

Abstract
While the early start and higher intensity of the 2012/13 influenza A virus (IAV) epidemic was not unprecedented, it was the first IAV epidemic season since the 2009 H1N1 influenza pandemic where the H3N2 subtype predominated. We directly sequenced the genomes of 154 H3N2 clinical specimens collected throughout the epidemic to better understand the evolution of H3N2 strains and to inform the H3N2 vaccine selection process. Phylogenetic analyses indicated that multiple co-circulating clades and continual antigenic drift in the haemagglutinin (HA) of clades 5, 3A, and 3C, with the evolution of a new 3C subgroup (3C-2012/13), were the driving causes of the epidemic. Drift variants contained HA substitutions and alterations in the potential N-linked glycosylation sites of HA. Antigenic analysis demonstrated that viruses in the emerging subclade 3C.3 and subgroup 3C-2012/13 were not well inhibited by antisera generated against the 3C.1 vaccine strains used for the 2012/13 (A/Victoria/361/2011) or 2013/14 (A/Texas/50/2012) seasons. Our data support updating the H3N2 vaccine strain to a clade 3C.2 or 3C.3-like strain or a subclade that has drifted further. They also underscore the challenges in vaccine strain selection, particularly regarding HA and neuraminidase substitutions derived during laboratory passage that may alter antigenic testing accuracy.
Introduction
Influenza viruses cause significant annual morbidity and mortality in the global human population [1]. Epidemics occur during the winter months, cycling roughly every six months between the northern and southern hemispheres. Two influenza A virus (IAV) subtypes, H3N2 and H1N1pdm09, and two influenza B virus (IBV) lineages, B/Yamagata and B/Victoria, have been circulating among humans since 2009. Epidemics caused by each of these subtypes/lineages vary from season to season; this is due, in part, to the selective advantage acquired by one subtype/lineage in a given season. Although the error-prone IAV RNA polymerase frequently generates nucleotide substitutions that can lead to a selective advantage in all eight genomic viral RNA segments (vRNAs), new epidemic variants are most frequently due to accumulated substitutions in the two surface glycoproteins, haemagglutinin (HA) and neuraminidase (NA). Substitutions in HA/NA can give rise to variants that escape host immunity from previous exposures or vaccinations and are selected in the non-naïve host population through a process called antigenic drift. Antigenic drift primarily occurs in epitopes recognised by antibodies that neutralise viral infectivity by blocking the interaction of HA with sialic acid receptors on host cell glycoproteins [2,3]. Antigenic drift necessitates frequent updating of the strains used in the influenza vaccine [3,4]. This requires global surveillance of the antigenic profile of circulating strains to inform the decisions made at biannual World Health Organization (WHO) meetings for the selection of influenza vaccine strains [4-6]. The acquisition of as few as one to five mutations in HA can necessitate an updated vaccine strain to optimally protect the public [7,8].
The H3N2 subtype caused a severe epidemic during the 2012/13 influenza season in North America and contributed to a longer than normal season, with increased morbidity also in Europe [9,10]. Human H3N2 viruses represent a very successful lineage that has circulated since the ‘Hong Kong Flu’ pandemic of 1968 [11,12]. New H3N2 strains evolve continually, resulting in annual epidemics that are periodically severe, such as the 2003/04 season (A/Fujian/411/2002-like viruses) [3,7]. The 2012/13 season in the United States (US) was notable in both greater epidemic severity and limited vaccine effectiveness, especially in the elderly [13-18]. Our incomplete understanding of the factors shaping the emergence of epidemic IAV/IBV variants impairs our ability to accurately predict strain fitness and select appropriate vaccine strains.
We hypothesised that the 2012/13 epidemic was caused by the emergence of a new HA variant that rapidly displaced previously circulating strains. To gain a better understanding of the genetic and molecular mechanisms central to the intensity and severity of the 2012/13 epidemic, we sequenced 154 coding-complete genomes from H3N2-positive nasopharyngeal swabs collected throughout the 2012/13 season from a large hospital network in Houston, Texas, US. This demographically diverse system of five hospitals provides a reasonable representation of the viruses circulating in the US during a non-pandemic seasonal influenza epidemic [19], which made it a suitable site for studying IAV genetic variation. Direct sequencing of primary swab specimens is critical because H3N2 viruses are known to rapidly acquire adaptive mutations that change their antigenicity when passaged in cell cultures or eggs [5,20]. It allows accurate analysis of HA and NA, the identification of substitutions, and the detection of reassortment among vRNAs encoding internal proteins that contribute to viral fitness.
