

Abstract
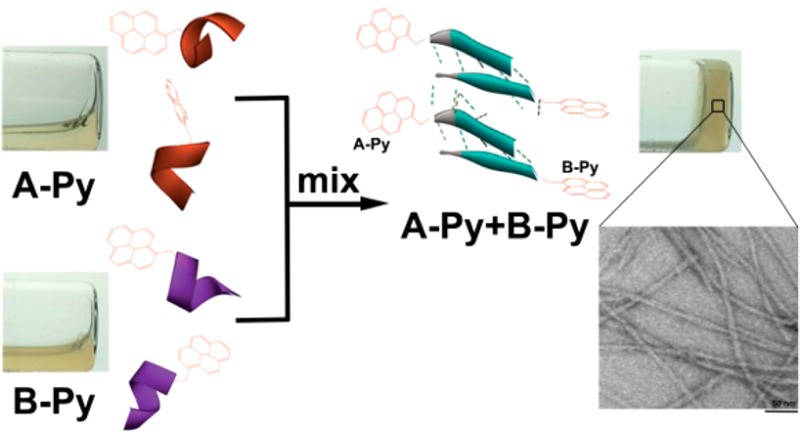
Isolated short peptides usually are unable to maintain their original secondary structures due to the lack of the restriction from proteins. Here we show that two complementary pentapeptides from a β-sheet motif of a protein, being connected to an aromatic motif (i.e., pyrene) at their C-terminal, self-assemble to form β-sheet like structures upon mixing. Besides enabling the self-assembly to result in supramolecular hydrogels upon mixing, aromatic–aromatic interactions promote the pentapeptides transform from α-helix to β-sheet conformation. As the first example of using aromatic–aromatic interactions to mimic the conformational restriction in a protein, this work illustrates a bioinspired way to generate peptide nanofibers with predefined secondary structures of the peptides by a rational design using protein structures as the blueprint.
The self-assembly of peptides to form nanoscale structures is common for naturally occurring1 and synthetic2 peptides. Insulin,1a,1c amyloids,1b defensin,1d and hormones3 are known natural peptides that self-assemble to form nanofibrils with beneficial or detrimental biological effects. Recently, the exploration of the biomedical applications of nanostructures formed by peptide amphiphiles2d,4 or oligopeptides5 has progressed significantly. Similarly, synthetic peptide derivatives that favor aromatic–aromatic interactions have received considerable attentions for the development of soft materials (e.g., hydrogels6) or nanoscale assemblies7 for potential biomedical applications, such as drug delivery,2c,8 regenerative medicine,9 antibacterial agents,10 and anticancer therapy.8b,11 Despite these advances, it remains difficult to control or predict the secondary structures formed by the self-assembling peptide derivatives (especially when the lengths of the peptides are short), a major obstacle for the further development of peptide soft biomaterials.
