

Abstract
Background
Extra-corpuscular hemoglobin is an endogenous factor enhancing inflammatory tissue damage, a process counteracted by the hemoglobin-binding plasma protein haptoglobin composed of alpha and beta subunits connected by disulfide bridges. Recent studies established that haptoglobin also binds and sequesters another pro-inflammatory mediator, HMGB1, via triggering CD163 receptor-mediated anti-inflammatory responses involving heme oxygenase-1 expression and IL-10 release. The molecular mechanism underlying haptoglobin-HMGB1 interaction remains poorly elucidated.
Methods
Haptoglobin β subunits were tested for HMGB1-binding properties, as well as efficacy in animal models of sterile liver injury (induced by intraperitoneal acetaminophen administration) or infectious peritonitis (induced by cecal ligation and puncture, CLP, surgery) using wild type (C57BL/6) or haptoglobin gene deficient mice.
Results
Structural-functional analysis demonstrated that the haptoglobin β subunit recapitulates the HMGB1-binding properties of full-length haptoglobin. Similar to HMGB1-haptoglobin complexes, the HMGB1-haptoglobin β complexes also elicited anti-inflammatory effects via CD163-mediated IL-10 release and heme oxygenase-1 expression. Treatment with haptoglobin β protein conferred significant protection in mouse models of polymicrobial sepsis as well as acetaminophen-induced liver injury, two HMGB1-dependent inflammatory conditions.
Conclusions
Haptoglobin β protein offers a novel therapeutic approach to fight against various inflammatory diseases caused by excessive HMGB1 release.
Keywords: Sepsis, acetaminophen intoxication, CD163, cytokine, haptoglobin (β), HMGB1
Introduction
Severe sepsis is the third leading cause of death after cardiovascular disease and cancer in the United States [1, 2] and is generally caused by dys-regulated immune reactions [3]. Extracellular high mobility group box 1 (HMGB1) has been identified as a key mediator in the inflammatory network of infectious as well as sterile inflammatory diseases [4]. As an evolutionarily ancient protein present in the nucleus of all cell lineages [4, 5], the 25 kDa HMGB1 molecule is highly conserved with 99% sequence identity in all mammals, and its functions depend on location, binding partners and redox states of the molecule. Upon cell activation or cell injury, nuclear HMGB1 is either released extracellularly or translocated to the cytoplasm, where it induces inflammasome activation and subsequent pyroptosis, and promotes autophagy by interacting with beclin-1[6] and counteracts the generation of apoptosis [7]. Extracellular HMGB1 triggers and sustains the inflammatory response by inducing cytokine release and by recruiting leukocytes. Actively secreted or passively released into the extracellular milieu, HMGB1 elicits cytokine, chemokine, neuroimmune and metabolic activities [5, 8].
CD163 is a scavenger receptor expressed on macrophages/monocytes and is previously known to remove extracellular hemoglobin-haptoglobin complexes [9]. We recently established that the plasma protein haptoglobin also binds and sequesters HMGB1 via CD163 [10]. Furthermore, the uptake of the haptoglobin-HMGB1 complexes elicits the release of anti-inflammatory cytokines and enzymes in a CD163-dependent fashion. Haptoglobin is a 100-kDa protein produced in the liver and secreted into the circulation as an acute phase protein [11]. Haptoglobin is composed of two or three α and β subunits linked by disulfide bonds; gene polymorphisms yield three common protein phenotypes, termed Hp1-1, Hp2-2, and Hp 2-1 [11] (Fig. 1A). In the present study, we localized the HMGB1-binding domain of haptoglobin to the β subunit (Hp β), a highly conserved and accessible molecule. Furthermore, we explored the therapeutic potential and protective mechanisms of haptoglobin β in animal models of lethal sepsis and sterile liver injury.
Figure 1. Haptoglobin (Hp) β binds HMGB1.


(A) Schematics of human haptoglobin. The Human haptoglobin gene has two alleles and gives rise to three phenotypes termed Hp1-1, Hp2-2, and Hp 2–1. All three phenotypes are comprised of α (α1 or α2) and β subunits linked by disulfide bonds [11].
(B–C) Surface plasmon resonance assay (SPR) of HMGB1 binding to haptoglobin β. Recombinant (B) or native (C) human haptoglobin β was immobilized on the sensor chip, and different concentrations of HMGB1 (0, 10, 50, 100 and 500 nM) were applied as analyte to the chip. Data is presented as response units over time (seconds). HMGB1 bound to haptoglobin β in a concentration-dependent manner and with high affinity, with an apparent Kd of 29 nM (to recombinant β) and 30 nM (to native β). Data shown are representative of three experiments.
(D) Depletion of endogenous HMGB1 by haptoglobin β-conjugated beads. Male C57BL/6 mice were subjected to cecal ligation and puncture surgery and euthanized at 24 hours afterwards by over-exposure to CO2. Sera (200 μl) from normal or CLP-septic mice were incubated with 50 μl of haptoglobin β-coupled beads or empty control beads at 37°C for 2 hours. Samples were then centrifuged at room temperature for 10 minutes. Supernatants were aspirated and subjected to western blot probed with anti-HMGB1 monoclonal antibodies (1 μg/ml). Beads were extensively washed with PBS containing 0.1% Triton X100; and eluates from the beads were subjected to western blot analysis probed with anti-HMGB1 antibodies (n = 5 mice per group). *: P <0.05 vs. empty beads.
Materials and methods
Materials
Human haptoglobin (from pooled human plasma, a mixture of Hp1-1, 2-1, 2-2, Cat #3536), triton X-114, imidazole, human macrophage-colony stimulating factor (M-CSF), acetaminophen and dexamethasone (Cat # D2915) were purchased from Sigma (St. Louis, MO). NHS-activated sepharose 4 fast flow beads were obtained from GE Healthcare (Cat #17-0906-01, Uppsala, Sweden). Trizol reagent was from Life Technologies (Carlsbad, CA). The RevertAid™ First Strand cDNA Synthesis Kit was from Fermentas (Vilnius, Lithuania). SYBR Premix Ex Taq™ II was from TaKaRa Bio Inco (Otsu, Japan). Thioglycollate medium was purchased from Becton Dickinson Co., (Sparks, MD). Primers for QPCR, trypsin-EDTA and carbenicillin were from Invitrogen Inc., (Carlsbad, CA). Isopropyl-D-thiogalactopyranoside (IPTG) was purchased from Pierce (Rockford, IL). Recombinant human CD163 protein, CD163 expression plasmid (SC117495, NM_004244.3) and MegaTran 1.0 transfection reagent were purchased from Origene (Rockville, MD). Antibodies for human heme oxigenase-1 (HO-1, Cat # ab52946) and CD163 (Cat # MCA1853) were from ABcam (Cambridge, MA) and Serotec (Raleigh, NC) respectively. Anti-human CD163-PE and anti-human CD80-PE antibodies were from BioLegend (San Diego, CA). FITC antibody labeling kit is from Thermo Scientific (Rockford, IL). Alanine transaminase (ALT) assay kits were from BIOO Scientific Corp., (Austin, TX).
Preparations of recombinant HMGB1
Recombinant disulfide HMGB1 was expressed in E. coli and purified to homogeneity as previously described [12, 13]. Fully reduced HMGB1 was generated as previously described [14]. HMGB1-FITC was prepared by using Pierce FITC Antibody Labeling Kit (Thermo Scientific, Rockford, lL).
Cloning and expression of human haptoglobin β subunit
The expression plasmid construct encoding human haptoglobin (accession number XM_042621) β subunit (245 amino acids corresponding to 162-406 residues of haptoglobin holo-protein) was made available by GeneCopoeia Inc., (Germantown, MD). The recombinant plasmid is tagged with 6 × histidine at the N-terminus. The correct DNA insert (757 bp) was confirmed by digesting the plasmid with restriction enzymes XmnI and NotI. The plasmid was transformed into E coli strain DH5α (Novagen, Madison, WI) and incubated with 2-YT medium containing carbenicillin (100 μg/ml). Fusion protein was isolated by affinity purification using the Nickel-charged 6 X histidine binding resin beads (Novagen). Protein eluate was dialyzed extensively against 20 mM Tris HCl (pH 8.0) to remove urea and excess salt. The identity of protein was confirmed by mass spectrometry (Stony Brook University, NY). The integrity of protein was verified by SDS-PAGE with Coomassie Blue staining, and the purity is predominantly over 85%. Contaminating LPS in protein preparations was removed by triton X-114 extraction method as described previously [13]. The LPS content in HMGB1, haptoglobin and haptoglobin β was measured by the Chromogenic Limulus Amebocyte Lysate Assay (Catalog # 50-647U, Lonza, Walkersville, MD). The LPS content in protein solutions is less than 10 pg/mg protein.