Methods
Hospital system and coding-complete viral genome sequencing
The Methodist Hospital System in Houston, Texas consists of five hospitals that serve a large population of ca 4 million people from the most ethnically diverse population in the United States (ca 32% Caucasian, 41% Hispanic/Latino, 20% African-American and 7% Asian) [21,22]. The IAV-positive human nasopharyngeal swab specimens used in this study were collected between 3 November 2012 and 8 February 2013. During this period, ca 20% of the specimens were from inpatients and 80% were from outpatients. The majority of inpatient isolates were obtained from patients admitted with influenza-like illness through the emergency department, who therefore did not have nosocomially acquired infections.
These viral samples were sequenced using the J. Craig Venter Institute's (JCVI's) high-throughput next-generation sequencing pipeline. Briefly, IAV vRNAs were isolated directly from the swab specimens, and the entire genome was amplified from 3 μl of RNA template using a multi-segment RT-PCR strategy (M-RTPCR) [23,24]. The amplicons were sequenced using the Ion Torrent PGM (Thermo Fisher Scientific, Waltham, Massachusetts, US) and/or the Illumina MiSeq v2 (Illumina, Inc., San Diego, California, US) instruments. When sequencing data from both platforms was available, the data were merged and assembled together; the resulting consensus sequences were supported by reads from both technologies.
Sequence curation
Reference sequences for IAV HA and NA nucleotide sequences were obtained from GenBank (National Center for Biotechnology Information) and the EpiFlu database (Global Initiative on Sharing All Influenza Data; GISAID). For phylogenetic analyses, three H3N2 reference sets were used: (i) all available strains presented by the Centers for Disease Control and Prevention (CDC) at the 2012/13 US vaccine composition meeting (available at [25]), (ii) all available strains derived from a phylogenetic tree in the 2013 southern hemisphere vaccine composition report [26] and (iii) a random selection of 50 isolates from public databases collected globally between 1 September 2012 and 8 February 2013. For the genome constellation analysis, all available coding-complete human H3N2 genomes in the EpiFlu database collected between 1 October 2011 and 20 February 2013 were downloaded on 6 June 2013.
Phylogenetic analyses
HA and NA nucleotide sequences and HA amino sequences were each aligned using MAFFT v7.158b with default settings [27-29] and trimmed using Qiagen's CLC Genomics Workbench v6.0. Four independent maximum likelihood (ML) dendrograms were inferred using GARLI [30,31], and the trees with the best log likelihood scores were analysed further with 500 bootstrap replicates. The ML tree and bootstrap values were combined using SumTrees in DendroPy [32]. For nucleotide sequences, the GTR G + I nucleotide substitution model with five rate categories was used, as determined by jModelTest v2.1.4 [33,34]. For protein sequences, the JTT G + I substitution model with four rate categories and empirically derived residue frequencies was used, as determined by ProtTest v2.4 [35].
Genome constellation analyses
Study and reference H3N2 genomes were parsed into gene segment-specific multi-FASTA files. MAFFT alignments for each segment were converted to a distance matrix using ANDES [36]. Dendrograms were generated using the complete linkage (farthest neighbour) hierarchical clustering technique in ANDES. Dendrograms were cut at a specified per cent identity and vRNA segments were assigned to groups according to their cluster at that cut-off. The segment group assignments were combined to obtain a genome constellation that was visualised using OrionPlot [37].
Viral isolation and propagation
A subset of viruses was selected to represent each co-circulating clade. Viruses were isolated from the primary swab specimen for subsequent propagation and antigenic analysis. Selected strains were passaged twice in Madin-Darby canine kidney (MDCK) cells to avoid selecting for mutations that frequently arise when IAV is isolated in embryonated chicken eggs. Supernatants from P2 viral stocks were clarified by centrifugation at 1,800 × g for 10 min at 4 °C, aliquoted, stored at −80 °C and sequenced.
