

ABSTRACT
Obesity is closely associated with metabolic diseases including type 2 diabetes. One hallmark characteristics of obesity is chronic inflammation that is coordinately controlled by complex signaling networks in adipose tissues. Compelling evidence indicates that reactive oxygen species (ROS) and its related signaling pathways play crucial roles in the progression of chronic inflammation in obesity. The pentose phosphate pathway (PPP) is an anabolic pathway that utilizes the glucoses to generate molecular building blocks and reducing equivalents in the form of NADPH. In particular, NADPH acts as one of the key modulators in the control of ROS through providing an electron for both ROS generation and scavenging. Recently, we have reported that glucose-6-phosphate dehydrogenase (G6PD), a rate-limiting enzyme of the PPP, is implicated in adipose tissue inflammation and systemic insulin resistance in obesity. Mechanistically, G6PD potentiates generation of ROS that augments pro-inflammatory responses in adipose tissue macrophages, leading to systemic insulin resistance. Here, we provide an overview of cell type- specific roles of G6PD in the regulation of ROS balance as well as additional details on the significance of G6PD that contributes to pro-oxidant NADPH generation in obesity-related chronic inflammation and insulin resistance.
KEYWORDS: adipocytes, adipose tissue inflammation, glucose-6-phophate dehydrogenase (G6PD), insulin sensitivity, macrophages, NADPH, obesity, reactive oxygen species (ROS)
Obesity is characterized by massively expanded white adipose tissue (WAT) and highly associated with metabolic diseases including type 2 diabetes. In obesity, nutritional stresses disrupt WAT architecture and function, and multiple pathways have been associated with unhealthy WAT expansion.1 Particularly, compelling evidence indicates that chronic inflammation promotes adipose tissue remodeling and dysfunction including insulin resistance and adipokine dysregulation in obesity.1,2 The link between obesity and adipose tissue inflammation has been derived after identification of tumor necrosis factor α (TNF α) in obese adipose tissue.3 The expression of TNF α is elevated in adipose tissues of different rodent models of obesity or diabetes and suppression of TNF α activity by its inhibitor ameliorates obesity-mediated insulin resistance.3 The reframing of obesity as an inflammatory condition was further supported by studies revealing dramatic accumulation of macrophages and pro-inflammatory responses in obese adipose tissues.4 Obese adipose tissues secrete several anti- and pro-inflammatory cytokines including interleukin 6, monocyte-chemoattractant protein 1, and interleukin 10.4,5 Among many cell types in adipose tissue, macrophages have been identified as the primary source of pro-inflammatory cytokines that confer vicious cycle of adipose tissue inflammation through having multiple impacts on other cells.1,4 In addition to aggravating inflammatory response, macrophage-derived cytokines abrogate insulin signaling in adipocytes through activation of IKKβ/NFκB and JNK pathways.5-7 There are also clinical reports uncovering insulin sensitizing effects of anti-inflammatory drugs including salicylate in diabetic patients.8
Among various intracellular signaling pathways, adipose tissue inflammation is linked to oxidative stress characterized by accumulation of reactive oxygen species (ROS).9 As visceral fat rapidly expands, adipose tissue generates higher level of ROS in several obese mouse models as compared with their lean littermates.10 Increased oxidative stress, in turn, potentiates expression and secretion of inflammatory cytokines by activating several transcriptional factors such as NFκB, c-Fos and c-Jun.11,12 In accordance with animal data, body mass index (BMI) closely correlates with the elevation of oxidative stress as well as adipose tissue inflammation in human.13,14
ROS is constantly generated by both enzymatic and non-enzymatic reactions in response to external and internal stimuli.15 Enzyme-mediated ROS production includes those involving NADPH oxidase, xanthine oxidase and uncoupled endothelial nitric oxide synthase (eNOS).15 The mitochondrial respiratory chain is a non-enzymatic source of ROS.15 In general, ROS production is buffered through cooperative activity of antioxidant enzymes such as superoxide dismutase, catalase and glutathione peroxidase.15 Balanced activity of ROS generation and scavenging is thus crucial to maintain adequate level of ROS in cells. However, oxidative stress accumulates as a result of excessive production and/or inadequate removal of ROS in various pathological conditions including obesity.10,16
As a cofactor and electron donor, NADPH is involved in many key metabolic processes including glycolysis, oxidative respiration, reductive biosynthesis of lipids and redox control (Fig. 1).9 Metabolically active cells such as adipocytes and hepatocytes utilize NADPH in de novo lipogenesis for the α-glycerol phosphate, fatty acids and triglyceride synthesis.9 The level of NADPH is sustained by different enzymatic systems, e.g. isocitrate dehydrogenase (IDH), which is expressed in both mitochondria and the cytosol, cytosolic malic enzyme (ME) and the pentose phosphate pathway (PPP).17 Recent studies have shown that dysregulation of NADPH-producing enzymes contributes to obesity and its related complications including lipid abnormalities and dysfunction of metabolic tissues.18,19 For instance, IDH transgenic mice exhibit obesity, hyperlipidemia, and fatty liver.18 On the other hand, ME1 deficiency contributes to reduction in obesity accompanied with decreases in fat mass, liver steatosis, and improvement of systemic glucose tolerance.19
Figure 1.