Although currently it remains impossible to predict the secondary structures of a peptide from the sequence alone, structural biology research has determined many protein structures (e.g., over 120 000 structures in protein data bank (PDB)12), which, as an invaluable bioinformatics resource, provides a candidate pool of peptides with known secondary structures. However, since the secondary structures are resulted from conformation restriction provided by multiple forces13 in proteins, the isolated short peptide sequence usually are unable to maintain their secondary structures exhibited in the proteins. Therefore, in order to use the protein structure to guide the design of self-assembling short peptides with predictable secondary structures, there is a need of facile strategy that defines the secondary structures of short peptides after their self-assembly. Inspired by nature,14 we decide to use aromatic–aromatic interactions to enhance intermolecular interactions for maintaining secondary structures of short peptides. Moreover, having relatively compact volumes, aromatic rings are effective motifs for enabling self-assembly,15 including the self-assembly of peptides,6a and even can generate spontaneous alignment of nanofibrils.16 In addition, being inherently directional,14 aromatic–aromatic interaction is fairly predictable and results in stable supramolecular hydrogels.6a,17
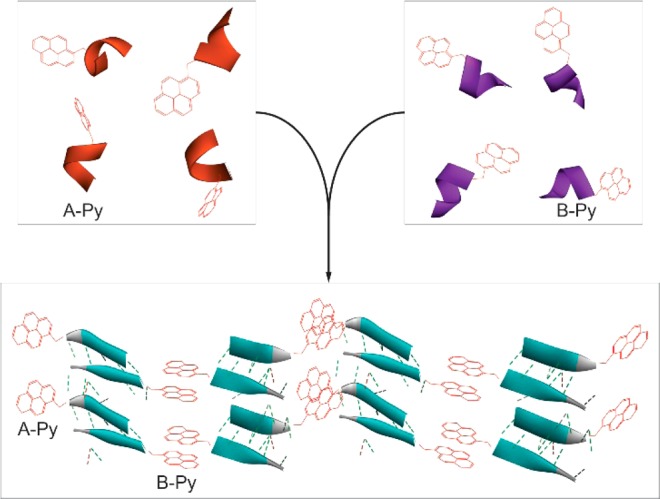
Based on the above rationale, we choose a decapeptidic sequence that forms a β-sheet at the intermolecular interface of the dimer of a protein (irisin).18 We connect pyrene (for aromatic–aromatic interactions) at the C-terminals of two complementary pentapeptidic segments (A and B), which forms seven intermolecular hydrogen bonds between them (Figure 1). This design produces two pyrene peptide conjugates (A-Py and B-Py). Our studies find that simply mixing A-Py with B-Py results in a supramolecular hydrogel that consists of nanofibrils, but each conjugate itself is unable to form a hydrogel. Fluorescent spectra confirm the aromatic–aromatic interaction between pyrene groups. Circular dichroism (CD) reveals that, while each conjugate mainly exists as α-helix, their complementary mixtures adopt β-sheet like secondary structures in the hydrogel (Scheme 1). Transmission electron microscopy (TEM) reveals that the intermolecular interactions between the complementary peptide derivatives promote the formation of nanofibrils with uniform widths. The enantiomers of A-Py and B-Py (i.e., DA-Py and DB-Py) also exhibit similar behaviors as those of A-Py and B-Py. As the first use of aromatic–aromatic interactions to mimic the conformational restriction in a protein and enable the transition from α-helical to β-sheet like structures of short peptides, this work illustrates a new, bioinspired approach to control molecular recognition in water19 and to generate supramolecular peptide nanofibers with predefined secondary structures by rationally using protein structures as the blueprint.
Figure 1.
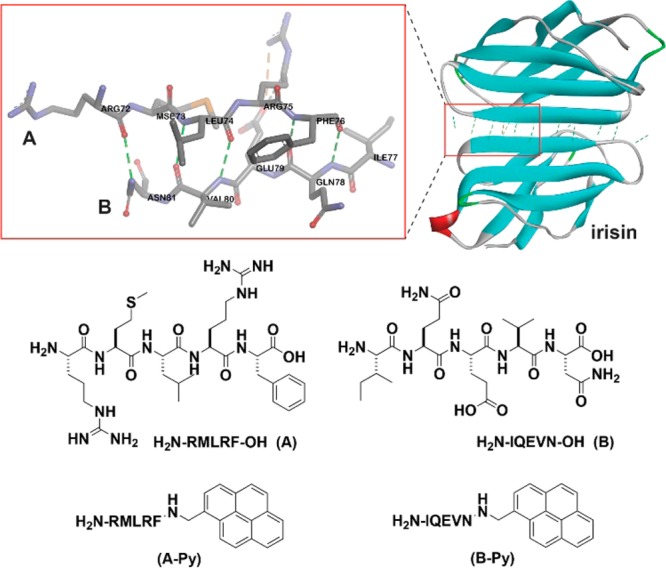
Pentapeptides and the corresponding C-terminal capped pentapeptides and the hydrogen bonds between the pentapeptide pair (A and B) at the interface of irisin dimer (adapted from the crystal structure of irisin (PDB: 4LSD(18)).