Purification of native haptoglobin β
Human haptoglobin samples were prepared in buffers containing 7 M Urea and 40 mM 1,4-Dithiothreitol (DTT, to break the disulfide bonds of the proteins). For isolation of β subunit in the haptoglobin preparations, haptoglobin species were separated by Gel-filtration chromatography using fast protein liquid chromatography (FPLC) technique (ÄKTA, GE Healthcare, Life Science, Pittsburgh, PA). Eluates were visualized by SDS-PAGE with Coomassie Blue staining. Fractions containing β subunits (25 KDa) were pooled and dialyzed against phosphate buffered saline and used in experiments.
Cell culture
Murine macrophage-like RAW 264.7 and human acute monocytic leukemia cell line THP-1 (American Type Culture Collection, ATCC, Rockville, MD) in culture plates were used at 90% confluence. Human primary monocytes were purified by density gradient centrifugation through Ficoll from blood donated by normal individuals in the Long Island Blood Bank (New York Blood Center, Melville, NY). Monocytes were allowed to differentiate into macrophages for 7 days in complete DMEM medium with 1ng/ml M-CSF. All experiments were carried out in Opti-MEM I medium. Primary mouse thioglycollate-elicited macrophages were obtained as previously described [15]. Briefly, each mouse was injected with 2 ml of thioglycollate broth (4%) intraperitoneally. Two days later, mice were euthanized and 5 ml of 11.6% sterile sucrose was injected into the peritoneal cavity. Peritoneal cavity lavage containing macrophages was collected by using BD insyte autoguard (BD bioscience, San Jose, CA). Cells were then passed through Cell Strainer (BD Falcon, Franklin Lakes, NJ) to remove debris, followed by three times of washing with RPMI medium. Mice residential macrophages were obtained by washing the peritoneal cavity with 5 ml sterile phosphate buffered saline. Cells were plated in culture dishes and used after seeding overnight.
Measurements of CD163, HO-1 expression and cytokines
Human acute monocytic leukemia (THP-1) cells were incubated with dexamethasone for 2 days to induce CD163 expression. Cells were exposed to HMGB1 in the presence or absence of haptoglobin or β subunit for 16 hours, the expression of HO-1 and CD163 in cell lysate was measured by western blot analysis as previously described [16, 17]. In some experiments, mouse HO-1 expression in cell lysate was measured by using ELISA kits (Enzo Life Science, Farmingdale, NY).
TNF, HMGB1, IL-6 and IL-10 released in the cell culture supernatants or systemically in mice were measured by commercially obtained ELISA kits (IBL international, Hamburg, Germany; or R&D System Inc., Minneapolis, MN). The expression profile of cytokines from primary human macrophages was determined by human cytokine array C1 (Raybiotech, Norcross, GA) according to manufacturer’s instructions. Twenty-two cytokines or chemokines were determined simultaneously.
RNA extraction and PCR
For mRNA measurements, mouse or human primary macrophages were seeded in 6-well culture plate. Stimulation with HMGB1 and haptoglobin or haptoglobin β was performed in Opti-MEM I medium. After treatment, cells were collected and total RNA was isolated by the Trizol reagent according to the manufacturer’s instructions. First-strand cDNA was synthesized with the RevertAid™ First Strand cDNA Synthesis Kit and analyzed by real-time quantitative PCR with SYBR Premix Ex Taq™ II. PCR was performed on the real-time PCR system ABI 7900HT (Applied Biosystems, Foster City, CA). The primer sequences of murine IL-6, TNF, IL-10, ARG-1, MMR, IL-12, CD163 and GAPDH are from previous reports [18–20]. Primers for human IL-6, IL-1β, MCP-1, TNF and 18S rRNA are obtained from previous publications [21, 22]. The primers were customer-made from Fisher Scientific Co. (Pittsburgh, PA). Data were analyzed on the basis of relative expression method using the formula ΔΔC expression = 2−ΔΔCt, where ΔΔCt = ΔCt (treated group) – ΔCt (control group), ΔCt = Ct (target gene) – Ct (GAPDH), and Ct = cycle at which the threshold is reached. Setting value of unstimulated group as 1, the gene expression of other groups was presented as folds of unstimulated group after normalization to GAPDH or 18s rRNA.
CD80 expression by flow cytometry
Flow cytometry was employed to examine the expression of CD80 on human primary macrophages. In brief, 1×106 of human primary macrophages were suspended in 100 μl PBS and incubated with HMGB1 (1 μg/ml) alone or plus haptoglobin (3 μg/ml) or haptoglobin β (1 μg/ml) for 30 minutes at 37°C. Cells were incubated with PBS containing Fc blocker rat anti-mouse CD16/CD32 (25 μg/ml, BD Pharmingen) for 10 min at 4°C and subsequently stained with anti-CD80-PE (10 μg/ml) for 45 minutes in the dark at 4°C. Cells were washed twice with PBS and immediately analyzed using an LSRII flow cytometer (BD Biosciences, Heidelberg, Germany) and FlowJo Version 9.0 (Tree Star, Inc., Ashland, OR).
Uptake of HMGB1 and haptoglobin (or β subunit) complexes by primary human macrophages
Human primary macrophages were incubated with FITC-HMGB1 (1 μg/ml) alone or plus haptoglobin 3 μg/ml) or haptoglobin β (1 μg/ml) for 30 minutes at 37°C. After washing with PBS to reduce non-specific binding, macrophages were stained with anti-CD163-PE antibody at 4°C for 45 minutes and then analyzed by a FACS Calibur flow cytometer. The optimal ratio of HMGB1 binding to haptoglobin or to haptoglobin β was determined by measuring the uptake of FITC-HMGB1 (1 μg/ml) with increasing amounts of haptoglobin or haptoglobin β (0–10 μg/ml), and we observed the optimal binding ratio is 1:1 for HMGB1 with haptoglobin or haptoglobin β.
Fluorescent Microscopy
Alexa fluor 555 labeled HMGB1 was performed according to the manufacturer’s instructions (Molecular Probes, Eugene, OR). To identify haptoglobin β-induced HMGB1 cell surface binding and uptake process, primary human macrophages on coverslips were pre-incubated with endocytosis inhibitor Dynasore (8 μM) [23] for 30 minutes at 37°C (for uptake) or 4°C (for cell surface binding) ; then the cells were incubated with Alexa fluor 555 labeled HMGB1 (1 μg/ml) in the presence or absence of haptoglobin β (1 μg/ml) for 2 hours at 37°C (for uptake) or 4°C (for cell surface binding). After incubation, cells were rinsed with phosphate buffered saline and fixed by using 4% PFA (Paraformaldehyde) for 30 minutes at room temperature and stained by 4′,6- diamidino-2-phenylinodole (DAPI) for nuclear staining and mounted on the slide using permanent mounting medium (Vecta mount). Images were taken using Carl Zeiss fluorescence microscope with 40 × objective.
Surface plasmon resonance analysis
Surface plasmon resonance analysis of binding of HMGB1 to haptoglobin β was conducted using the BIAcore 3000 instrument as previously described [10, 15, 24]. For HMGB1 and haptoglobin β binding analyses, purified human native or recombinant haptoglobin β protein (1 μM, in 10 mM sodium acetate, pH 4.5) was immobilized onto the CM5 sensor chip. To evaluate binding, a series of concentrations of analytes (HMGB1 0–500 nM), were passed over the sensor chip. The association of analyte and ligand was recorded respectively by surface plasmon resonance. Results were analyzed using the software BIAeval 3.2 (BIAcore Inc. NJ). For the binding studies of CD163 to the complex of recombinant haptoglobin β and HMGB1, human CD163 protein was coated on the sensor chip. Increasing concentrations of the complexes of HMGB1 and or haptoglobin β (formed at 1:1 molar ratio) were flowed over the sensor chip; and analyses were performed in a similar manner as described above.