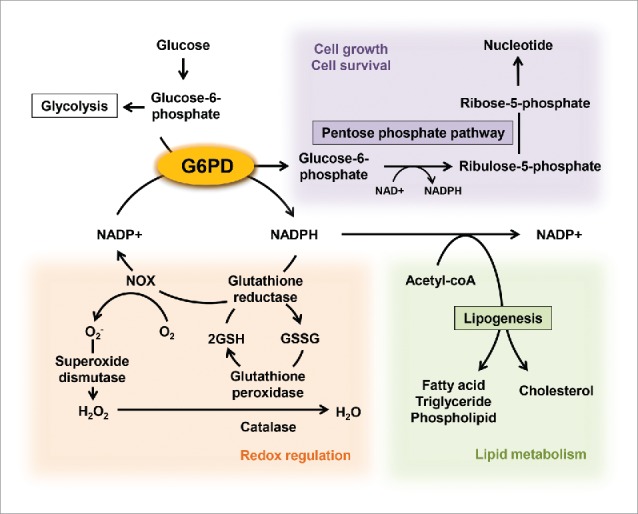
Roles of G6PD in the regulation of cellular metabolisms. G6PD, a rate limiting enzyme of the pentose phosphate pathway, have multiple impacts on a variety of cellular metabolisms through producing NADPH and ribulose-5-phosphate, the latter providing intermediates used for nucleic acid production. NADPH supports the NADPH oxidase (NOX)-mediated ROS generation. On the other hand, glutathione reductase also uses NADPH to reduce oxidized glutathione (GSSG) to reduced glutathione (GSH) for use by glutathione peroxidase that reduces H2O2 to H2O.
Additionally, many reports underscore that endogenous NADPH-generating systems serve dual, opposing roles in maintaining ROS balance.20-22 For instance, antioxidant systems including the glutathione and thioredoxin pathways rely heavily on NADPH for sustaining their activity. NADPH is also the primary substrate for ROS generation by NADPH oxidase (NOX) and inducible nitric oxide synthase (iNOS). NOX and iNOS transfer electrons from NADPH to oxygen and L-argine, generating superoxide and nitric oxide, respectively.23 Of note, NOX and iNOS have been implicated as pivotal regulators of ROS generation in obese adipose tissue.24 Several reports demonstrate increments in the expression of NADPH oxidase subunits in WAT of KKAy obese mice.8 iNOS is also upregulated and conspires with NADPH oxidase to activate adipose tissue inflammation and subsequent insulin resistance in obesity.25 Reciprocally, NADPH oxidase inhibitors mitigate ROS production, pro-inflammatory response and subsequent insulin resistance in obese mice.26 On the other hand, both expression and activity of enzymes involved in the antioxidant glutathione system tend to be suppressed in obese adipose tissues, which further augments oxidative stress in obesity.10,27 Both transgene expression of antioxidant genes and antioxidant molecules improve TNF α-induced insulin resistance in adipocytes as well as glucose intolerance in obese subjects.28
Among several NADPH-generating systems, the PPP largely accounts for most of cytoplasmic NADPH regeneration compared with other enzymatic systems.20 The PPP is largely divided into 2 phases: the oxidative generation of NADPH and the nonoxidative interconversion of sugars, the latter providing intermediates used for nucleotide biosynthesis or glycolytic pathway.29 Glucose-6-phosphate dehydrogenase (G6PD) is a rate-limiting enzyme of the PPP and controls the entry of G6P into the PPP. G6PD catalyzes the conversion of G6P to 6-phosphogluconolactone and the formation of NADPH from NADP+. Therefore, G6PD activity is a key determinant of the cytosolic NADP+/NADPH ratio and consequently contributes to a variety of metabolic pathways that utilize NADPH as a cofactor (Fig. 1).21
Multiple lines of evidence suggest that G6PD-derived NADPH has a contradictory impact on ROS level in different cell types (Table 1).12,30-40 Previous reports highlight that cells with intrinsic susceptibility to ROS predominantly use G6PD-derived NADPH for antioxidant defense in response to oxidative stress. In the case of red blood cells (RBCs), there is constant production of ROS by spontaneous reaction and oxygen oxidation of ferrous iron (Fe2+) to ferric iron (Fe3+). One of the distinct characteristics of RBCs is their large dependence on G6PD-derived NADPH to activate the antioxidant glutathione system as compared with other cells.31 Since RBCs are not equipped with other NADPH-producing systems, when G6PD becomes dysfunctional, these RBCs become highly susceptible to oxidative stress.31 Therefore, the most common complications of G6PD deficiency in human is acute hemolytic anemia in response to oxidizing stimuli including microbial infection.32 Neurons are another cell type that is intrinsically vulnerable to oxidative stress and their antioxidant system is significantly alleviated in response to G6PD deficiency.33 Given that neurons express antioxidant scavengers and enzymes at a very low concentration/activity, reduction of NADPH level by G6PD deficiency appears to affect ROS scavenging system more than ROS production that is mediated by both NADPH-dependent and independent pathways.34-36 Moreover, several neurodegenerative diseases are characterized by downregulation of both expression and activity of G6PD in parallel with a decrement of neuronal antioxidant response.37,38
Table 1.