Scheme 1. Aromatic–Aromatic Interaction Enables Self-Assembly and α-Helix to β-Sheet Transition.
The decapeptidic sequence from irisin18 is Arg-Met-Leu-Arg-Phe-Ile-Gln-Glu-Val-Asn (RMLRFIQEVN), which self-associates to form an antiparallel β-sheet in irisin dimer (Figure 1). To avoid the self-association of the decapeptide, we divide it into two pentapeptides: RMLRF (A) and IQEVN (B). According to the crystal structure of irisin,18A and B form seven intermolecular hydrogen bonds. Because RMLRFIQEVN forms the antiparallel β-sheet, it is unlikely that RMLRF (A) (or IQEVN (B)) prefers self-dimerization. To introduce and monitor the aromatic–aromatic interactions, we conjugate pyrene, a molecule that gives characteristic excimer fluorescence,20 at the C-terminal of A and B. The studies of the mixture of A and B, A-Py and B-Py provide insights on how the aromatic–aromatic interactions dictate the secondary structures and self-assembly of these peptides.
Solid phase peptide synthesis (SPPS)21 affords A and B in excellent yields (90%, Scheme S1). The synthesis of A-Py and B-Py requires the combination of SPPS and liquid phase synthesis, and the overall yield is about 80%. As shown in Figure 2, all of the four molecules are unable to form homotypic hydrogels. A or B is a transparent and colorless solution. With the conjugation of pyrene, A-Py or B-Py forms a light yellow solution. The mixture of A and B still remains as a transparent, colorless solution. The mixture of A-Py and B-Py (5.0 mM each) forms a stable hydrogel within 5 min. This heterotypic hydrogelation is also supported by rheology (Figure S7) and 1H NMR (Figure S8) experiments. The fast hydrogelation upon the mixing of A-Py and B-Py indicates strong interaction between A-Py and B-Py. These results reveal that the conjugation of pyrene to the peptides at C-terminal allows the conjugates to self-assemble in water.
Figure 2.

Optical images (without or under UV irradiation) of the solutions or hydrogels formed by 10 mM of A, B, A-Py, and B-Py, respectively or the mixture of 5.0 mM A and 5.0 mM B, 5.0 mM A-Py and 5.0 mM B-Py in PBS buffer at pH= 7.4.
The TEM images of A or B at 10 mM or the mixture of 5.0 mM of A and B hardly exhibit any features (Figure 3), confirming that A or B alone or their mixture is unable to self-assemble in water, agreeing with the results shown in Figure 2. In contrast, the TEM image of the mixture of A-Py and B-Py shows nanofibers with uniform diameters of 8 ± 2 nm, which differ from the TEM image of A-Py or B-Py at 10 mM that exhibits polymorphism, that is, nanofibers with different diameters and aggregates (for A-Py) or nanofibers together with aggregates (for B-Py). These TEM images confirm that the conjugation of pyrene enables the self-assembly of the pentapeptides from irisin in water to form well-defined nanofibers.
Figure 3.

TEM images of the solutions of A, B, A-Py, and B-Py at 10 mM or the mixture of 5.0 mM A and 5.0 mM B, and the hydrogel of the mixture of 5.0 mM A-Py and 5.0 mM B-Py. All are in PBS buffer and pH = 7.4 (scale bar = 100 nm).