Influenza virus rescue using gene synthesis and reverse genetics
To avoid spurious substitutions and generate a uniform virus population, positive (A/Perth/16/2009(H3N2) and A/Victoria/361/2011(H3N2)) and negative (A/Indiana/8/2011(H3N2v)) control viruses for antigenic testing were generated using gene synthesis [38] and a modified reverse genetics system [23,24] and were confirmed by sequencing. Briefly, 6:2 reassortant viruses (designated with an ‘r’ before the strain name, e.g. rA/Perth/16/2009) were rescued from plasmids encoding the six internal protein vRNAs from the strains indicated in Table 1 (e.g. A/Puerto Rico/8/1934) and the linear synthesised HA and NA genes of the desired H3N2 viruses.
Table 1. Antigenic properties of study samples measured by HI assays using ferret or sheep antisera raised against contemporary influenza A(H3N2) strains.
| HA GenBank or EpiFlua accession number | HA clade | Antigens used to generate ferret sera | Antigens used to generate sheep sera | |||||
|---|---|---|---|---|---|---|---|---|
| rA/Perth/16/2009 | rA/Victoria/361/2011-likeb (cell-grown) | rA/Victoria/361/2011 (egg-grown) | rA/Indiana/8/2011 (H3N2v) | A/Victoria/361/2011 | A/Texas/50/2012 | |||
| Reference antigensc | ||||||||
| rA/Perth/16/2009 (cell-grown) | GQ293081.1 | 1 | 320 | 320 | 113 | <20 | 453 | 1,280 |
| rA/Victoria/361/2011 (cell-grown) | EPI349103 | 3C.1 | 640 | 453 | 538 | 20 | 905 | >2,560 |
| rA/Victoria/361/2011 (egg-grown) | EPI353906 | 3C.1 | 226 | 320 | 905 | 40 | 1,280 | >2,560 |
| rA/Indiana/8/2011 (H3N2v) | JN638733.1 | N/A | <20 | <20 | <20 | 320 | <80 | <80 |
| Test antigens | ||||||||
| A/Texas/JMM_3/2012 | CY134748.1 | 5 | 226 | 453 | 113 | <20 | 320 | 1,810 |
| A/Texas/JMM_48/2012 | CY135076.1 | 3A | 226 | 57 | 160 | <20 | <80 | 905 |
| A/Texas/JMM_20/2012 | CY134868.1 | 3C.2 | 320 | 226 | 320 | <20 | 320 | 1,810 |
| A/Texas/JMM_37/2012 | CY134996.1 | 3C.3 | 80 | <20 | 28 | <20 | <80 | <80 |
| A/Texas/JMM_4/2012 | CY134756.1 | 3C-2012/13 | 80 | <20 | 40 | <20 | <80 | <80 |
HA: haemagglutinin; NA: neuraminidase; N/A: not applicable.
The authors gratefully acknowledge the originating (WHO Collaborating Centre for Reference and Research on Influenza in North Melbourne, Victoria, Australia) and submitting (Centers for Disease Control and Prevention, Atlanta, Georgia, US) laboratories that contributed the cell-and egg-grown A/Victoria/361/2011 sequences to GISAID's EpiFlu database.
rA/South_Australia/3/2011, which has the same amino acid sequence as A/Victoria/361/2011 (cell-grown) except for one change in the signal peptide.
These reassortant viruses are named for the strain from which the HA and NA were obtained; the six internal genes match A/Puerto Rico/8/1934(H1N1), A/New York/238/2005(H3N2) or A/New York/1682/2009(H1N1pdm09) sequences.
Highlighted cells indicate two-way HI results, where the specified serum is tested using the same (or a very similar) antigen to that used to generate the serum. An ‘r’ before the strain name indicates the virus was generated using gene synthesis and reverse genetics. Results from one experimental replicate are presented and are representative of the results obtained from at least two additional experimental replicates performed on a different day. All HA controls were between 3 and 8 HA units.
