hepatic pathways in the regulation of glucose metabolism in liver. Thus, the metabolism of the enigmatic diabetic liver may be explained by a new paradigm whereby hyperinsulinemia directly drives hepatic lipogenesis and lipid accumulation while indirect mechanisms, including excess circulating FFAs, block insulin’s ability to suppress HGP. Several questions remain unanswered, however, particularly with regard to the mechanism by which free fatty acids are increased during insulin resistance and obesity and how they act as a dominant regulator of HGP independent of direct action of insulin (Outstanding Questions Box). It will be essential to unravel these underlying mechanisms to devise effective strategies to combat hepatic insulin resistance and improve the metabolic dysfunction that is rampant in modern society.
Outstanding Questions Box.
What are the relative contributions of SREBP1c and ChREBP to de novo lipogenesis and hepatic steatosis?
How does the insulin-resistant liver become insensitive to the direct regulation of HGP during T2D?
How does the adipose tissue become insensitive to insulin action during metabolic disease?
What is the mechanism driving hyperglucagonemia during metabolic disease?
Trends Box.
Insulin action in liver coordinately regulates lipid synthesis and glucose production by cell autonomous and non-autonomous mechanisms
Liver insulin action is required for lipid synthesis and the development of fatty liver
Insulin’s ability to suppress hepatic glucose production is determined by both direct and indirect mechanisms including the contribution of adipose tissue lipolysis and gluconeogenic regulation by glucagon
Defining the molecular mechanisms responsible for insulin’s selective control of lipid synthesis and glucose production may allow for the development of therapeutic agents that reduce hyperglycemia without inducing hypertriglyceridemia
Acknowledgments
The authors apologize to researchers whose relevant studies were not discussed due to space. The authors would like to thank David Steger for careful reading of this manuscript. P.M.T. is currently supported by the National Institutes of Health (K01 DK111715) and research funding from Pfizer, Inc. M.A.L. is supported by the JPB foundation. M.J.B. is currently an employee of Pfizer, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ramnanan CJ, et al. Physiologic action of glucagon on liver glucose metabolism. Diabetes, obesity & metabolism. 2011;13(Suppl 1):118–125. doi: 10.1111/j.1463-1326.2011.01454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin HV, Accili D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 2011;14:9–19. doi: 10.1016/j.cmet.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rizza RA. Pathogenesis of fasting and postprandial hyperglycemia in type 2 diabetes: implications for therapy. Diabetes. 2010;59:2697–2707. doi: 10.2337/db10-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lambert JE, et al. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwarz JM, et al. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. The American journal of clinical nutrition. 2003;77:43–50. doi: 10.1093/ajcn/77.1.43. [DOI] [PubMed] [Google Scholar]
- 6.Donnelly KL, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petersen KF, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A. 2007;104:12587–12594. doi: 10.1073/pnas.0705408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 2008;7:95–96. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 9.Leavens KF, Birnbaum MJ. Insulin signaling to hepatic lipid metabolism in health and disease. Crit Rev Biochem Mol Biol. 2011;46:200–215. doi: 10.3109/10409238.2011.562481. [DOI] [PubMed] [Google Scholar]
- 10.Haas JT, et al. Hepatic insulin signaling is required for obesity-dependent expression of SREBP-1c mRNA but not for feeding-dependent expression. Cell Metab. 2012;15:873–884. doi: 10.1016/j.cmet.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biddinger SB, et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008;7:125–134. doi: 10.1016/j.cmet.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Semple RK, et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest. 2009;119:315–322. doi: 10.1172/JCI37432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taniguchi CM, et al. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 14.Gross DN, et al. The role of FOXO in the regulation of metabolism. Curr Diab Rep. 2009;9:208–214. doi: 10.1007/s11892-009-0034-5. [DOI] [PubMed] [Google Scholar]
- 15.Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217–222. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ono H, et al. Hepatic Akt activation induces marked hypoglycemia, hepatomegaly, and hypertriglyceridemia with sterol regulatory element binding protein involvement. Diabetes. 2003;52:2905–2913. doi: 10.2337/diabetes.52.12.2905. [DOI] [PubMed] [Google Scholar]
- 17.Horie Y, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113:1774–1783. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leavens KF, et al. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab. 2009;10:405–418. doi: 10.1016/j.cmet.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He L, et al. The critical role of AKT2 in hepatic steatosis induced by PTEN loss. Am J Pathol. 2010;176:2302–2308. doi: 10.2353/ajpath.2010.090931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horton JD, et al. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimomura I, et al. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J Biol Chem. 1999;274:30028–30032. doi: 10.1074/jbc.274.42.30028. [DOI] [PubMed] [Google Scholar]