The emission spectrum of 10 mM of A-Py (Figure 4A) shows a stronger peak around 400 nm (pyrene monomer) than that around 470 nm (pyrene excimers22), indicating that monomeric A-Py dominates at 10 mM. B-Py exhibits the peak around 400 nm and the peak centered at 470 nm to be comparable in intensity, suggesting that monomeric and dimeric B-Py coexist in the solution at 10 mM. The mixture of A-Py and B-Py, at 5.0 mM, shows a large broad peak centered at 470 nm, confirming that pyrene–pyrene interactions dominate in the mixture and agreeing with that the mixture of A-Py and B-Py forms a hydrogel. These results agree well with the TEM images (Figure 3). As shown in Figure 4B, the CD spectrum of solution of A-Py (10 mM) exhibits two negative peaks at 202 and 228 nm, suggesting α-helix like conformation. The solution of B-Py (10 mM) shows two small negative peaks at 199 and 220 nm, also suggesting α-helix. The weaker CD signal of B-Py than that of A-Py is likely due to the pyrene–pyrene interactions in the solution of B-Py. In the case of B-Py, the self-assembly of these α-helices likely prefers antiparallel arrangement, which decreases the CD signal, which agrees with the fluorescent spectra. The mixture of A-Py and B-Py shows a quite different CD spectrum that it contains a positive peak at 202 nm and a negative peak around 220 nm, which is similar to the CD spectrum of β-sheet like structure. The above CD spectra indicate that the molecules themselves tend to form α-helix like structure, while their mixture forms β-sheet like structures.
Figure 4.
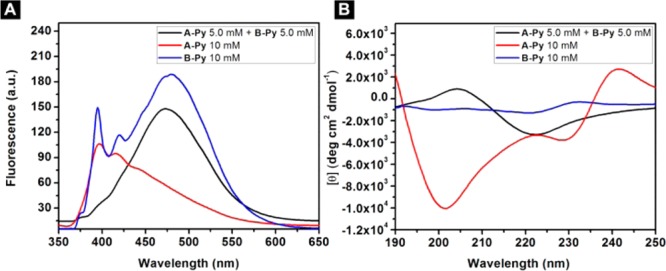
(A) Emission spectra (λex = 330 nm) and (B) CD spectra of the solutions of A-Py (10 mM), B-Py (10 mM) and the hydrogel of the mixture of A-Py (5.0 mM) and B-Py (5.0 mM) in PBS buffer at pH = 7.4.
The decrease of the concentration of the mixture of A-Py and B-Py from 2.5 mM to 0.040 mM results in the increase of the peaks around 375 and 400 nm (Figure 5A). The ratio between the intensity of the peaks at 470 and 375 almost drops to 0 at 0.040 mM (Figure S9), which confirms that the transition point is around 0.040 mM. These results confirm the aromatic–aromatic interaction between pyrene in water. As shown in Figure 5B, with the decrease of the concentration from 2.5 mM to 0.16 mM, the CD spectra of the mixtures of A-Py and B-Py show the increase of the intensity of the transition between 200 and 220 nm, indicating less β-sheet features. When concentration drops to 0.040 mM, positive peak around 200 nm becomes negative, which indicates that there is more α-helix like structures in the solution. These results support that the aromatic–aromatic interactions promote the conversion from α-helices to β-sheet of the pentapeptides (Scheme 1).
Figure 5.
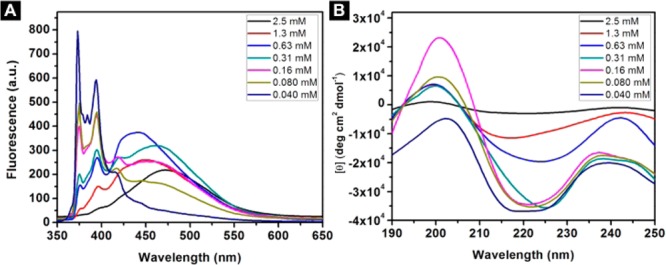
(A) Emission spectra (λex = 330 nm) and (B) circular dichroism (CD) spectra of the solutions of the mixtures of A-Py and B-Py from 2.5 mM to 0.040 mM in PBS buffer at pH = 7.4.