Antisera generation/acquisition and haemagglutination inhibition
Ferret antisera were generated by inoculating ferrets (two individuals per virus) intranasally with ca 1 × 106 TCID50 of clarified virus supernatant in 1 ml, and antisera were collected 30 days after inoculation. These ferret experiments were conducted by an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited company, BIOQUAL, Inc. (Rockville, Maryland, US). Antisera from the two ferrets inoculated with the same strain were pooled 1:1 and treated with three times their volume of receptor-destroying enzyme (RDE; Hardy Diagnostics, Santa Maria, California, US) overnight at 37 °C. RDE was inactivated at 56 °C for 30 min, and the RDE-treated antisera were diluted with six times the original antisera volume using phosphate-buffered saline. Lyophilised sheep antisera raised against A/Victoria/361/2011 and A/Texas/50/2012 were provided by the Center for Biologics Evaluation and Research (CBER), Food and Drug Administration (FDA), Department of Health and Human Services, US, and were also RDE-treated. Haemagglutination inhibition (HI) assays were performed [39] to determine the ability of various antisera to inhibit binding of the IAV isolates to guinea pig red blood cells.
Modelling H3 N-linked glycosylation
All potential HA N-linked glycosylation sites among contemporary H3N2 clades in this study were deduced from sequons and compared with the HA of A/Finland/486/2004(H3N2), for which the H3 structure has been solved [40]. Comparing the asparagine (N) residues of predicted sequons with the three-dimensional H3 structure confirmed that they would all be on the protein surface. While crystal structures can often detect the core of N-linked glycans, the actual composition and structure of the high-mannose and complex sugars is poorly understood. Here, we use the high-mannose glycan deduced from small-angle X-ray scattering data for the N165-linked glycan of X31 (H3 structural numbering) [41] as a ‘type glycan’ modelled onto the A/Finland/483/2004 H3 structure (Protein Data Bank ID: 2YP3 [40]) using the AllosMod server [41] which implements an all-atom modelling approach allowing for limited protein conformational flexibility. In brief, AllosMod uses CHARMM [42,43] force field parameters to derive preliminary glycan geometries and then optimises combined protein–glycan structures with MODELLER [44]. The all-atom structural model was visualised in PyMOL v1.7.1.7 [45]. Scripts that implement this modelling and visualisation protocol are available at [46].
Results
Phylogenetic analyses to understand H3N2 evolution
The coding-complete genomes of 154 IAV H3N2 specimens were sequenced and their genome sequences were submitted to GenBank (Table 2).
Table 2. GenBank accession numbers of coding-complete influenza A(H3N2) genomes sequenced for this study, Texas, 3 November 2012–8 February 2013 (n = 154).
Phylogenetic analyses of HA nucleotide sequences showed that the majority of circulating H3N2 strains arose from three major H3 clades: 5, 3A, and 3C (Figure 1A; clade nomenclature was adopted from US CDC [25]).
Figure 1. Phylogenetic relationships among the influenza A(H3N2) study samples, vaccine strains, and selected references inferred from maximum likelihood analyses of the HA nucleotide (A), HA protein (B), and NA nucleotide (C) sequences, Texas, 3 November 2012-8 February 2013 (n = 154).
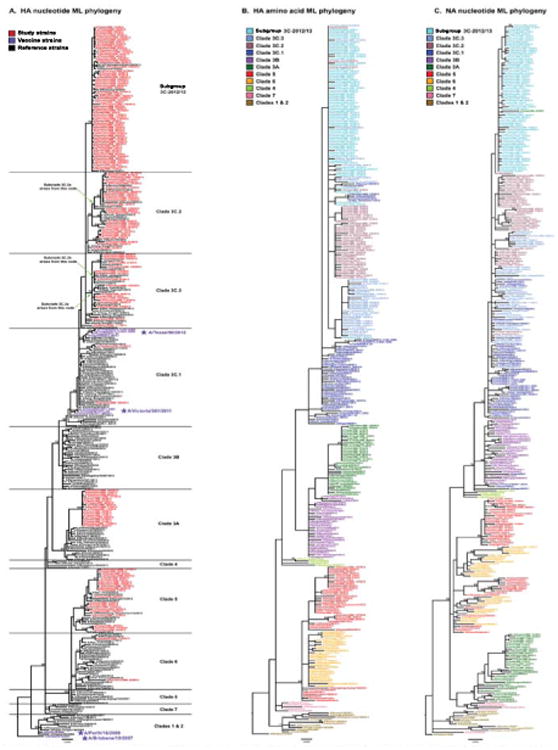
CDC: Centers for Disease Control and Prevention; HA: haemagglutinin; ML: maximum likelihood; NA: neuraminidase; WHO: World Health Organization
Bootstrap values for nodes with ≥70% support following 500 replicates are provided. Clades are classified following the CDC nomenclature [25] as closely as possible and are defined based on the HA nucleotide phylogeny (A). A new subgroup, designated 3C-2012/13, appears to have arisen during the 2012/13 influenza season. The nodes from which the new WHO-designated 3C.2a, 3C.3a, and 3C.3b subclades subsequently arise are marked on the phylogeny to place our 2012/13 study in the context of more recent seasons; placement was based on additional ML and neighbour-joining phylogenetic analyses using available H3 sequence data through 2014 (data not shown). For the HA protein (B) and NA nucleotide phylogenies (C), strain names are coloured by HA nucleotide clade definitions, showing that the HA protein and NA nucleotide clades largely match the nucleotide-based HA clade definitions. However, some interleaving of 3C subclades is observed in the HA protein tree, indicating that these subclades diverge largely through synonymous nucleotide mutations. The interleaving of colours in the NA nucleotide tree signifies intrasubtypic reassortment between the HA and NA gene segments. Scale bars indicate the average number of nucleotide changes per site.