- 22.Owen JL, et al. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc Natl Acad Sci U S A. 2012;109:16184–16189. doi: 10.1073/pnas.1213343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li S, et al. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A. 2010;107:3441–3446. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moon YA, et al. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012;15:240–246. doi: 10.1016/j.cmet.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Filhoulaud G, et al. Novel insights into ChREBP regulation and function. Trends Endocrinol Metab. 2013;24:257–268. doi: 10.1016/j.tem.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 26.Kim MS, et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J Clin Invest. 2016;126:4372–4386. doi: 10.1172/JCI81993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benhamed F, et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest. 2012;122:2176–2194. doi: 10.1172/JCI41636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iizuka K, et al. Deficiency of carbohydrate response element-binding protein (ChREBP). reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A. 2004;101:7281–7286. doi: 10.1073/pnas.0401516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dentin R, et al. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes. 2006;55:2159–2170. doi: 10.2337/db06-0200. [DOI] [PubMed] [Google Scholar]
- 30.Gross DN, et al. The role of FoxO in the regulation of metabolism. Oncogene. 2008;27:2320–2336. doi: 10.1038/onc.2008.25. [DOI] [PubMed] [Google Scholar]
- 31.Rena G, et al. Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. Embo J. 2002;21:2263–2271. doi: 10.1093/emboj/21.9.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rena G, et al. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274:17179–17183. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- 33.Deng X, et al. FoxO1 inhibits sterol regulatory element-binding protein-1c (SREBP-1c). gene expression via transcription factors Sp1 and SREBP-1c. J Biol Chem. 2012;287:20132–20143. doi: 10.1074/jbc.M112.347211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang W, et al. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem. 2006;281:10105–10117. doi: 10.1074/jbc.M600272200. [DOI] [PubMed] [Google Scholar]
- 35.Zhang W, et al. Integrated Regulation of Hepatic Lipid and Glucose Metabolism by Adipose Triacylglycerol Lipase and FoxO Proteins. Cell reports. 2016;15:349–359. doi: 10.1016/j.celrep.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haeusler RA, et al. Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nat Commun. 2014;5:5190. doi: 10.1038/ncomms6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Titchenell PM, et al. Direct Hepatocyte Insulin Signaling Is Required for Lipogenesis but Is Dispensable for the Suppression of Glucose Production. Cell Metab. 2016;23:1154–1166. doi: 10.1016/j.cmet.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Porstmann T, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wan M, et al. Postprandial hepatic lipid metabolism requires signaling through Akt2 independent of the transcription factors FoxA2, FoxO1, and SREBP1c. Cell Metab. 2011;14:516–527. doi: 10.1016/j.cmet.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yecies JL, et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011;14:21–32. doi: 10.1016/j.cmet.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun Z, et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat Med. 2012;18:934–942. doi: 10.1038/nm.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore MC, et al. Regulation of hepatic glucose uptake and storage in vivo. Advances in nutrition. 2012;3:286–294. doi: 10.3945/an.112.002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cherrington AD, et al. Insulin action on the liver in vivo. Biochem Soc Trans. 2007;35:1171–1174. doi: 10.1042/BST0351171. [DOI] [PubMed] [Google Scholar]
- 44.Sindelar DK, et al. A comparison of the effects of selective increases in peripheral or portal insulin on hepatic glucose production in the conscious dog. Diabetes. 1996;45:1594–1604. doi: 10.2337/diab.45.11.1594. [DOI] [PubMed] [Google Scholar]
- 45.Edgerton DS, et al. Insulin’s direct effects on the liver dominate the control of hepatic glucose production. J Clin Invest. 2006;116:521–527. doi: 10.1172/JCI27073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cherrington AD, et al. The direct and indirect effects of insulin on hepatic glucose production in vivo. Diabetologia. 1998;41:987–996. doi: 10.1007/s001250051021. [DOI] [PubMed] [Google Scholar]