Nuclear magnetic resonance (NMR) analyses to determine the interacting amino acid residues between HMGB1 and haptoglobin β
Recombinant Δ30-HMGB1 (HMGB1 expressed minus 30 C terminal amino acids) of various redox states was produced in complex with haptoglobin β; and the nature of interaction was examined by NMR. Standard HSQC (Heteronuclear Single Quantum Correlation) -based 3D triple resonance (1H, 15N and 13C) was used to assign the backbone residues as described previously [10, 25]. 3D HNCO and HNCACO experiments were used to sequentially assign the backbone NH via the carboxyl group. The CBCACONH and HNCACB experiments were used to assign the backbone NH via the backbone Cα and side-chain Cβ groups. 15N and/or 13C-labelled recombinant proteins were expressed in minimal media and purified by ion exchange and size exclusion chromatography. NMR samples were prepared at 10–100μM concentrations in PBS with a final concentration of 10% D2O (Deuterium oxide, 2H2O). Chemical shift perturbation studies were carried out using either a Bruker Avance III 600 or 800MHz NMR system with standard 5mm NMR tubes or with triple (TCI) resonance micro cryoprobes at 25°C (298K). All data was processed using Topspin 3.1 (Bruker) and analyzed using CCPN (Collaborative Computing Project for NMR) analysis.
CD163 shRNA lentiviral transduction of human macrophages
Ficoll gradient separated human primary monocytes were differentiated for 7 days in complete DMEM medium containing 10% heat-inactivated human serum, 2 mM glutamine, 100U/ml penicillin, 100 μg/ml streptomycin and 2 ng/ml M-CSF in 24-well Primaria tissue culture plates at 5×105 cells/well [26]. After screening of five human CD163 (NM_004244) Mission™ shRNA lentiviral clones by using FACS analysis, the clone that showed the best knock down efficiency (TRCN0000421748) was selected. The selected clone was transduced into human macrophages by spinoculation according to the manufacturer’s instructions (Sigma).
Animal experiments
Male C57BL/6 mice were obtained from Jackson Laboratory (Bar Harbor, ME) and allowed to acclimate for 7 days before use in the experiment. All animal procedures were approved by the Institutional Animal Care and Use Committee. Mice were housed in the animal facility of The Feinstein Institute for Medical Research under standard temperature, light and dark cycles.
Haptoglobin or CD163 gene deficient and wild type mice
Haptoglobin−/− C57BL/6 mice were generated and provided by Dr. Dominique P.V. de Klein (Utrecht, Netherlands). CD163−/− C57BL/6 mice were purchased from KOMP repository (University of California-Davis, Davis, CA).
Since the gene deficient mice are all derived from C57BL/6 mice, small colonies of wild type C57BL/6 (Jackson Laboratory) were maintained under the same conditions as the gene deficient mice. We performed genotyping on genomic DNA obtained from tail snips as reported previously [10, 27].
Removal of HMGB1 using haptoglobin β-conjugated beads from sera of septic mice
Recombinant human haptoglobin β was cross-linked to NHS-activated sepharose 4 fast flow beads according to the manufacturer’s instructions (GE Healthcare). Approximately 16 mg haptoglobin β was bound to each ml of drained beads. Male C57BL/6 mice were subjected to cecal ligation and puncture surgery and euthanized at 24 hours afterwards by over-exposure to CO2. Sera from normal or septic mice (200 μl) were pre-cleared by NHS-activated sepharose 4 fast flow beads, and were then incubated with 50 μl of haptoglobin β-containing or control beads at 37°C for 2 hours. Samples were then centrifuged at room temperature for 5 minutes to separate beads and supernatants, after extensive wash with PBS containing 0.1% Triton X100, eluates from the beads were subjected to western blot analysis probed with anti-HMGB1 monoclonal antibodies (1 μg/ml) [28].
Acetaminophen liver toxicity model
Male C57BL/6 mice (8–12 weeks old) were fasted overnight and received intraperitoneal injection of acetaminophen (400 mg/kg) according to our previous reports [14, 29]. Mice were given injections of haptoglobin β intraperitoneally (IP) once a day for 5 days and survival was monitored for two weeks. In some experiments, male wild type (C57BL/6) mice had administration of acetaminophen (400 mg/kg) and received haptoglobin β treatment (200 μg/mouse IP injected at 2 and 7 hours post-acetaminophen injection); and mice were euthanized at 24 hour post acetaminophen administration. Hepatotoxicity was evaluated by serum alanine aminotransferase (ALT) activity using color endpoint assay kits. For histological evaluation, harvested livers were fixed in 10% formalin and embedded in paraffin. Five μm sections were cut and stained with hematoxylin and eosin (H&E) performed by AML laboratory (Baltimore, MD). The liver histology was evaluated in a blinded fashion, and clinical scores were calculated based on the amount of necrosis and inflammation (cell swelling, loss of tissue structure, and congestion) using a previously reported method with modifications [30]. Score 0 = no evidence of necrosis or inflammation as assessed from three to four representative sections from each animal; score 1 = mild necrosis or inflammation (<25% of the total area examined); score 2 = notable necrosis and inflammation (25–50% of the total area); score 3 = severe necrosis and inflammation (>50% area).
Cecal ligation and puncture (CLP)
Wild type (C57BL/6) or haptoglobin−/− mice (male, 8–12 weeks of age) were subjected to CLP procedure [26]. Survival was monitored for 2 weeks. Administration of haptoglobin β or vehicle control was given intraperitoneally once a day for 3 days starting at 24 hours post-CLP surgery, and survival was monitored for 2 weeks. In some experiments, male wild type mice were subjected to CLP surgery and received haptoglobin β treatment (200 μg/mouse injected intraperitoneally at 24 and 36 hours post-CLP surgery), and mice were euthanized at 48 hours post CLP. Sera were isolated for analyses.
Statistical analysis
All data were presented as means ± SEM unless otherwise stated. Differences between treatment groups were determined by student’s t test. Differences between treatment groups in animal survival studies were determined using 2-tailed Fisher’s exact test. P values less than 0.05 were considered statistically significant.
Results
Identification of the haptoglobin β subunit as an HMGB1 binding protein
We recently reported that haptoglobin binds HMGB1 with high affinity and sequesters HMGB1 toxicity both in vitro and in vivo [10]. With three forms of gene polymorphisms termed Hp1-1, Hp2-2, and Hp 2-1, haptoglobins are comprised of α and two β subunits linked by disulfide bonds [11] (Fig 1A). To study the molecular basis of haptoglobin binding to HMGB1, we chose the β subunit because it is present in all forms of human haptoglobin and also it is more accessible relative to the α subunit since it is located outside on the schematics of molecular arrangement of haptoglobin, as revealed by using high-resolution scanning transmission electron microscopy [11, 31] (Fig 1A). Haptoglobin β subunits were tested for HMGB1 binding properties using various assays. We first used surface plasmon resonance analysis which provides sensitive and quantitative measurements of molecular interactions [15]. We observed a dose-dependent binding of recombinant haptoglobin β subunit to HMGB1 with an apparent Kd of 29 nM (Fig. 1B). Similarly, native haptoglobin β isolated from human plasma haptoglobin also effectively bound HMGB1 in a dose-dependent fashion with a Kd value of 30 nM (Fig. 1C), indicating that the haptoglobin β subunit is capable of capturing the HMGB1 protein.
To further assess the HMGB1-capturing capacity of haptoglobin β in a biological environment, we conjugated recombinant haptoglobin β protein to chromatographic beads and subsequently used it to bind HMGB1 in the serum of septic animals. C57BL/6 mice were subjected to CLP surgery to induce peritonitis and sepsis as described in Methods. Haptoglobin β-conjugated or empty (control) beads were added to sera of these septic mice. As expected, haptoglobin β-conjugated beads captured significant amounts of HMGB1 from serum samples of CLP-septic mice ex vivo as compared to empty beads (Fig. 1D).
Characterization of haptoglobin β binding sites in HMGB1
HMGB1 contains two DNA-binding domains (termed “box A” and “box B”) and an acidic C-terminal sequence comprising exclusively glutamic and aspartic acids. HMGB1 expresses three inherent cysteine residues and their redox states determine the biological function of the extracellular HMGB1 molecule [8]. Fully reduced HMGB1 expresses chemotactic function; while mild oxidation generates disulfide HMGB1, which induces cytokine release via TLR4 signaling pathway; further oxidation of any of the cysteines creates sulfonyl HMGB1 with no known pro-inflammatory function [32]. We recently demonstrated that full-length haptoglobin binds fully reduced as well as disulfide HMGB1, but not sulfonyl HMGB1 [10]. To characterize the haptoglobin β binding sites in HMGB1, nuclear magnetic resonance (NMR) studies were performed. The amino acids residues in HMGB1 affected by binding of haptoglobin β were identified by Heteronuclear Single Quantum Correlation (HSQC) analysis. As shown in figure 2, NMR analysis from Δ30-HMGB1 revealed significant chemical perturbations of amino acids upon binding to haptoglobin β. There are predominantly eight amino acid residues located in or close to the HMGB1 box A region that interact with haptoglobin β subunit, including those of residues F18, T22, R24, E25, K28, H31, A54 and K55. We observed that haptoglobin β binds both fully reduced (data not shown) as well as disulfide HMGB1 (Fig. 2A–B).