The authors gratefully acknowledge the 87 originating and submitting laboratories who directly contributed sequences used in the phylogenetic analyses to GISAID: ADImmune Corporation, Taiwan; Alabama Department of Public Health, Bureau of Clinical Laboratories; Arkansas Children's Hospital; Austin Health, Australia; California Department of Health Services; Canterbury Health Services, New Zealand; US Centers for Disease Control and Prevention; CRR virus Influenza region Sud, France; Delaware Public Health Lab; Gart Naval General Hospital, United Kingdom; Georgia Public Health Laboratory; Government Virus Unit, Hong Kong; Health Protection Agency, England; Health Protection Inspectorate, Estonia; Hospital Clinic, Spain; Institute of Epidemiology and Infectious Diseases AMS of Ukraine; Institute of Epidemiology Disease Control and Research (IEDCR) & Bangladesh National Influenza Centre (NIC); Institute of Immunology and Virology Torlak, Serbia; Institute of Medical and Veterinary Science (IMVS), Australia; Institute of Public Health, Montenegro; Institute de Salud Publica de Chile; Institute Nacional de Saude, Portugal; Institut Pasteur de Dakar, Senegal; Institut Pasteur de Madagascar; Iowa State Hygienic Laboratory; Istituto Superiore di Sanit, Italy; Kansas Department of Health and Environment; Kentucky Division of Laboratory Services; Laboratory for Virology, National Institute of Public Health, Slovenia; Laboratory of Influenza and ILI, Belarus; Landspitali - University Hospital, Iceland; Louisiana Department of Health and Hospitals; Maine Health and Environmental Testing Laboratory; Maryland Department of Health and Mental Hygiene; Melbourne Pathology, Australia; Michigan Department of Community Health; Ministry of Health of Ukraine; Minnesota Department of Health; Monash Medical Centre, Australia; Montana Laboratory Services Bureau; Montana Public Health Laboratory; National Centre of Infectious and Parasitic Diseases, Bulgaria; National Institute for Communicable Disease, South Africa; National Institute for Health and Welfare, Finland; National Institute for Medical Research, United Kingdom; National Institute of Health, Korea; National Microbiology Laboratory, Health Canada; Nebraska Public Health Lab; Nevada State Health Laboratory; New Hampshire Public Health Laboratories; New Jersey Department of Health & Senior Services; New York City Department of Health; New York Medical College; New York State Department of Health; Norwegian Institute of Public Health; Oregon Public Health Laboratory; Pathwest QE II Medical Centre, Australia; Pennsylvania Department of Health; Prince of Wales Hospital, Australia; Queensland Health Scientific Services, Australia; Republic Institute for Health Protection, Macedonia; Rhode Island Department of Health; Robert Koch-Institute, Germany; San Antonio Metropolitan Health, Texas; South Dakota Public Health Lab; Southern Nevada Public Health Lab; Spokane Regional Health District, Washington; Statens Serum Institute, Denmark; State of Hawaii Department of Health; State of Idaho Bureau of Laboratories; Swedish Institute for Infectious Disease Control; Texas Department of State Health Services-Laboratory Services; University of Vienna, Austria; U.S. Air Force School of Aerospace Medicine; Utah Department of Health; VACSERA, Egypt; Victorian Infectious Diseases Reference Laboratory, Australia; Virginia Division of Consolidated Laboratories; Westmead Hospital, Australia; West Virginia Office of Laboratory Services; WHO Chinese National Influenza Center; WHO Collaborating Centre for Reference and Research on Influenza, Australia; WHO National Influenza Centre, Norway; WHO National Influenza Centre, National Institute of Medical Research (NIMR), United Kingdom; WHO National Influenza Centre Russian Federation; Wisconsin State Laboratory of Hygiene; and Wyoming Public Health Laboratory.