The TEM images (Figure S10) of the mixtures at lower concentrations show that the mixture of A-Py and B-Py contains uniform nanofibers until the concentration below 0.080 mM. Besides, with the decrease of the concentration, the widths of nanofibers in the mixture of A-Py and B-Py remain constant, which confirms that the binding between A-Py and B-Py at the concentration above 0.040 mM. The mixture of A-Py and B-Py has a low critical micelle concentration (CMC) value at 8 μM while the molecules alone show much higher CMC (575 and 338 μM for A-Py and B-Py, respectively), more than 40-folds than that of the mixture (Figure S11). These results agree with the fluorescence results and TEM images. Moreover, we replace the l-amino acid residues in A-Py and B-Py with d-amino acid residues (Scheme S2) to generate two enantiomers, DA-Py and DB-Py. The mixture of DA-Py and DB-Py forms a hydrogel while DA-Py or DB-Py alone remains as a solution (Figure S12). TEM reveals that the nanofibers in the hydrogel have the uniform widths of 8 ± 2 nm. Similarly, the CD spectra of the mixture of DA-Py and DB-Py exhibit β-sheet like feature and DA-Py or DB-Py alone shows α-helix like structure (Figure S13). Thus, the blueprint from the protein structures is applicable for designing nanofibers of d-peptide derivatives by simply replacing l-amino acids with d-amino acids.
In conclusion, we have rationally developed self-assembling molecules by connecting an aromatic motif to the C-terminal of two pentapeptides that are part of a β-sheet motif of a protein. We also find that the regiochemistry, the distance between the pyrene and the pentapeptides, and sequence complementation are important (see Supporting Information) for the self-assembly and hydrogelation. This work illustrates a facile approach to design self-assembling peptides for generating soft materials that have predefined secondary structures. Although the aromatic motif used here is pyrene, other aromatic motifs or self-assembling enablers may provide the restriction for reconstituting the secondary structures of peptide segments observed in protein structures. Inspired by this work, our future direction might be replacing pyrene by other enabling motif for aqueous self-assembly, such as alkyl chains23 or hydrophobic amino acid residues,24 to create other types of supramolecular interactions.
Acknowledgments
This work was partially supported by NIH (R01CA142746), NSF (MRSEC-1420382), and W. M. Keck Foundation.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b11512.
Synthetic scheme, NMR and LC–MS, CD spectra for all the molecules, and additional controls (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Jimenez J. L.; Nettleton E. J.; Bouchard M.; Robinson C. V.; Dobson C. M.; Saibil H. R. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 9196. 10.1073/pnas.142459399. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hardy J.; Selkoe D. J. Science 2002, 297, 353. 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]; c Hutchison K. G. J. Pharm. Pharmacol. 1985, 37, 528. 10.1111/j.2042-7158.1985.tb03060.x. [DOI] [PubMed] [Google Scholar]; d Chu H. T.; Pazgier M.; Jung G.; Nuccio S. P.; Castillo P. A.; de Jong M. F.; Winter M. G.; Winter S. E.; Wehkamp J.; Shen B.; Salzman N. H.; Underwood M. A.; Tsolis R. M.; Young G. M.; Lu W. Y.; Lehrer R. I.; Baumler A. J.; Bevins C. L. Science 2012, 337, 477. 