- 47.Wan M, et al. A noncanonical, GSK3-independent pathway controls postprandial hepatic glycogen deposition. Cell Metab. 2013;18:99–105. doi: 10.1016/j.cmet.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perry RJ, et al. Hepatic Acetyl CoA Links Adipose Tissue Inflammation to Hepatic Insulin Resistance and Type 2 Diabetes. Cell. 2015;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu M, et al. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med. 2012;18:388–395. doi: 10.1038/nm.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsumoto M, et al. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 2007;6:208–216. doi: 10.1016/j.cmet.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 51.Titchenell PM, et al. Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nat Commun. 2015;6:7078. doi: 10.1038/ncomms8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O-Sullivan I, et al. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat Commun. 2015;6:7079. doi: 10.1038/ncomms8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dong XC, et al. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008;8:65–76. doi: 10.1016/j.cmet.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Okamoto H, et al. Restoration of liver insulin signaling in Insr knockout mice fails to normalize hepatic insulin action. J Clin Invest. 2005;115:1314–1322. doi: 10.1172/JCI23096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Myers MG, Jr, Olson DP. Central nervous system control of metabolism. Nature. 2012;491:357–363. doi: 10.1038/nature11705. [DOI] [PubMed] [Google Scholar]
- 56.Pocai A, et al. A brain-liver circuit regulates glucose homeostasis. Cell Metab. 2005;1:53–61. doi: 10.1016/j.cmet.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 57.Konner AC, et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5:438–449. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 58.Lam TK, et al. Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nat Med. 2005;11:320–327. doi: 10.1038/nm1201. [DOI] [PubMed] [Google Scholar]
- 59.Ramnanan CJ, et al. Brain insulin action augments hepatic glycogen synthesis without suppressing glucose production or gluconeogenesis in dogs. J Clin Invest. 2011;121:3713–3723. doi: 10.1172/JCI45472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramnanan CJ, et al. Evidence against a physiologic role for acute changes in CNS insulin action in the rapid regulation of hepatic glucose production. Cell Metab. 2012;15:656–664. doi: 10.1016/j.cmet.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12:141–151. doi: 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Myers SR, et al. Effects of small changes in glucagon on glucose production during a euglycemic, hyperinsulinemic clamp. Metabolism. 1991;40:66–71. doi: 10.1016/0026-0495(91)90194-2. [DOI] [PubMed] [Google Scholar]
- 63.Miller RA, et al. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494:256–260. doi: 10.1038/nature11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ozcan L, et al. Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell Metab. 2012;15:739–751. doi: 10.1016/j.cmet.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dentin R, et al. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature. 2007;449:366–369. doi: 10.1038/nature06128. [DOI] [PubMed] [Google Scholar]
- 66.Kawamori D, et al. Insulin signaling in alpha cells modulates glucagon secretion in vivo. Cell Metab. 2009;9:350–361. doi: 10.1016/j.cmet.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee Y, et al. Hyperglycemia in rodent models of type 2 diabetes requires insulin-resistant alpha cells. Proc Natl Acad Sci U S A. 2014;111:13217–13222. doi: 10.1073/pnas.1409638111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee Y, et al. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes. 2011;60:391–397. doi: 10.2337/db10-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mittelman SD, et al. Indirect effect of insulin to suppress endogenous glucose production is dominant, even with hyperglucagonemia. J Clin Invest. 1997;100:3121–3130. doi: 10.1172/JCI119867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lefebvre PJ, et al. Inhibiting or antagonizing glucagon: making progress in diabetes care. Diabetes, obesity & metabolism. 2015;17:720–725. doi: 10.1111/dom.12480. [DOI] [PubMed] [Google Scholar]
- 71.Roden M, et al. Effects of free fatty acid elevation on postabsorptive endogenous glucose production and gluconeogenesis in humans. Diabetes. 2000;49:701–707. doi: 10.2337/diabetes.49.5.701. [DOI] [PubMed] [Google Scholar]
- 72.Rebrin K, et al. Free fatty acid as a link in the regulation of hepatic glucose output by peripheral insulin. Diabetes. 1995;44:1038–1045. doi: 10.2337/diab.44.9.1038. [DOI] [PubMed] [Google Scholar]
- 73.Rebrin K, et al. Causal linkage between insulin suppression of lipolysis and suppression of liver glucose output in dogs. J Clin Invest. 1996;98:741–749. doi: 10.1172/JCI118846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mittelman SD, Bergman RN. Inhibition of lipolysis causes suppression of endogenous glucose production independent of changes in insulin. Am J Physiol Endocrinol Metab. 2000;279:E630–637. doi: 10.1152/ajpendo.2000.279.3.E630. [DOI] [PubMed] [Google Scholar]
- 75.Castro AV, et al. Obesity, insulin resistance and comorbidities? Mechanisms of association. Arquivos brasileiros de endocrinologia e metabologia. 2014;58:600–609. doi: 10.1590/0004-2730000003223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim JK, et al. PKC-theta knockout mice are protected from fat-induced insulin resistance. J Clin Invest. 2004;114:823–827. doi: 10.1172/JCI22230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim JK, et al. Inactivation of fatty acid transport protein 1 prevents fat-induced insulin resistance in skeletal muscle. J Clin Invest. 2004;113:756–763. doi: 10.1172/JCI18917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim JK, et al. Prevention of fat-induced insulin resistance by salicylate. J Clin Invest. 2001;108:437–446. doi: 10.1172/JCI11559. [DOI] [PMC free article] [PubMed] [Google Scholar]