Dual roles of G6PD in maintaining ROS balance in a cell type- and tissue-specific manner.
| Tissue/Cell type | Pro or Anti oxidative role | Reference |
|---|---|---|
| Bovine aortic endothelial cells | Anti | Leopold JA et al. (2007) Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat. Med. 13, 189–197 |
| β cells | Anti | Zhang Z et al. (2010) High glucose inhibits glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and β-cell apoptosis. FASEB J 24, 1497–1505 |
| Kidney | Anti | Xu Y et al. (2010) Glucose-6-phosphate dehydrogenase-deficient mice have increased renal oxidative stress and increased albuminuria. FASEB J 24, 609–616 |
| Cardiomyocyte | Anti | Jain M et al. (2003) Glucose-6-phosphate dehydrogenase modulates cytosolic redox status and contractile phenotype in adult cardiomyocytes. Circ. Res. 93, e9-l 6 |
| Aorta | Pro | Matsui R et al. (2005) Glucose-6 phosphate dehydrogenase deficiency decreases the vascular response to angiotensin IL Circulation 112, 257–263 |
| Liver | Pro | Gupte RS et al. (2009) Synergistic activation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase by Src kinase elevates superoxide in type 2 diabetic, Zucker fa/fa, rat liver. Free Radie Biol Med. 47, 219–228 |
| Heart | Pro | Serpillon S et al. (2009) Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived NADPH. Am J Physiol Heart Circ Physiol. 297, Hl 53–62 |
| Adipocyte | Pro | Park J et al. (2006) Increase in glucose-6-phosphate dehydrogenase in adipocytes stimulates oxidative stress and inflammatory signals. Diabetes 55, 2939–2949 |
| β cell | Pro | Lee JW and Choi AH et al. (2011) G6PD upregulation promotes pancreatic β-cell dysfunction. Endocrinology 152, 793–803 |
| Macrophage | Pro | Ham M et al. (2013) Macrophage glucose-6-phosphate dehydrogenase stimulates proinflammatory responses with oxidative stress. Mal Cell Biol. 33, 2425–2435 |
Conversely, pro-oxidant pathways in certain types of cell are more sensitive to changes in the level of G6PD-derived NADPH compared with antioxidant pathways. Under stressful conditions, G6PD prominently potentiates ROS generation in several cell types including macrophages, granulocytes, and myocardial cells.14,41 In the case of myocardial cells, pharmacological inhibition of G6PD results in the suppression of ROS generation when exposed to environmental cues causing heart failure.42,43 Particularly, the pro-oxidant role of G6PD manifests in pro-inflammatory responses of macrophages.14,44 Mononuclear cells isolated from G6PD-deficient patients secrete fewer pro-inflammatory cytokines than those from normal subjects.44 Additionally, G6PD is upregulated and augments ROS production and subsequent pro-inflammatory responses in macrophages when challenged with obesity-related stimuli such as free fatty acids and lipopolysaccharide (LPS).12 In line with in vitro data, G6PD expression increases in adipose tissues of both obese mice and obese human individuals, which significantly correlates with increased oxidative stress, adipose tissue inflammation and insulin resistance.12,42,43 Apparently, macrophages utilize G6PD-derived NADPH to produce ROS that stimulates NADPH oxidase and iNOS expression through activation of NFκB and p38, potentiating vicious cycle of ROS production in obesity.6,12,42,43 In addition to ROS production, NFκB and p38 promptly induce expression and secretion of pro-inflammatory cytokines in macrophages and mediate adipose tissue inflammation.14 Suppression of G6PD expression/activity downregulates NADPH oxidase and iNOS, ROS production and mitigates pro-inflammatory response in classically activated macrophages.12,39,40