10.1126/science.1218831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Nagy K. J.; Giano M. C.; Jin A.; Pochan D. J.; Schneider J. P. J. Am. Chem. Soc. 2011, 133, 14975. 10.1021/ja206742m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Debnath S.; Roy S.; Ulijn R. V. J. Am. Chem. Soc. 2013, 135, 16789. 10.1021/ja4086353. [DOI] [PubMed] [Google Scholar]; c Cheetham A. G.; Zhang P. C.; Lin Y. A.; Lock L. L.; Cui H. G. J. Am. Chem. Soc. 2013, 135, 2907. 10.1021/ja3115983. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hartgerink J. D.; Beniash E.; Stupp S. I. Science 2001, 294, 1684. 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- Maji S. K.; Perrin M. H.; Sawaya M. R.; Jessberger S.; Vadodaria K.; Rissman R. A.; Singru P. S.; Nilsson K. P. R.; Simon R.; Schubert D.; Eisenberg D.; Rivier J.; Sawchenko P.; Vale W.; Riek R. Science 2009, 325, 328. 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yu Y. C.; Berndt P.; Tirrell M.; Fields G. B. J. Am. Chem. Soc. 1996, 118, 12515. 10.1021/ja9627656. [DOI] [Google Scholar]; b Kunitake T. Angew. Chem., Int. Ed. Engl. 1992, 31, 709. 10.1002/anie.199207091. [DOI] [Google Scholar]
- a Zhang S. G.; Holmes T.; Lockshin C.; Rich A. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 3334. 10.1073/pnas.90.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhao X. B.; Pan F.; Xu H.; Yaseen M.; Shan H. H.; Hauser C. A. E.; Zhang S. G.; Lu J. R. Chem. Soc. Rev. 2010, 39, 3480. 10.1039/b915923c. [DOI] [PubMed] [Google Scholar]; c Haines L. A.; Rajagopal K.; Ozbas B.; Salick D. A.; Pochan D. J.; Schneider J. P. J. Am. Chem. Soc. 2005, 127, 17025. 10.1021/ja054719o. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Huang B. Q.; Hirst A. R.; Smith D. K.; Castelletto V.; Hamley I. W. J. Am. Chem. Soc. 2005, 127, 7130. 10.1021/ja050412d. [DOI] [PubMed] [Google Scholar]; e Childers W. S.; Mehta A. K.; Lu K.; Lynn D. G. J. Am. Chem. Soc. 2009, 131, 10165. 10.1021/ja902332s. [DOI] [PubMed] [Google Scholar]; f Bowerman C. J.; Nilsson B. L. J. Am. Chem. Soc. 2010, 132, 9526. 10.1021/ja1025535. [DOI] [PubMed] [Google Scholar]
- a Ma M. L.; Kuang Y.; Gao Y.; Zhang Y.; Gao P.; Xu B. J. Am. Chem. Soc. 2010, 132, 2719. 10.1021/ja9088764. [DOI] [PubMed] [Google Scholar]; b Zhang Y.; Gu H. W.; Yang Z. M.; Xu B. J. Am. Chem. Soc. 2003, 125, 13680. 10.1021/ja036817k. [DOI] [PubMed] [Google Scholar]
- a Reches M.; Gazit E. Science 2003, 300, 625. 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]; b Anzini P.; Xu C. F.; Hughes S.; Magnotti E.; Jiang T.; Hemmingsen L.; Demeler B.; Conticello V. P. J. Am. Chem. Soc. 2013, 135, 10278. 10.1021/ja404677c. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Culshaw J. L.; Cheng G.; Schmidtmann M.; Hasell T.; Liu M.; Adams D. J.; Cooper A. I. J. Am. Chem. Soc. 2013, 135, 10007. 10.1021/ja403987j. [DOI] [PubMed] [Google Scholar]
- a Caponi P.-F.; Ulijn R. V. In Smart Materials for Drug Delivery; The Royal Society of Chemistry: 2013; Vol. 1, p 232. [Google Scholar]; b Gao Y.; Kuang Y.; Guo Z. F.; Guo Z. H.; Krauss I. J.; Xu B. J. Am. Chem. Soc. 2009, 131, 13576. 10.1021/ja904411z. [DOI] [PubMed] [Google Scholar]
- Mahler A.; Reches M.; Rechter M.; Cohen S.; Gazit E. Adv. Mater. 2006, 18, 1365. 10.1002/adma.200501765. [DOI] [Google Scholar]