Figure 2. Nuclear magnetic resonance (NMR) analysis of disulfide Δ30-HMGB1 binding with human haptoglobin β.

(A). Upper: 15N-1H- Heteronuclear Single Quantum Coherence (HSQC) spectra for Δ30-HMGB1 in the presence or absence of haptoglobin β. HMGB1 residues (8 amino acids) with significant chemical shift perturbations due to haptoglobin β binding are labeled. Lower: enlarged 15N-HSQC spectra of Δ30-HMGB1 free (black) and in complex with haptoglobin β (red). The 8 residues showing chemical shift changes seen with disulfide Δ30-HMGB1 upon complex formation with haptoglobin β are circled. Non-overlapping black and red resonance spectra indicate a significant chemical shift; hence, an interaction between proteins via that amino acid.
(B) Chemical shift perturbation of disulfide Δ30-HMGB1 in complex with haptoglobin β is plotted as a function of residue number of HMGB1. The majority of interacting amino acids are located within HMGB1 Box A. Data are representative from 4 independent analyses.
Haptoglobin β protein inhibits HMGB1-induced cytokine release from cultured macrophages
Like full-length haptoglobin, haptoglobin β dose-dependently inhibited HMGB1-induced TNF secretion with up to 90% inhibition at 50 μg/ml haptoglobin β supplementation (Fig. 3A). The effective haptoglobin β concentration that inhibited 50% of the TNF release (EC50) was 10 μg/ml, which is similar or slightly superior to the capacity of full-length haptoglobin (20 μg/ml [10]). In primary human macrophage cultures, the addition of haptoglobin β effectively inhibited HMGB1-induced secretion of IL-6, MIG and MCP-2, but not of GRO; which suggest specificity of the responses (Fig. 3B). Overall, these results indicate that haptoglobin β and haptoglobin similarly and selectively attenuate HMGB1-induced cytokine and chemokine release in macrophages.
Figure 3. Sequestration of HMGB1 by haptoglobin β subunit in vitro.


(A) RAW 264.7 cells in 96-well culture plates were stimulated with HMGB1 in combination with various amounts of recombinant haptoglobin β for 16 hours. TNF released in the supernatants were measured. The effective concentration of haptoglobin β that inhibited 50% of released TNF (EC50) was approximately at 10 μg/ml (n = 5 experiments). *: p<0.05 vs. HMGB1 alone.
(B) Primary human macrophages in culture plates were stimulated with HMGB1 (1 μg/ml) plus haptoglobin or haptoglobin β at 50 μg/ml for 16 hours. Cytokine released in the supernatants were analyzed by using human cytokine antibody array. The addition of haptoglobin or haptoglobin β inhibited HMGB1-induced IL-6, MIG, MCP-2 (but not GRO) release from macrophages. Data shown are from three separate experiments. *: p<0.05 vs. HMGB1 alone.
(C) Haptoglobin (and β subunit) inhibits HMGB1-induced M1 marker expression. Human primary macrophages (106 cells per assay) were suspended in 100 μl PBS and incubated with HMGB1 (1 μg/ml) alone or plus haptoglobin (3 μg/ml) or haptoglobin β (1 μg/ml) for 30 minutes at 37°C. Cells were incubated for 10 min at 4°C with PBS containing Fc blocker rat anti-mouse CD16/CD32 (25 μg/ml) and subsequently stained with anti-CD80-PE (10 μg/ml) for 45 minutes in the dark at 4°C. Cells were washed twice with PBS and immediately analyzed on a FACS Calibur flow cytometer (n = 5 repeats). *: P<0.05 vs. HMGB1 alone.
Haptoglobin and haptoglobin β reduces HMGB1-induced M1 polarization in macrophages
Depending on the microenvironment, macrophages can polarize into at least two major functional subtypes, the classically activated subtype (M1) representing pro-inflammatory, cytotoxic activities and the alternatively activated subtype (M2) mediating anti-inflammatory and repairing activities [33]. Previous reports have shown that HMGB1 facilitates macrophage reprogramming towards a pro-inflammatory M1-like phenotype in experimental autoimmune myocarditis [34]. To examine whether HMGB1-haptoglobin (or haptoglobin β) complexes shift macrophage M1/M2 polarization, we measured the expression profile of M1 and M2 markers in both primary mouse and human macrophage cultures stimulated with either HMGB1-, or HMGB1-haptoglobin (or haptoglobin β) complexes. HMGB1 alone significantly stimulated the mRNA expression of primary M1 markers (TNF= 43, IL-6= 36 and IL-12= 99 folds over basal levels) in primary mouse macrophages. HMGB1 bound to either haptoglobin or its β subunit had reduced M1 marker expression (over 50% reduction) as compared to HMGB1 stimulation alone. Haptoglobin (or haptoglobin β) stimulation alone did not have intrinsic effects on mouse macrophage polarization (Table 1). Similar findings were observed in primary human macrophages (Table 2). Flow cytometry revealed that after 24-hour incubation, the HMGB1-induced expression of the primary M1 marker, CD80, was also diminished by over 50% in the presence of haptoglobin or haptoglobin β; confirming that haptoglobin (or its β subunit) reduces HMGB1-induced M1 polarization in macrophages (Fig. 3C).
Table 1.
mRNA expression of murine macrophage M1 and M2 markers in mouse macrophages.
| HMGB1 | Hp | Hpβ | HMGB1+Hp | HMGB1+ Hpβ | ||
|---|---|---|---|---|---|---|
| M1 | TNF | 43 ± 15 | 6 ± 3 | 6 ± 2 | 19 ± 8* | 17 ± 4* |
| IL-6 | 36 ± 11 | 6 ± 4 | 6 ± 4 | 15 ± 6* | 18 ± 13* | |
| IL-12 | 99 ± 45 | 9 ± 3 | 12 ± 4 | 37 ± 11* | 29 ± 8* | |
| M2 | IL-10 | 3 ± 0.4 | 0.8 ± 0.1 | 1.6 ± 0.5 | 9 ± 2* | 23 ± 2* |
| ARG-1 | 0.5 ± 0.1 | 0.5 ± 0.1 | 1 ± 0.2 | 0.4 ± 0.1 | 1.1 ± 0.3 | |
| MMR | 0.9 ± 0.5 | 0.8 ± 0.3 | 0.9 ± 0.2 | 0.9 ± 0.2 | 0.9 ± 0.2 | |
| CD163 | 0.6 ± 0.2 | 0.3 ± 0.1 | 1.2 ± 0.2 | 0.3 ± 0.1 | 0.3 ± 0.1 |
Thioglycollate-elicited peritoneal mouse macrophages in a 6-well culture plate were stimulated with HMGB1, haptoglobin or β alone; or HMGB1(1 μg/ml) plus haptoglobin (3 μg/ml) or haptoglobin β (1 μg/ml) for 8 hours in Optimem I medium. Total RNA was isolated using the Trizol reagent. The expression of mRNA in the cells was measured by real-time quantitative PCR. House-keeping gene GAPDH was used as a control for equal loading and integrity of RNA. The gene expression of unstimulated group after normalization to GAPDH is set as 1. Data are presented as folds over unstimulated. N=6–8 experiments.
P<0.05 vs. HMGB1 alone.
Table 2.
mRNA expression of M1 markers in human macrophages.
| HMGB1 | Hp | Hpβ | HMGB1+Hp | HMGB1+ Hpβ | |
|---|---|---|---|---|---|
| TNF | 10 ± 3 | 2 ± 0.4 | 1 ± 0.5 | 5 ± 1.6* | 3 ± 0.3* |
| IL-6 | 44 ± 8 | 5 ± 2 | 2 ± 1 | 23 ± 7* | 17 ± 7* |
| IL-1β | 268 ± 44 | 5 ± 1.7 | 6 ± 1.7 | 47 ± 12* | 38 ± 8.4* |
| MCP1 | 15 ± 3 | 2 ± 0.7 | 2 ± 1.3 | 6 ± 2.1* | 5 ± 1.7* |
Primary human macrophages in a 6-well culture plate were stimulated with HMGB1, haptoglobin or β alone; or HMGB1 (1 μg/ml) plus haptoglobin (3 μg/ml) or haptoglobin β (1 μg/ml) for 8 hours in Optimem I medium. Total RNA was isolated using the Trizol reagent. The expression of mRNA in the cells was measured by real-time quantitative PCR. The expression of 18S rRNA was used as a control for equal loading and integrity of RNA. The gene expression of unstimulated group after normalization to 18S rRNA is set as 1. Data are presented as folds over unstimulated. N=6 separate experiments.