Very recently, the importance of G6PD to adipose tissue dysfunction is substantiated in diet-induced obesity.45 In agreement with cell culture and ex vivo data, G6PD-deficient mutant mice (G6PDmut) display amelioration of adipose tissue inflammation with a concomitant improvement in dysfunction of adipose tissue in obesity. The expression of pro-inflammatory genes decreases whereas the expression of adiponectin, one of the beneficial adipokines, increases in adipose tissue of high fat diet fed G6PDmut mice.45 Additionally, ROS production and NADPH oxidase expression in adipose tissue are significantly decreased in G6PDmut mice.45 Together, such changes in adipose tissue result in the improvement of whole body insulin sensitivity and glucose tolerance.45 One of the distinct phenotypes of G6PDmut mice, which differs from other knockout mice lacking NADPH-producing enzymes, is that there are no significant changes in the lipid synthesis and bodyweight in diet-induced obesity. These phenotypes are of interest as they imply that a decrease in G6PD-derieved NADPH appears to selectively diminish ROS generation without having impacts on lipid synthesis.45 Molecular mechanisms involved in such selective effect are under investigation. Importantly, adoptive transfer of G6PDmut bone marrow mitigated obesity-induced ROS generation, adipose tissue inflammation, and systemic insulin resistance in obesity. Previously, it has been demonstrated that G6PD is upregulated in both adipocyte and stromal vascular fraction isolated from obese adipose tissue.12,39 Moreover, G6PD overexpression in vitro stimulates oxidative stress and pro-inflammatory response in adipocytes.39 Given that G6PD expression is retained in the adipocytes of recipient mice, the results of bone marrow transplantation suggest the relative importance of macrophage G6PD in adipose tissue inflammation and consequent changes in whole body energy homeostasis.45 In obesity, there is a significant increase in the recruitment of macrophages to adipose tissue, which in turn aggravates adipose tissue inflammation. Thus, a large influx of G6PDmut bone marrow derived macrophages into adipose tissue would overcome the deleterious effects of increased G6PD expression in obese adipocytes.45
In conclusion, our recent findings bring to light hematopoietic G6PD as a potential candidate for pharmacological inhibition in the context of obesity (Fig. 2). Although the pathophysiological roles of G6PD manifest in obesity, the underlying molecular mechanisms involved in obesity-induced G6PD expression remain to be elucidated. Previously, we demonstrate that nutrient excess and pro-inflammatory cues increase G6PD expression in both adipocytes and macrophages in vitro.12,39 Furthermore, several studies suggest that hypoxia and enzymes mediating histone modification actively control G6PD expression/activity in various cell types such as neurons and muscle cells.46-48 Because adipose tissue simultaneously faces those signaling pathways in obesity, further studies are needed to precisely delineate the signaling pathways leading to upregulation of G6PD in obesity. In addition, another open question is what molecular events are responsible for dual, opposing effects of G6PD deficiency on ROS balance in different cell types. A better understanding of such regulation would be important for identifying therapeutic targets for diseases associated with G6PD-induced dysregulation of ROS control.
Figure 2.
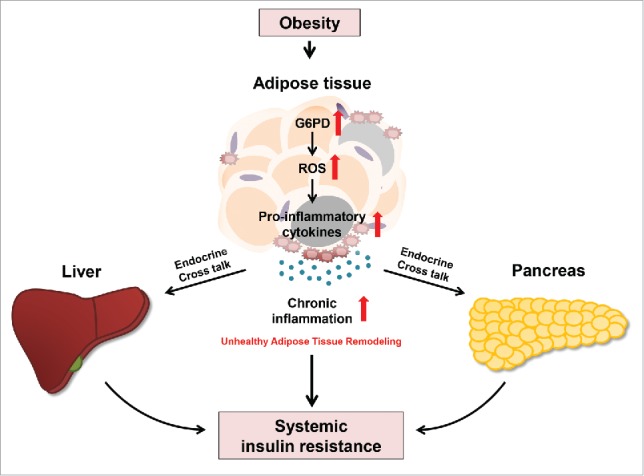
Mechanism by which G6PD drives adipose tissue inflammation and systemic insulin resistance in obesity. Obese adipose tissue manifests in oxidative stress and secretion of pro-inflammatory cytokines in response to metabolic stresses, which is associated with G6PD activation. In obesity, G6PD expression increases and stimulates accumulation of oxidative stress by inducing ROS generation. Increased ROS activates transcriptional factors involved in the expression of pro-inflammatory cytokines. In turn, adipose tissue inflammation elevates and leads to systemic insulin resistance in obesity.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Creative Research Initiative Program of the National Research Foundation (NRF) funded by the Korean government (the Ministry of Science, ICT & Future Planning, 2011–0018312).