- Xing B. G.; Yu C. W.; Chow K. H.; Ho P. L.; Fu D. G.; Xu B. J. Am. Chem. Soc. 2002, 124, 14846. 10.1021/ja028539f. [DOI] [PubMed] [Google Scholar]
- a Li J. Y.; Gao Y.; Kuang Y.; Shi J. F.; Du X. W.; Zhou J.; Wang H. M.; Yang Z. M.; Xu B. J. Am. Chem. Soc. 2013, 135, 9907. 10.1021/ja404215g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang H. M.; Yang Z. M. Soft Matter 2012, 8, 2344. 10.1039/C2SM06923G. [DOI] [Google Scholar]; c Yang Z. M.; Xu K. M.; Guo Z. F.; Guo Z. H.; Xu B. Adv. Mater. 2007, 19, 3152. 10.1002/adma.200701971. [DOI] [Google Scholar]; d Pires R. A.; Abul-Haija Y. M.; Costa D. S.; Novoa-Carballal R.; Reis R. L.; Ulijn R. V.; Pashkuleva I. J. Am. Chem. Soc. 2015, 137, 576. 10.1021/ja5111893. [DOI] [PubMed] [Google Scholar]; e Zhou J.; Du X. W.; Yamagata N.; Xu B. J. Am. Chem. Soc. 2016, 138, 3813. 10.1021/jacs.5b13541. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Wang H. M.; Feng Z. Q. Q.; Wu D. D.; Fritzsching K. J.; Rigney M.; Zhou J.; Jiang Y. J.; Schmidt-Rohr K.; Xu B. J. Am. Chem. Soc. 2016, 138, 10758. 10.1021/jacs.6b06075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. Nucleic Acids Res. 2000, 28, 235. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehn J.-M.Supramolecular Chemistry: Concepts and Perspectives; Wiley-VCH: New York, 1995. [Google Scholar]
- Burley S. K.; Petsko G. A. Science 1985, 229, 23. 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- a Biedermann F.; Schneider H. J. Chem. Rev. 2016, 116, 5216. 10.1021/acs.chemrev.5b00583. [DOI] [PubMed] [Google Scholar]; b Schneider H. J. Acc. Chem. Res. 2013, 46, 1010. 10.1021/ar3000579. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Du X. W.; Gao Y.; Shi J. F.; Xu B. J. Am. Chem. Soc. 2014, 136, 2970. 10.1021/ja4127399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Du X. W.; Zhou J.; Shi J. F.; Xu B. Chem. Rev. 2015, 115, 13165. 10.1021/acs.chemrev.5b00299. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yan X. Z.; Wang F.; Zheng B.; Huang F. H. Chem. Soc. Rev. 2012, 41, 6042. 10.1039/c2cs35091b. [DOI] [PubMed] [Google Scholar]; c Yu G. C.; Yan X. Z.; Han C. Y.; Huang F. H. Chem. Soc. Rev. 2013, 42, 6697. 10.1039/c3cs60080g. [DOI] [PubMed] [Google Scholar]
- Schumacher M. A.; Chinnam N.; Ohashi T.; Shah R. S.; Erickson H. P. J. Biol. Chem. 2013, 288, 33738. 10.1074/jbc.M113.516641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesides G. M. Interface Focus 2015, 5, 10. 10.1098/rsfs.2015.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z.; Singh N. J.; Lim J.; Pan J.; Kim H. N.; Park S.; Kim K. S.; Yoon J. J. Am. Chem. Soc. 2009, 131, 15528. 10.1021/ja906855a. [DOI] [PubMed] [Google Scholar]
- Chan W. C.; White P. D.. Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Oxford University Press Inc.: New York, 2000. [Google Scholar]
- Bains G. K.; Kim S. H.; Sorin E. J.; Narayanaswami V. Biochemistry 2012, 51, 6207. 10.1021/bi3005285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister A.; Drescher S.; Garamus V. M.; Karlsson G.; Graf G.; Dobner B.; Blume A. Langmuir 2008, 24, 6238. 10.1021/la800166h. [DOI] [PubMed] [Google Scholar]
- Berdugo C.; Miravet J. F.; Escuder B. Chem. Commun. 2013, 49, 10608. 10.1039/c3cc45623d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.