P<0.05 vs. HMGB1 alone.
The induced mRNA expression of M2 markers in primary mouse macrophage cultures by HMGB1 or HMGB1-haptoglobin (or β) complexes was very modest as compared to M1 markers, and qualitatively similar between the stimulation pathways with the exception for IL-10. HMGB1-haptoglobin β complexes induced a 23 fold increase of IL-10 mRNA as compared to basal levels (Table 1). Taken together, haptoglobin and its β subunit mainly play a role in the M1/M2 switch balance by down-regulating HMGB1-induced M1 markers.
HMGB1-haptoglobin β complexes bind CD163 to induce HO-1 and IL-10 expression
HMGB1-haptoglobin complexes mediate anti-inflammatory effects by binding to CD163 receptor, which elicits heme oxygenase-1 (HO-1) and IL-10 synthesis [10]. We hypothesized that HMGB1-haptoglobin β complexes may use the same molecular pathway to activate anti-inflammatory responses. Indeed, HMGB1-haptoglobin β complexes (1:1 molar ratio) bound CD163 in a concentration-dependent manner and with high affinity (apparent Kd value of 70 nM ) as revealed by surface plasmon resonance analysis; and with increased affinity as compared to HMGB1-haptoglobin complexes (Kd value of 130 nM) [10](Fig. 4A). HMGB1-haptoglobin β complexes also induced a similar increase of IL-10 release and HO-1 expression, as compared to HMGB1 stimulation alone in human monocytic THP-1 cells (Fig. 4B).
Figure 4. HMGB1 and haptoglobin (or β subunit) complexes signal through CD163 to induce HO-1 and IL-10 expression in macrophages.



(A) Surface plasmon resonance assay (SPR) of HMGB1 and haptoglobin β complexes to CD163. Recombinant human CD163 was coated on the sensor chip, the complexes of HMGB1 and haptoglobin β (1:1 molar ratio) at concentrations of 0, 32, 65, 125 or 250 nM were flow over the chip and the binding to CD163 (response units) was recorded. The Kd of HMGB1 and haptoglobin β complexes to CD163 is approximately 70 nM. Data are representative of 3 separate experiments.
(B) Human monocytic THP-1 cells (in 24-well plate) were cultured with dexamethasone (2.5×10−7 M) for 2 days to induce CD163 expression. Cells were then stimulated with HMGB1 alone (5 μg/ml) in combination with haptoglobin (15 μg/ml) or haptoglobin β (5 μg/ml) at 37°C for 16 hours. After incubation, cell cultures were centrifuged and supernatants were collected to measure IL-10 release. The expression of HO-1 and β-actin in cell lysate was measured by western blot. Data are expressed as folds of the unstimulated group after normalization to β-actin (n = 3 experiments). *: p<0.05 vs. HMGB1 alone.
(C–D) Knock down CD163 abolishes HMGB1 and haptoglobin (or haptoglobin β) complexes-induced HO-1 and IL-10 expression in macrophages. Primary human macrophages were transduced with specific shRNA lentiviral particles targeting CD163 or vector alone (control). At 72 hours after transduction, cells (in 24-well plate) were stimulated with HMGB1 (1 μg/ml) with or without haptoglobin (3 μg/ml) or haptoglobin β (1 μg/ml) for 16 hours. Cell cultures were centrifuged. The expression of CD163, HO-1 and β-actin in cell lysate was measured by western blot (C). Supernatants were collected to measure IL-10 release (D). Data are presented as folds of unstimulated group after normalization to β actin (n = 3 experiments). *: P<0.05 vs. WT.
(E) Residential peritoneal macrophages from CD163−/− and wild type control mice were stimulated with HMGB1 (1 μg/ml) alone, plus haptoglobin (3 μg/ml) or haptoglobin β (1 μg/ml) for 16 hours. IL-10 released and HO-1 expression in cell lysate were measured using ELISA kits (n = 3 experiments). *: P<0.05 vs. WT.
Reduction of CD163 expression diminishes IL-10 and HO-1 synthesis induced by HMGB1-haptoglobin β complexes
To further assess the importance of CD163 in HMGB1-haptoglobin β complexes-mediated signaling, we knocked down CD163 expression in primary human macrophages using CD163-specific shRNA. Transfection of shRNA resulted in over 90% reduction of CD163 protein expression at 72 hours (Fig. 4C, upper), which was associated with significant impairment of HO-1 expression and IL-10 release following stimulation with HMGB1-haptoglobin β or HMGB1-haptoglobin complexes (Fig. 4C–D).
Lack of CD163 eliminates HMGB1-haptoblobin β complexes-induced IL-10 release and HO-1 activation
Macrophages from CD163 gene deficient mice were used to further evaluate the role of CD163 for HMGB1-haptoglobin β complexes biology. HMGB1 induced similar amounts of IL-10 release from CD163 gene deficient or knock down macrophages as compared to wild type cells indicating an intact functional capacity to non-CD163-dependent receptor activation via TLR4 (Fig. 4D–E). However, IL-10 release and HO-1 expression induced by HMGB1 and haptoglobin (or haptoglobin β) complexes were both completely abolished in CD163−/− macrophages; in contrast to wild type cells (Fig. 4E). Together, these data indicate that CD163 plays a critical role in the elimination of systemic HMGB1 via macrophage internalization of HMGB1 and haptoglobin (or haptoglobin β) complexes.
CD163 mediates endocytic uptake of HMGB1-haptoglobin β complexes
To test the possibility that haptoglobin β subunit mediates HMGB1 endotocytosis in a CD163-dependent fashion, we assessed the uptake of fluorochrome-labeled HMGB1 (FITC-HMGB1) in human primary macrophages. Endocytosis in macrophages of HMGB1-haptoglobin complexes has previously been shown to be a dynamin-dependent process [10]. Flow cytometry studies demonstrated that 2% of the human macrophages stained positive after exposure to FITC-HMGB1 alone, 17% stained positive after FITC-HMGB1-haptoglobin complexes exposure and 44% after incubation with FITC-HMGB1-haptoglobin β subunit complexes (Fig. 5A). Knock down of CD163 expression almost completely eliminated the uptake of HMGB1-haptoglobin (or haptoglobin β) complexes (Fig. 5A). This non-haptoglobin-dependent uptake of HMGB1 is most likely mediated via other HMGB1 receptors, possibly RAGE because it has previously been implicated in HMGB1 uptake [35].
Figure 5. Uptake and cell surface binding of HMGB1 and haptoglobin (or β) complexes.


(A) The uptake of HMGB1 and haptoglobin (or haptoglobin β) complexes was measured by flow cytometry analysis. CD163 shRNA lentiviral or vector alone (WT) transduced primary human macrophages were incubated with FITC-labeled HMGB1 (1 μg/ml) plus haptoglobin (3 μg/ml) or haptoglobin β (1μg/ml) for 30 minutes. Uptake of FITC-HMGB1 was performed by staining for CD163 PE and FITC on cells. Data are representative from 5 separate experiments.
(B–C) Uptake or cell surface binding of HMGB1-haptoglobin β complexes. Human macrophages on cover slips were incubated with Alexa 555 labeled HMGB1 (red) alone or in complex with haptoglobin β for 2 hours at 37°C (B) or 4°C (C). Dynasore (DYN) was pre-incubated with cells for 30 min before the addition of HMGB1 and/or haptoglobin β. Endocytic uptake or cell surface binding of HMGB1 was visualized via Carl Zeiss fluorescence microscope. Nuclei were counterstained with DAPI (blue). Lower panel: Corresponding phase contrast image of cells. Scale bar = 10 μm. Data are representative from 6 independent experiments.