References
- [1].Huh JY, Park YJ, Ham M, Kim JB. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol Cells 2014; 37(5):365-71. Available from https://www.ncbi.nlm.nih.gov/pubmed/24781408; PMID:24781408; https://doi.org/ 10.14348/molcells.2014.0074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest 2011; 121(6):2094-101. Available from https://www.ncbi.nlm.nih.gov/pubmed/21633177; PMID:21633177; https://doi.org/ 10.1172/JCI45887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993; 259(5091):87-91. Available from https://www.ncbi.nlm.nih.gov/pubmed/7678183; PMID:7678183; https://doi.org/ 10.1126/science.7678183 [DOI] [PubMed] [Google Scholar]
- [4].Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, et al.. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003; 112(12):1821-30. Available from https://www.ncbi.nlm.nih.gov/pubmed/14679177; PMID:14679177; https://doi.org/ 10.1172/JCI200319451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 2006; 116(7):1793-801. Available from https://www.ncbi.nlm.nih.gov/pubmed/16823477; PMID:16823477; https://doi.org/ 10.1172/JCI29069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest 2011; 121(6):2111-7. Available from https://www.ncbi.nlm.nih.gov/pubmed/21633179; PMID:21633179; https://doi.org/ 10.1172/JCI57132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett 2008; 582(1):97-105. Available from https://www.ncbi.nlm.nih.gov/pubmed/18053812; PMID:18053812; https://doi.org/ 10.1016/j.febslet.2007.11.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ebstein W. Invited comment on W. Ebstein: On the therapy of diabetes mellitus, in particular on the application of sodium salicylate. J Mol Med (Berl) 2002; 80(10):618; discussion 619. Available from https://www.ncbi.nlm.nih.gov/pubmed/12530411; PMID:12530411; https://doi.org/ 10.1007/s00109-002-0383-x [DOI] [PubMed] [Google Scholar]
- [9].Park J, Chung JJ, Kim JB. New evaluations of redox regulating system in adipose tissue of obesity. Diabetes Res Clin Pract 2007; 77 Suppl 1:S11-6. Available fromAvailable from https://www.ncbi.nlm.nih.gov/pubmed/17452057; https://doi.org/ 10.1016/j.diabres.2007.01.037 [DOI] [PubMed] [Google Scholar]
- [10].Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 2004; 114(12):1752-61. Available from https://www.ncbi.nlm.nih.gov/pubmed/15599400; PMID:15599400; https://doi.org/ 10.1172/JCI21625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kanamoto T, Rimayanti UHO and Kiuchi Y. Platelet-derived growth factor receptor alpha is associated with oxidative stress-induced retinal cell death. Curr Eye Res 2011; 36(4):336-40. Available from https://www.ncbi.nlm.nih.gov/pubmed/21405954; PMID:21405954; https://doi.org/ 10.3109/02713683.2011.556301 [DOI] [PubMed] [Google Scholar]
- [12].Ham M, Lee JW, Choi AH, Jang H, Choi G, Park J, Kozuka C, Sears DD, Masuzaki H, Kim JB. Macrophage glucose-6-phosphate dehydrogenase stimulates proinflammatory responses with oxidative stress. Mol Cell Biol 2013; 33(12):2425-35. Available from https://www.ncbi.nlm.nih.gov/pubmed/23572562; PMID:23572562; https://doi.org/ 10.1128/MCB.01260-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Keaney JF Jr, Larson MG, Vasan RS, Wilson PW, Lipinska I, Corey D, Massaro JM, Sutherland P, Vita JA, Benjamin EJ, et al.. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol 2003; 23(3):434-9. Available from https://www.ncbi.nlm.nih.gov/pubmed/12615693; PMID:12615693; https://doi.org/ 10.1161/01.ATV.0000058402.34138.11 [DOI] [PubMed] [Google Scholar]
- [14].Olusi SO. Obesity is an independent risk factor for plasma lipid peroxidation, depletion of erythrocyte cytoprotectic enzymes in humans. Int J Obes Relat Metab Disord 2002; 26(9):1159-64. Available from https://www.ncbi.nlm.nih.gov/pubmed/12187391; PMID:12187391; https://doi.org/ 10.1038/sj.ijo.0802066 [DOI] [PubMed] [Google Scholar]
- [15].Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 2013; 12(12):931-47. Available from https://www.ncbi.nlm.nih.gov/pubmed/24287781; PMID:24287781; https://doi.org/ 10.1038/nrd4002 [DOI] [PubMed] [Google Scholar]