The backbone of the phylogeny had a ladder-like structure with relatively short terminal branches indicative of seasonal IAV antigenic drift arising from immune escape. While multiple older clade 5 strains were co-circulating in Houston, all circulating clade 3A strains were derived from a single monophyletic group. Importantly, a large number of the study specimens (66/154 = 42.9%) contained viruses that formed an emerging monophyletic group, which formed a new 3C subgroup that we designated 3C-2012/13. The majority of 3C viruses fell into subclades 3C.2 and 3C.3 or subgroup 3C-2012/13, while only two strains fell within subclade 3C.1, which was the subclade of the vaccine seed strains chosen for the 2012/13 and 2013/14 seasons (A/Victoria/361/2011 and A/Texas/50/2012, respectively). Analysis of H3N2 viruses worldwide from the onset of 2012/13 season in the northern hemisphere through June 2014 demonstrated that 3C.3 and 3C-2012/13 viruses also circulated in Europe and Asia (e.g. 3C.3-like A/Roma/11/2014 and 3C-2012/13-like A/Saint-Petersburg/170/2013; data not shown), suggesting that influenza surveillance in Houston, Texas, provided a reasonable sampling of circulating strains. However, it appears the 3C-2012/13 subgroup circulated through 2013 and then died out (data not shown). A larger phylogenetic analysis that included strains from recent seasons also placed our 2012/13 study strains in the context of the newest WHO-designated subclades for 3C.2 (i.e. 3C.2a) and 3C.3 (i.e. 3C.3a and 3C3.b) [47]. These new subclades are related to, but didn't arise directly from the 2012/13 viruses circulating in Texas; their placements based on this larger analysis are labelled on the left of the HA nucleotide phylogeny (Figure 1A).
Strains from all three major clades (5, 3A, and 3C) were co-circulating in Houston throughout the early, middle, and late 2012/13 epidemic (data not shown), indicating that strong temporal segregation of viral clades was not occurring within a single IAV season (Figure 1A). This shows that the emergence of a genetically novel virus early in the season did not rapidly displace other co-circulating strains.
Analysis of HA protein sequences for the same dataset showed that there were relatively few residue changes across all HA clades (Figure 2). This is particularly true for the HA proteins of the subclade 3C.3 and subgroup 3C-2012/13, which is illustrated by the interleaving of colours based on the nucleotide clade assignments (Figure 1B). This HA protein phylogeny emphasises how subclades 3C.2 and 3C.3, as well as subgroup 3C-2012/13, are more closely related to each other than to clade 3C.1 viruses.
Figure 2. HA amino acid differences across among representative influenza A(H3N2) strains for various HA clades, Texas, 3 November 2012–8 February 2013 (n = 5).

HA: haemagglutinin; Met: methionine; +/- indicates whether the residue is associated with the gain (+) or loss (−) of a potential glycosylation site.
a S45N creates a potential glycosylation site (SSS to NSS), with glycosylation occurring at N45.
b S124N removes a potential glycosylation site (NES to NEN), with glycosylation occurring at N122 (antigenic site A).
c T128N and T128A remove a potential glycosylation site (NWT to NWN or NWA), with glycosylation occurring at N126 (antigenic site A).
d N144D removes a potential glycosylation site (NSS to DSS), with glycosylation occurring at N144 (antigenic site A).
All residue changes that exist in HA among the study samples (yellow), vaccine strains (purple), and clade consensus sequences (white) are provided. All sequences that match the A/Perth/16/2009 strain (clade 1) are coloured in pink, while residues that differ from that strain are shown in blue (first residue difference) or green (second residue difference). Residues associated with the gain (+) or loss (−) of a potential glycosylation site are indicated. Antigenic site assignments are derived from several sources [60-63]. P0 indicates the original clinical swab specimen, while P2 indicates the second-passage viral stock grown in MDCK cells.