In agreement with these findings, fluorescent (Alexa 555)-conjugated HMGB1 was endocytosed by primary human macrophages when incubated for 2 hours at 37°C as compared to untreated cells (Fig. 5B). This cellular uptake of Alexa 555-HMGB1 was further enhanced by the addition of haptoglobin β, but attenuated by pre-treatment with dynasore, an endocytosis inhibitor. In comparison, cell surface binding of fluorescent (Alexa 555)-conjugated HMGB1 by primary human macrophages was further enhanced by the addition of haptoglobin β as compared to untreated cells. In contrast, this binding was not significantly altered by pre-treatment with dynasore (Fig. 5C). Together, the results indicate that CD163 mediates endocytic uptake of HMGB1-haptoglobin β complexes.
Protective effects of haptoglobin β administration in mice model of sepsis
To assess the therapeutic capacity of haptoglobin β, polymicribial Gram-negative sepsis was induced in both wild type and haptoglobin−/− mice. As shown in figure 6A, intraperitoneal administration of haptoglobin β (daily doses of 200 μg/mouse/day for 3 days starting 24 hours after CLP surgery), significantly improved 2-week survival in wild type mice (85% survival in haptoglobin β-treated vs. 50% in vehicle-treated group; n=22 mice per group, P<0.05). The benefits of the haptoglobin β administration were achieved despite the delayed start and short duration of the therapy. In agreement with these findings, haptoglobin β administration to CLP-induced septic wild type mice significantly decreased systemic pro-inflammatory cytokine levels including HMGB1, IL-6, and TNF as compared to vehicle-treated mice. In contrast, anti-inflammatory IL-10 serum levels increased in response to the haptoglobin β therapy (Fig. 6B). We previously showed that haptoglobin gene deficient (−/−) mice are more susceptible to lethal sepsis and develop significantly elevated plasma HMGB1 levels [10]. Here we show that in haptoglobin−/− CLP-septic mice, the administration of haptoglobin β conferred dose-dependent and significant protection against sepsis lethality when given daily doses of 100 μg/mouse for 3 days starting 24 hours after CLP surgery (84 % survival in high dose haptoglobin β-treated vs. 37 % in vehicle-treated group; n=22 mice per group, P<0.05, Fig. 6C). Haptoglobin β-treated animals were considerably more alert and active than vehicle-treated mice. Ten times less dose of haptoglobin β was less effective. These results demonstrated that the haptoglobin β subunit conferred effective protection against CLP-sepsis lethality in both wild type and haptoglobin−/− mice.
Figure 6. Beneficial effects of haptoglobin β subunit in experimental sepsis.

(A–C) Administration of haptoglobin β subunit confers protection against lethal sepsis in wild type (A) and haptoglobin−/− mice (C) partly through reducing systemic cytokine accumulation and increase IL-10 release (B).
(A) C57BL/6 mice (male, 8–10 weeks of age) were subjected to CLP surgery. Recombinant human haptoglobin (Hp) β was administered at 200 μg per mouse (in 200 μl PBS injected intraperitoneally) once a day for 3 days starting at 24 hours after CLP surgery. Mice in the control group received vehicle (PBS) only. Animal survival was monitored for two weeks (n = 22 per group). *: P < 0.05 vs. vehicle control group.
(B) Male wild type (C57BL/6) mice were subjected to CLP surgery and received haptoglobin β (200 μg/mouse) or vehicle (PBS) IP injection at 24 and 36 hours post-CLP surgery, and were euthanized at 48 hour post CLP. Serum levels of HMGB1, IL-6, TNF and IL-10 were measured (n = 8 or 10 mice per group). *: P <0.05 vs. CLP-vehicle group.
(C) Haptoglobin−/− mice (male, 8–12 weeks of age) were subjected to CLP. Mice received administration of recombinant haptoglobin β (or vehicle) daily for 3 days at 10 or 100 μg/mouse beginning at 24 hours after CLP surgery. Animal survival was monitored for 2 weeks (n = 22 mice per group). *: P<0.05 vs. vehicle control group.
Protective effects of haptoglobin β administration in mice model of acute liver injury caused by acetaminophen intoxication
To further investigate the capacity of exogenous haptoglobin to neutralize HMGB1 activity in vivo, we utilized a murine model of sterile liver injury using acetaminophen-induced hepatotoxicity. Hepatic inflammation and lethality caused by acetaminophen toxicity are highly dependent upon HMGB1 [15, 36, 37]. Therapeutic administration of haptoglobin β significantly improved survival from acetaminophen-induced mortality (2-week survival in acetaminophen control group = 35% versus haptoglobin β treated group = 80%, P<0.05, n=20 mice per group. Fig. 7A). Biomarkers for liver injury (alanine aminotransferase, ALT) and for inflammation (HMGB1, TNF and IL-6) were all significantly reduced in the haptoglobin β-treated mice as compared to the control vehicle-treated group, while haptoglobin β treatment increased levels of anti-inflammatory cytokine IL-10 systemically (Fig. 7B). Histological evaluation revealed decreased inflammatory cell infiltration and improved inflammatory scores in haptoglobin β-treated mice as compared to the controls (Fig. 7C). The extent of necrotic lesions, disclosed by the loss of overall cell structural integrity, was also reduced by haptoglobin β treatment (Fig. 7C). Taken together, these results demonstrated that the exogenous haptoglobin β protein ameliorated systemic inflammation from both a sterile cause (acetaminophen) and an infectious one (CLP-sepsis).
Figure 7. Beneficial effects of haptoglobin β in acetaminophen intoxication.

(A) C57BL/6 mice (male, 8–12 weeks old) were fasted overnight and received intraperitoneal injection of acetaminophen (APAP, 400 mg/kg, in 200 μl volume) to induced liver toxicity. Mice were given injections of haptoglobin β (200 μg per mouse, IP) or vehicle (PBS) control once a day for 5 days starting at 2 hours post-acetaminophen injection, and survival was monitored for two weeks (n = 20 per group). *: P < 0.05 vs. vehicle control group.
(B) Male wild type mice (C57BL/6) were fasted overnight and received intraperitoneal injection of acetaminophen (400 mg/kg, in 200 μl volume). Mice were given injections of haptoglobin β (200 μg per mouse, IP) or vehicle (PBS) control at 2 and 7 hours post-acetaminophen injection, and were euthanized at 24 hours after acetaminophen administration (n = 8 or 10 mice per group). Liver and serum were collected for analysis. Serum levels of ALT, HMGB1, IL-6, TNF and IL-10 were measured. *: P < 0.05 vs. vehicle control group.
(C) Slices of liver were fixed in 10% formalin and stained with hematoxylin and eosin. Data shown are representative from 9 mice per group. Arrow indicates necrotic region. Clinical scores are calculated (Methods). Scale bar = 50 μm. *: P<0.05 vs. vehicle controls. Magnifications: ×200.
Discussion
The present study revealed that haptoglobin β recapitulates the immune-modulatory and protective effects of full-length haptoglobin in animal models of CLP-induced sepsis and acetaminophen-induced liver injury (Fig. 8). These results indicate that haptoglobin β can bind HMGB1 to counter-regulate pro-inflammatory properties by eliciting the production of anti-inflammatory enzymes (heme oxygenase-1) and cytokines in a CD163-dependent fashion and by inhibiting the induction of pro-inflammatory mediators. Like full-length haptoglobin, administration of haptoglobin β conferred protection against lethal sepsis and liver injury.
Figure 8. Model of haptoglobin β subunit as an endogenous inhibitor of HMGB1.
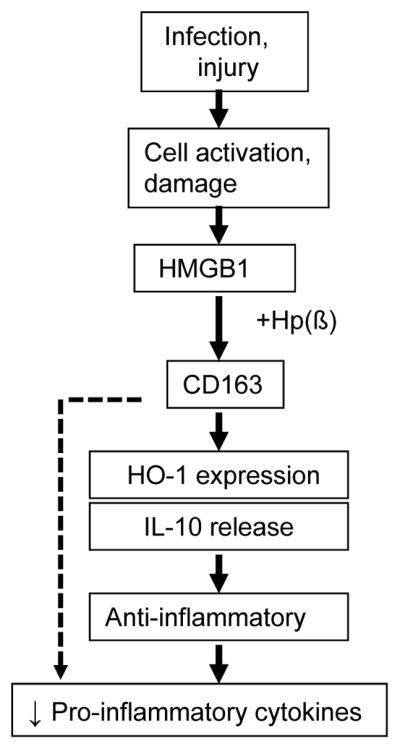
During infection or injury, HMGB1, a critical mediator in the final common pathway of inflammation, is actively secreted or passively released outside of the cells. Haptoglobin binds HMGB1 and inhibits its toxicity; and haptoglobin β subunit alone is sufficient to recapitulate the effects of the full-length haptoglobin. HMGB1-haptoglobin β complexes act via CD163 and elicit an anti-inflammatory response by stimulating HO-1 expression and IL-10 release; and by inhibiting pro-inflammatory cytokine release. Thus, haptoglobin β acts as an endogenous inhibitor of HMGB1 via a novel CD163-dependent anti-inflammatory pathway.