- [16].Le Lay S, Simard G, Martinez MC, Andriantsitohaina R. Oxidative stress and metabolic pathologies: from an adipocentric point of view. Oxid Med Cell Longev 2014; 2014:908539. Available from https://www.ncbi.nlm.nih.gov/pubmed/25143800; PMID:25143800; https://doi.org/ 10.1155/2014/908539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhou T, Zhou KK, Lee K, Gao G, Lyons TJ, Kowluru R, Ma JX. The role of lipid peroxidation products and oxidative stress in activation of the canonical wingless-type MMTV integration site (WNT) pathway in a rat model of diabetic retinopathy. Diabetologia 2011; 54(2):459-68. Available from https://www.ncbi.nlm.nih.gov/pubmed/20978740; PMID:20978740; https://doi.org/ 10.1007/s00125-010-1943-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Koh HJ, Lee SM, Son BG, Lee SH, Ryoo ZY, Chang KT, Park JW, Park DC, Song BJ, Veech RL, et al.. Cytosolic NADP+-dependent isocitrate dehydrogenase plays a key role in lipid metabolism. J Biol Chem 2004; 279(38):39968-74. Available from https://www.ncbi.nlm.nih.gov/pubmed/15254034; PMID:15254034; https://doi.org/ 10.1074/jbc.M402260200 [DOI] [PubMed] [Google Scholar]
- [19].Al-Dwairi A, Pabona JM, Simmen RC, Simmen FA. Cytosolic malic enzyme 1 (ME1) mediates high fat diet-induced adiposity, endocrine profile, and gastrointestinal tract proliferation-associated biomarkers in male mice. PLoS One 2012; 7(10):e46716. Available from https://www.ncbi.nlm.nih.gov/pubmed/23056418; PMID:23056418; https://doi.org/ 10.1371/journal.pone.0046716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Stanton RC. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012; 64(5):362-9. Available from https://www.ncbi.nlm.nih.gov/pubmed/22431005; PMID:22431005; https://doi.org/ 10.1002/iub.1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hecker PA, Leopold JA, Gupte SA, Recchia FA, Stanley WC. Impact of glucose-6-phosphate dehydrogenase deficiency on the pathophysiology of cardiovascular disease. Am J Physiol Heart Circ Physiol 2013; 304(4):H491-500. Available from https://www.ncbi.nlm.nih.gov/pubmed/23241320; PMID:23241320; https://doi.org/ 10.1152/ajpheart.00721.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res 2010; 107(9):1058-70. Available from https://www.ncbi.nlm.nih.gov/pubmed/21030723; PMID:21030723; https://doi.org/ 10.1161/CIRCRESAHA.110.223545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007; 87(1):245-313. Available from https://www.ncbi.nlm.nih.gov/pubmed/17237347; PMID:17237347; https://doi.org/ 10.1152/physrev.00044.2005 [DOI] [PubMed] [Google Scholar]
- [24].Mittal M, Gu XQ, Pak O, Pamenter ME, Haag D, Fuchs DB, Schermuly RT, Ghofrani HA, Brandes RP, Seeger W, et al.. Hypoxia induces Kv channel current inhibition by increased NADPH oxidase-derived reactive oxygen species. Free Radic Biol Med 2012; 52(6):1033-42. Available from https://www.ncbi.nlm.nih.gov/pubmed/22222468 [DOI] [PubMed] [Google Scholar]
- [25].Lee JW, Choi AH, Ham M, Kim JW, Choe SS, Park J, Lee GY, Yoon KH, Kim JB. G6PD up-regulation promotes pancreatic beta-cell dysfunction. Endocrinology 2011; 152(3):793-803. Available from https://www.ncbi.nlm.nih.gov/pubmed/21248143; PMID:21248143; https://doi.org/ 10.1210/en.2010-0606 [DOI] [PubMed] [Google Scholar]
- [26].Meng R, Zhu DL, Bi Y, Yang DH, Wang YP. Apocynin improves insulin resistance through suppressing inflammation in high-fat diet-induced obese mice. Mediators Inflamm 2010; 2010:858735. Available from https://www.ncbi.nlm.nih.gov/pubmed/21403905; PMID:21403905; https://doi.org/ 10.1155/2010/858735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Curtis JM, Grimsrud PA, Wright WS, Xu X, Foncea RE, Graham DW, Brestoff JR, Wiczer BM, Ilkayeva O, Cianflone K, et al.. Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes 2010; 59(5):1132-42. Available from https://www.ncbi.nlm.nih.gov/pubmed/20150287; PMID:20150287; https://doi.org/ 10.2337/db09-1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006; 440(7086):944-8. Available from https://www.ncbi.nlm.nih.gov/pubmed/16612386; PMID:16612386; https://doi.org/ 10.1038/nature04634 [DOI] [PubMed] [Google Scholar]