The NA nucleotide tree also formed a backbone characteristic of IAV that shows evidence of limited genetic drift in NA (Figure 1C). The NA phylogeny also showed three main groupings for the study samples, which roughly corresponded to the HA clade assignments 5, 3A, and 3C. However, colouring the NA tree using the HA clade assignments demonstrated that intrasubtypic reassortment between H3 and N2 occurs readily, as indicated by the interleaving of HA clade colours (Figure 1C). This was primarily found among 3C subclades and between clades 5 and 6. The most pronounced intrasubtypic reassortment was seen in A/Texas/JMM_27/2012, which has an HA belonging to clade 3A and an NA that groups with 3C-2012/13 viruses.
Genome constellation analyses to identify reassortment
To better understand the extent of intrasubtypic reassortment occurring among seasonal H3N2 viruses, we performed genome constellation analyses using 198 genomes (154 from our study samples and 44 additional strains). A 98% nucleotide identity cut-off value provided a genome constellation resolution that matched that of the major HA clade designations (i.e. 1, 5, 6, 3A, 3B, and 3C) (Figure 3A).
Figure 3. Genome constellations for coding-complete influenza A(H3N2) sequences from the 2012/13 influenza epidemic for all clades (A) and for clade 3 (B), Texas, 3 November 2012–8 February 2013 (n =154).

a These study samples form their own HA monophyly within clade 5.
b These study samples form their own HA monophyly within clade 3C.2.
c Of the 13 strains in this constellation that are also in the HA phylogeny, only one strain falls within clade 3C.2.
d These study samples form their own HA monophyly within clade 3C.3.
e Please note that each column is evaluated independently and cluster numbering is arbitrary.
- Genome constellation 1 (orange) segments were defined as sharing 98% nucleotide identity with the A/Perth/16/2009 vaccine strain. Twelve unique genome constellations were identified at the 98% nucleotide identity cut-off, eight of which were observed in the study samples.
- Clade 3 genome constellation 1 (purple) segments were defined as sharing 99% nucleotide identity with the A/Victoria/361/2011 vaccine strain. 10 unique genome constellations were identified at the 99% nucleotide identity cut-off, nine of which were observed in the study samples.
The authors gratefully acknowledge the 32 originating and submitting laboratories who directly contributed sequences used in the constellation analysis to GISAID: Alaska State Virology Lab; Arizona Department of Health Services; Austin Health, Australia; California Department of Health Services; Canterbury Health Services, New Zealand; US Centers for Disease Control and Prevention; Institute of Medical and Veterinary Science (IMVS), Australia; Institut Pasteur New Caledonia; Iowa State Hygienic Laboratory; John Hunter Hospital, Virology Unit, Clinical Microbiology, Australia; Kentucky Division of Laboratory Services; Melbourne Pathology, Australia; Michigan Department of Community Health; New Mexico Department of Health; New York State Department of Health; Papua New Guinea Institute of Medical Research; Pathwest QE II Medical Centre, Australia; Pennsylvania Department of Health; Puerto Rico Department of Health; Queensland Health Scientific Services, Australia; Research Institute of Tropical Medicine, Philippines; Rhode Island Department of Health; Royal Hobart Hospital, Australia; Southern Nevada Public Health Lab; Spokane Regional Health District, Washington; State of Hawaii Department of Health; Texas Department of State Health Services-Laboratory Services; USAMC-AFRIMS Department of Virology, Cambodia; Utah Department of Health; Victorian Infectious Diseases Reference Laboratory, Australia; WHO Collaborating Centre for Reference and Research on Influenza, Australia; and WHO National Influenza Centre, National Institute of Medical Research (NIMR), United Kingdom.