Numerous reports have established the critical pathogenic role of HMGB1 in animal models of infectious and sterile inflammatory diseases [4, 5]. For instance, it was recently demonstrated that trauma-induced hemorrhagic shock is accompanied systemic accumulation of disulfide HMGB1 which mediates secondary lung injury in a TLR4-dependent fashion [38]. The report demonstrates intestinal epithelial cells to be the key source for HMGB1 released after TLR4 activation. We propose that the initial response to the bleeding may consume haptoglobin, leading to increased HMGB1 systemically, since the haptoglobin binding to hemoglobin is extremely strong being in the femtomolar range, while the haptoglobin-HMGB1 binding has a Kd value in the nanomolar range [10, 39]. HMGB1 unbound from haptoglobin-HMGB1 complexes may then act systemically as a potent endogenous activator of TLR4 receptors present on multiple cell types, leading to further HMGB1 release. The TLR4 ligand that activates epithelial cells to release HMGB1 is HMGB1 itself [4]. It is also important to consider that HMGB1 might be therapeutically captured by administering haptoglobin or β subunit, which not only prevents HMGB1-mediated TLR4 activation, but also elicits an anti-inflammatory response.
In addition, macrophage polarization is another mechanism of innate immune regulation that can be modulated by HMGB1 and haptoglobin complexes. The polarization into M1 macrophages results in promotion of T helper (Th) 1 inflammatory response; whereas the polarization into M2 macrophages contributes to Th2 responses that facilitates tissue repair [40]. Previous studies demonstrated that HMGB1 facilitates macrophage M1-like phenotype switching [41, 42], which can be attenuated by monoclonal anti-HMGB1 antibodies [34]. Here we provide evidence to support the possibility that HMGB1-haptoglobin (β) complexes might regulate the M1/M2 macrophage phenotype balance by inhibiting HMGB1-mediated polarization of the inflammatory M1 subtype.
Hemoglobin also binds to the β chain of haptoglobin, and only the β chain of the haptoglobin is involved in CD163 receptor recognition [31, 43, 44]. The crystal structure of porcine haptoglobin and hemoglobin complexes confirmed this interaction [45]. A minimum of 80 amino acids in the haptoglobin β chain are required for hemoglobin-binding and maintaining the anti-oxidant activity of haptoglobin [46]. We are currently investigating the regions of haptoglobin β that bind HMGB1 and whether they overlap with the hemoglobin-binding sites.
As compared to humans, mice have a lower haptoglobin baseline levels in sera and subtle differences in hemoglobin metabolism [47, 48]. The species differences in haptoglobin and subsequent HMGB1 responses warrant further studies. Nonetheless, these considerations do not affect the general conclusions that can be deduced from our observations that haptoglobin β modulates HMGB1 activity.
In summary, we have discovered haptoglobin β subunit acts as an active HMGB1 antagonist which is capable of binding and inhibiting the pro-inflammatory properties of HMGB1 in a CD163-dependent fashion. Notably, mice express relative lower haptoglobin levels, which might be easily depleted by hemoglobin upon mild hemolysis [47, 49, 50], and can be readily replenished by administering exogenous haptoglobin or β subunit. It might be a long journey to transform the experimental therapeutic results in animal models to bedside in patients. However, there is substantial clinical success in the use of plasma-purified haptoglobin to treat several inflammatory diseases [51, 52] in conjunction with extracorporeal detoxification, massive blood transfusions, and attenuation of thermal injury and hemoglobin-induced kidney toxicity (reviewed in [53]). It would thus be important to assess results of clinical trials with haptoglobin β protein to treat severe systemic inflammatory conditions caused by excessive systemic accumulation of HMGB1.
Acknowledgments
This work was supported by grants from the NIH, RO1GM62508 (to KJT), RO1AT005076 and R01GM063075 (to HW), and RO1GM098446 (to HY). DJA would like to acknowledge support from the Wellcome Trust and the University of Liverpool Technology Directorate voucher scheme.
Footnotes
Conflict of interest statement
The authors have no conflict of interests to declare.
References
- 1.Angus DC, Wax RS. Epidemiology of sepsis: an update. Critical care medicine. 2001;29:S109–16. doi: 10.1097/00003246-200107001-00035. [DOI] [PubMed] [Google Scholar]
- 2.Lagu T, Rothberg MB, Shieh MS, Pekow PS, Steingrub JS, Lindenauer PK. Hospitalizations, costs, and outcomes of severe sepsis in the United States 2003 to 2007. Crit Care Med. 2012;40:754–61. doi: 10.1097/CCM.0b013e318232db65. [DOI] [PubMed] [Google Scholar]
- 3.Deutschman CS, Tracey KJ. Sepsis: Current Dogma and New Perspectives. Immunity. 2014;40:463–75. doi: 10.1016/j.immuni.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annual review of immunology. 2011;29:139–62. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsung A, Tohme S, Billiar TR. High-mobility group box-1 in sterile inflammation. J Intern Med. 2014;276:425–43. doi: 10.1111/joim.12276. [DOI] [PubMed] [Google Scholar]
- 6.Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. The Journal of cell biology. 2010;190:881–92. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu X, Messer JS, Wang Y, et al. Cytosolic HMGB1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J Clin Invest. 2015;125:1098–110. doi: 10.1172/JCI76344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. Journal of leukocyte biology. 2013;93:865–73. doi: 10.1189/jlb.1212662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, Moestrup SK. Identification of the haemoglobin scavenger receptor. Nature. 2001;409:198–201. doi: 10.1038/35051594. [DOI] [PubMed] [Google Scholar]
- 10.Yang H, Wang H, Levine YA, et al. Identification of CD163 as an antiinflammatory receptor for HMGB1-haptoglobin complexes. JCI insight. 2016;1 doi: 10.1172/jci.insight.85375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yueh SC, Lai YA, Chen WL, Hsu HH, Mao SJ. An improved method for haptoglobin 1-1, 2-1, and 2-2 purification using monoclonal antibody affinity chromatography in the presence of sodium dodecyl sulfate. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2007;845:210–7. doi: 10.1016/j.jchromb.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, Bloom O, Zhang M, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science (New York, NY) 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 13.Li J, Wang H, Mason JM, et al. Recombinant HMGB1 with cytokine-stimulating activity. Journal of immunological methods. 2004;289:211–23. doi: 10.1016/j.jim.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 14.Yang H, Lundback P, Ottosson L, et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1) Molecular medicine. 2012;18:250–9. doi: 10.2119/molmed.2011.00389. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Yang H, Wang H, Ju Z, et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med. 2015;212:5–14. doi: 10.1084/jem.20141318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian J, Avalos AM, Mao SY, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nature immunology. 2007;8:487–96. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 17.Moreno JA, Munoz-Garcia B, Martin-Ventura JL, et al. The CD163-expressing macrophages recognize and internalize TWEAK: potential consequences in atherosclerosis. Atherosclerosis. 2009;207:103–10. doi: 10.1016/j.atherosclerosis.2009.04.033. [DOI] [PubMed] [Google Scholar]
- 18.Gao S, Wang L, Liu W, Wu Y, Yuan Z. The synergistic effect of homocysteine and lipopolysaccharide on the differentiation and conversion of raw264. 7 macrophages. J Inflamm (Lond) 2014;11:13. doi: 10.1186/1476-9255-11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamazaki T, Mori M, Arai S, et al. Circulating AIM as an indicator of liver damage and hepatocellular carcinoma in humans. PLoS One. 2014;9:e109123. doi: 10.1371/journal.pone.0109123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang GX, Yu S, Gran B, et al. Role of IL-12 receptor beta 1 in regulation of T cell response by APC in experimental autoimmune encephalomyelitis. Journal of immunology (Baltimore, Md : 1950) 2003;171:4485–92. doi: 10.4049/jimmunol.171.9.4485. [DOI] [PubMed] [Google Scholar]