- [29].Riganti C, Aldieri E, Bergandi L, Fenoglio I, Costamagna C, Fubini B, Bosia A, Ghigo D. Crocidolite asbestos inhibits pentose phosphate oxidative pathway and glucose 6-phosphate dehydrogenase activity in human lung epithelial cells. Free Radic Biol Med 2002; 32(9):938-49. Available from https://www.ncbi.nlm.nih.gov/pubmed/11978496; PMID:11978496; https://doi.org/ 10.1016/S0891-5849(02)00800-6 [DOI] [PubMed] [Google Scholar]
- [30].Nobrega-Pereira S, Fernandez-Marcos PJ, Brioche T, Gomez-Cabrera MC, Salvador-Pascual A, Flores JM, Vina J, Serrano M. G6PD protects from oxidative damage and improves healthspan in mice. Nat Commun 2016; 7:10894. Available from https://www.ncbi.nlm.nih.gov/pubmed/26976705; PMID:26976705; https://doi.org/ 10.1038/ncomms10894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Leite AA, Barretto OC. Erythrocyte glucose-6-phosphate dehydrogenase activity assay and affinity for its substrate under “physiological” conditions. Braz J Med Biol Res 1998; 31(12):1533-5. Available from https://www.ncbi.nlm.nih.gov/pubmed/9951548; PMID:9951548; https://doi.org/ 10.1590/S0100-879X1998001200004 [DOI] [PubMed] [Google Scholar]
- [32].Galvez R, Ribera V, Gonzalez-Escalada JR, Souto A, Canovas ML, Castro A, Herrero B, de Los Angeles Maqueda M, Castilforte M, Marco-Martinez JJ, et al.. Analgesic efficacy of zoledronic acid and its effect on functional status of prostate cancer patients with metastasis. Patient Prefer Adherence 2008; 2:215-24. Available from https://www.ncbi.nlm.nih.gov/pubmed/19920966; PMID:19920966; https://doi.org/ 10.2147/PPA.S2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fanucchi MV, Bracher A, Doran SF, Squadrito GL, Fernandez S, Postlethwait EM, Bowen L, Matalon S. Post-exposure antioxidant treatment in rats decreases airway hyperplasia and hyperreactivity due to chlorine inhalation. Am J Respir Cell Mol Biol 2012; 46(5):599-606. Available from https://www.ncbi.nlm.nih.gov/pubmed/22162906; PMID:22162906; https://doi.org/ 10.1165/rcmb.2011-0196OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Raps SP, Lai JC, Hertz L, Cooper AJ. Glutathione is present in high concentrations in cultured astrocytes but not in cultured neurons. Brain Res 1989; 493(2):398-401. Available from https://www.ncbi.nlm.nih.gov/pubmed/2765907; PMID:2765907; https://doi.org/ 10.1016/0006-8993(89)91178-5 [DOI] [PubMed] [Google Scholar]
- [35].Makar TK, Nedergaard M, Preuss A, Gelbard AS, Perumal AS, Cooper AJ. Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: evidence that astrocytes play an important role in antioxidative processes in the brain. J Neurochem 1994; 62(1):45-53. Available from https://www.ncbi.nlm.nih.gov/pubmed/7903354; PMID:7903354; https://doi.org/ 10.1046/j.1471-4159.1994.62010045.x [DOI] [PubMed] [Google Scholar]
- [36].Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol 2000; 62(6):649-71. Available from https://www.ncbi.nlm.nih.gov/pubmed/10880854; PMID:10880854; https://doi.org/ 10.1016/S0301-0082(99)00060-X [DOI] [PubMed] [Google Scholar]
- [37].Russell RL, Siedlak SL, Raina AK, Bautista JM, Smith MA, Perry G. Increased neuronal glucose-6-phosphate dehydrogenase and sulfhydryl levels indicate reductive compensation to oxidative stress in Alzheimer disease. Arch Biochem Biophys 1999; 370(2):236-9. Available from https://www.ncbi.nlm.nih.gov/pubmed/10510282; PMID:10510282; https://doi.org/ 10.1006/abbi.1999.1404 [DOI] [PubMed] [Google Scholar]
- [38].Palmer AM. The activity of the pentose phosphate pathway is increased in response to oxidative stress in Alzheimer's disease. J Neural Transm (Vienna) 1999; 106(3-4):317-28. Available from https://www.ncbi.nlm.nih.gov/pubmed/10392540; PMID:10392540; https://doi.org/ 10.1007/s007020050161 [DOI] [PubMed] [Google Scholar]