- 21.Bouhlel MA, Derudas B, Rigamonti E, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–43. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 22.Kiselak EA, Shen X, Song J, et al. Transcriptional regulation of an axonemal central apparatus gene, sperm-associated antigen 6, by a SRY-related high mobility group transcription factor, S-SOX5. J Biol Chem. 2010;285:30496–505. doi: 10.1074/jbc.M110.121590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saenz R, Futalan D, Leutenez L, et al. TLR4-dependent activation of dendritic cells by an HMGB1-derived peptide adjuvant. J Transl Med. 2014;12:211. doi: 10.1186/1479-5876-12-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang H, Tracey KJ. Targeting HMGB1 in inflammation. Biochimica et biophysica acta. 2010;1799:149–56. doi: 10.1016/j.bbagrm.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi HW, Tian M, Song F, et al. Aspirin’s Active Metabolite Salicylic Acid Targets High Mobility Group Box 1 to Modulate Inflammatory Responses. Mol Med. 2015 doi: 10.2119/molmed.2015.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang H, Ochani M, Li J, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arredouani MS, Kasran A, Vanoirbeek JA, Berger FG, Baumann H, Ceuppens JL. Haptoglobin dampens endotoxin-induced inflammatory effects both in vitro and in vivo. Immunology. 2005;114:263–71. doi: 10.1111/j.1365-2567.2004.02071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qin S, Wang H, Yuan R, et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. The Journal of experimental medicine. 2006;203:1637–42. doi: 10.1084/jem.20052203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antoine DJ, Jenkins RE, Dear JW, et al. Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. Journal of hepatology. 2012;56:1070–9. doi: 10.1016/j.jhep.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Desmet VJ, Knodell RG, Ishak KG, Black WC, Chen TS, Craig R, Kaplowitz N, Kiernan TW, Wollman J. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis [Hepatology 1981;1:431–435] Journal of hepatology. 2003;38:382–6. doi: 10.1016/s0168-8278(03)00005-9. [DOI] [PubMed] [Google Scholar]
- 31.Wejman JC, Hovsepian D, Wall JS, Hainfeld JF, Greer J. Structure of haptoglobin and the haptoglobin-hemoglobin complex by electron microscopy. Journal of molecular biology. 1984;174:319–41. doi: 10.1016/0022-2836(84)90341-3. [DOI] [PubMed] [Google Scholar]
- 32.Antoine DJ, Harris HE, Andersson U, Tracey KJ, Bianchi ME. A systematic nomenclature for the redox states of high mobility group box (HMGB) proteins. Molecular medicine. 2014 doi: 10.2119/molmed.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Porcheray F, Viaud S, Rimaniol AC, et al. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol. 2005;142:481–9. doi: 10.1111/j.1365-2249.2005.02934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su Z, Zhang P, Yu Y, et al. HMGB1 Facilitated Macrophage Reprogramming towards a Proinflammatory M1-like Phenotype in Experimental Autoimmune Myocarditis Development. Sci Rep. 2016;6:21884. doi: 10.1038/srep21884. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Xu J, Jiang Y, Wang J, et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014;21:1229–39. doi: 10.1038/cdd.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Antoine DJ, Williams DP, Kipar A, Laverty H, Park BK. Diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatotoxicity. Molecular medicine. 2010;16:479–90. doi: 10.2119/molmed.2010.00126. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Lea JD, Clarke JI, McGuire N, Antoine DJ. Redox-Dependent HMGB1 Isoforms as Pivotal Co-Ordinators of Drug-Induced Liver Injury: Mechanistic Biomarkers and Therapeutic Targets. Antioxidants & redox signaling. 2016;24:652–65. doi: 10.1089/ars.2015.6406. [DOI] [PubMed] [Google Scholar]
- 38.Sodhi CP, Jia H, Yamaguchi Y, et al. Intestinal Epithelial TLR-4 Activation Is Required for the Development of Acute Lung Injury after Trauma/Hemorrhagic Shock via the Release of HMGB1 from the Gut. Journal of immunology (Baltimore, Md : 1950) 2015 doi: 10.4049/jimmunol.1402490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lim SK, Kim H, bin Ali A, et al. Increased susceptibility in Hp knockout mice during acute hemolysis. Blood. 1998;92:1870–7. [PubMed] [Google Scholar]
- 40.Barros MH, Hauck F, Dreyer JH, Kempkes B, Niedobitek G. Macrophage polarisation: an immunohistochemical approach for identifying M1 and M2 macrophages. PLoS One. 2013;8:e80908. doi: 10.1371/journal.pone.0080908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karuppagounder V, Giridharan VV, Arumugam S, et al. Modulation of Macrophage Polarization and HMGB1-TLR2/TLR4 Cascade Plays a Crucial Role for Cardiac Remodeling in Senescence-Accelerated Prone Mice. PLoS One. 2016;11:e0152922. doi: 10.1371/journal.pone.0152922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tian S, Zhang L, Tang J, Guo X, Dong K, Chen SY. HMGB1 exacerbates renal tubulointerstitial fibrosis through facilitating M1 macrophage phenotype at the early stage of obstructive injury. Am J Physiol Renal Physiol. 2015;308:F69–75. doi: 10.1152/ajprenal.00484.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nielsen MJ, Moestrup SK. Receptor targeting of hemoglobin mediated by the haptoglobins: roles beyond heme scavenging. Blood. 2009;114:764–71. doi: 10.1182/blood-2009-01-198309. [DOI] [PubMed] [Google Scholar]
- 44.Nielsen MJ, Petersen SV, Jacobsen C, Thirup S, Enghild JJ, Graversen JH, Moestrup SK. A unique loop extension in the serine protease domain of haptoglobin is essential for CD163 recognition of the haptoglobin-hemoglobin complex. The Journal of biological chemistry. 2007;282:1072–9. doi: 10.1074/jbc.M605684200. [DOI] [PubMed] [Google Scholar]
- 45.Andersen CBF, Torvund-Jensen M, Nielsen MJ, et al. Structure of the haptoglobin-haemoglobin complex. Nature. 2012;489:456–9. doi: 10.1038/nature11369. [DOI] [PubMed] [Google Scholar]
- 46.Melamed-Frank M, Lache O, Enav BI, Szafranek T, Levy NS, Ricklis RM, Levy AP. Structure-function analysis of the antioxidant properties of haptoglobin. Blood. 2001;98:3693–8. doi: 10.1182/blood.v98.13.3693. [DOI] [PubMed] [Google Scholar]
- 47.Peacock AC, Gelderman AH, Ragland RH, Hoffman HA. Haptoglobin levels in serum of various strains of mice. Science (New York, NY) 1967;158:1703–4. doi: 10.1126/science.158.3809.1703. [DOI] [PubMed] [Google Scholar]
- 48.Etzerodt A, Kjolby M, Nielsen MJ, Maniecki M, Svendsen P, Moestrup SK. Plasma clearance of hemoglobin and haptoglobin in mice and effect of CD163 gene targeting disruption. Antioxidants & redox signaling. 2013;18:2254–63. doi: 10.1089/ars.2012.4605. [DOI] [PubMed] [Google Scholar]
- 49.Boretti FS, Buehler PW, D’Agnillo F, et al. Sequestration of extracellular hemoglobin within a haptoglobin complex decreases its hypertensive and oxidative effects in dogs and guinea pigs. J Clin Invest. 2009;119:2271–80. doi: 10.1172/JCI39115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nielsen MJ, Andersen CB, Moestrup SK. CD163 binding to haptoglobin-hemoglobin complexes involves a dual-point electrostatic receptor-ligand pairing. J Biol Chem. 2013;288:18834–41. doi: 10.1074/jbc.M113.471060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Irwin DC, Baek JH, Hassell K, et al. Hemoglobin-induced lung vascular oxidation, inflammation, and remodeling contribute to the progression of hypoxic pulmonary hypertension and is attenuated in rats with repeated-dose haptoglobin administration. Free Radic Biol Med. 2015;82:50–62. doi: 10.1016/j.freeradbiomed.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schaer CA, Laczko E, Schoedon G, Schaer DJ, Vallelian F. Chloroquine interference with hemoglobin endocytic trafficking suppresses adaptive heme and iron homeostasis in macrophages: the paradox of an antimalarial agent. Oxid Med Cell Longev. 2013;2013:870472. doi: 10.1155/2013/870472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schaer DJ, Vinchi F, Ingoglia G, Tolosano E, Buehler PW. Haptoglobin, hemopexin, and related defense pathways-basic science, clinical perspectives, and drug development. Front Physiol. 2014;5:415. doi: 10.3389/fphys.2014.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]