- [39].Park J, Choe SS, Choi AH, Kim KH, Yoon MJ, Suganami T, Ogawa Y, Kim JB. Increase in glucose-6-phosphate dehydrogenase in adipocytes stimulates oxidative stress and inflammatory signals. Diabetes 2006; 55(11):2939-49. Available from https://www.ncbi.nlm.nih.gov/pubmed/17065329; PMID:17065329; https://doi.org/ 10.2337/db05-1570 [DOI] [PubMed] [Google Scholar]
- [40].Park J, Rho HK, Kim KH, Choe SS, Lee YS, Kim JB. Overexpression of glucose-6-phosphate dehydrogenase is associated with lipid dysregulation and insulin resistance in obesity. Mol Cell Biol 2005; 25(12):5146-57. Available from https://www.ncbi.nlm.nih.gov/pubmed/15923630; PMID:15923630; https://doi.org/ 10.1128/MCB.25.12.5146-5157.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hothersall JS, Gordge M, Noronha-Dutra AA. Inhibition of NADPH supply by 6-aminonicotinamide: effect on glutathione, nitric oxide and superoxide in J774 cells. FEBS Lett 1998; 434(1-2):97-100. Available from https://www.ncbi.nlm.nih.gov/pubmed/9738459; PMID:9738459; https://doi.org/ 10.1016/S0014-5793(98)00959-4 [DOI] [PubMed] [Google Scholar]
- [42].Gupte RS, Vijay V, Marks B, Levine RJ, Sabbah HN, Wolin MS, Recchia FA, Gupte SA. Upregulation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase activity increases oxidative stress in failing human heart. J Card Fail 2007; 13(6):497-506. Available from https://www.ncbi.nlm.nih.gov/pubmed/17675065; PMID:17675065; https://doi.org/ 10.1016/j.cardfail.2007.04.003 [DOI] [PubMed] [Google Scholar]
- [43].Gupte SA, Levine RJ, Gupte RS, Young ME, Lionetti V, Labinskyy V, Floyd BC, Ojaimi C, Bellomo M, Wolin MS, et al.. Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J Mol Cell Cardiol 2006; 41(2):340-9. Available from https://www.ncbi.nlm.nih.gov/pubmed/16828794; PMID:16828794; https://doi.org/ 10.1016/j.yjmcc.2006.05.003 [DOI] [PubMed] [Google Scholar]
- [44].Sanna F, Bonatesta RR, Frongia B, Uda S, Banni S, Melis MP, Collu M, Madeddu C, Serpe R, Puddu S, et al.. Production of inflammatory molecules in peripheral blood mononuclear cells from severely glucose-6-phosphate dehydrogenase-deficient subjects. J Vasc Res 2007; 44(4):253-63. Available from https://www.ncbi.nlm.nih.gov/pubmed/17361089; PMID:17361089; https://doi.org/ 10.1159/000100903 [DOI] [PubMed] [Google Scholar]
- [45].Ham M, Choe SS, Shin KC, Choi G, Kim JW, Noh JR, Kim YH, Ryu JW, Yoon KH, Lee CH, et al.. Glucose-6-Phosphate Dehydrogenase Deficiency Improves Insulin Resistance With Reduced Adipose Tissue Inflammation in Obesity. Diabetes 2016; 65(9):2624-38. Available from https://www.ncbi.nlm.nih.gov/pubmed/27284106; PMID:27284106; https://doi.org/ 10.2337/db16-0060 [DOI] [PubMed] [Google Scholar]
- [46].Chettimada S, Gupte R, Rawat D, Gebb SA, McMurtry IF, Gupte SA. Hypoxia-induced glucose-6-phosphate dehydrogenase overexpression and -activation in pulmonary artery smooth muscle cells: implication in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2015; 308(3):L287-300. Available from https://www.ncbi.nlm.nih.gov/pubmed/25480333; PMID:25480333; https://doi.org/ 10.1152/ajplung.00229.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gao L, Mejias R, Echevarria M, Lopez-Barneo J. Induction of the glucose-6-phosphate dehydrogenase gene expression by chronic hypoxia in PC12 cells. FEBS Lett 2004; 569(1-3):256-60. Available from https://www.ncbi.nlm.nih.gov/pubmed/15225644; PMID:15225644; https://doi.org/ 10.1016/j.febslet.2004.06.004 [DOI] [PubMed] [Google Scholar]
- [48].Makarona K, Caputo VS, Costa JR, Liu B, O'Connor D, Iskander D, Roper D, Robertson L, Bhatnagar N, Terpos E, et al.. Transcriptional and epigenetic basis for restoration of G6PD enzymatic activity in human G6PD-deficient cells. Blood 2014; 124(1):134-41. Available from https://www.ncbi.nlm.nih.gov/pubmed/24805191; PMID:24805191; https://doi.org/ 10.1182/blood-2014-02-553792 [DOI] [PubMed] [Google Scholar]