This yielded 12 genome constellations and demonstrated that vRNAs coding for internal proteins are also involved in intrasubtypic reassortment. While all constellations shared highly similar PA and M segments, seven monophyletic HA 3C.2 samples had reassorted PB2 segments (constellation 12 in Figure 3A) that grouped with the PB2 of HA clade 3A (constellations 2 and 4 in Figure 3A). Similarly, while most HA clade 5 and 6 strains had unique PB2 and PB1 segments (blue boxes in the PB2 and PB1 columns of Figure 3A), three monophyletic HA clade 5 strains had retained the A/Perth/16/2009-like PB2 and PB1 segments (orange boxes for PB2 and PB1 in constellation 7 in Figure 3A). Monophyletic HA strains sharing common intrasubtypic reassortment genome constellations demonstrated that these reassortment events are likely to occur once, creating viral strains that transmit and spread to new individuals, rather than occurring as multiple independent reassortment events. NA diversity was also observed among clade 5 and 6 viruses, which can also be appreciated by the interleaving colours when HA clade designations were mapped onto the NA phylogeny (Figure 1C).
To stringently explore the diversity among clade 3C viruses, a genome constellation analysis containing only the clade 3C genomes was performed using a 99% nucleotide identity cut-off value (Figure 3B). For the most part, the four 3C subclades/subgroups each contained their own unique genome constellations (Figure 3B). We found that most 3C-2012/13 viruses contained a unique NS segment compared with other 3C viruses (constellation 9 in Figure 3B), and all 3C-2012/13 viruses had a unique NP segment (constellations 8 and 9 in Figure 3B). In addition, seven monophyletic HA 3C.2 specimens contained unique PB2, PA, HA, and M segments at the 99% cut-off (constellation 10 in Figure 3B), and five monophyletic HA 3C.3 samples had incorporated a unique NA (constellation 6 in Figure 3B).
Overall, genome constellation analysis revealed that intrasubtypic reassortment occurs frequently in human H3N2 viruses. In this collection alone, intrasubtypic reassortment resulted in the expansion of antigenic drift variants into new genome constellations, the generation of new HA/NA combinations, and the acquisition of a unique NS vRNA.
Assessment of antigenic evolution in a single epidemic season
HI assays were performed [39] to determine the extent of antigenic drift among the H3 proteins during the 2012/13 epidemic. Two-way HI measurements, which paired an antigen with the antiserum raised against that antigen, provided HI titres that were typical for anti-H3N2 ferret and sheep antisera (Table 1). The specificity of the assay was shown by the negative control serum raised against a zoonotic H3N2 variant virus, (H3N2)v, which is antigenically similar to viruses that circulated in humans in the early to mid-1990s. Analysis of isolates propagated from specimens selected from the major clades showed that ferret and sheep polyclonal antisera against the recommended vaccine strains (A/Victoria/361/2011 and A/Texas/50/2012) prevented the binding of clade 5, 3C.1, and 3C.2 isolates to the sialic acid receptors on guinea pig red blood cells (Table 1). However, the emerging 3C.3 and 3C-2012/13 viruses escaped inhibition by these vaccine sera (> 16-fold reduction) (Table 1). This illustrates that the few amino acid differences in the H3 proteins (e.g. residues 128, 142, and 145 using H3 structural numbering) between the emergent 3C.3 and 3C-2012/13 viruses vs the 3C.1 viruses have enabled escape from polyclonal antibody responses to natural infections or vaccinations.
The virus isolates tested in HI were sequenced and had the same HA amino acid sequences as the original swab specimens in all but two cases (Figure 2), while the NA vRNAs of 3C.3 and 3C-2012/13 viruses acquired a mutation (i.e. D151N/G) that has been shown to reduce NA activity [48], illustrating the difficulty in accurately analysing the true antigenicity of viruses circulating in humans using in vitro assays and the importance of directly sequencing the viral population in swab specimens rather than viruses isolated from eggs or tissue culture. Furthermore, the HA of many contemporary H3N2 viruses bind inefficiently to guinea pig RBCs and the cognate NA aids in binding to receptors on the RBCs. Addition of oseltamivir in the HA assay to reduce NA binding reduced the HA titre so much for these viruses that HI assays could not be performed in the presence of oseltamivir.
Examination of the HA and NA residue changes (Figure 2, Figure 4) demonstrated that a few simultaneously evolving amino acid substitutions, some causing the gain or loss of sequons for N-linked glycans, differentiated the HA molecules among strains in the 2012/2013 epidemic.
Figure 4. Location of antigenic sites on the H3 monomer, along with key clade 3C substitutions and glycosylation sites.
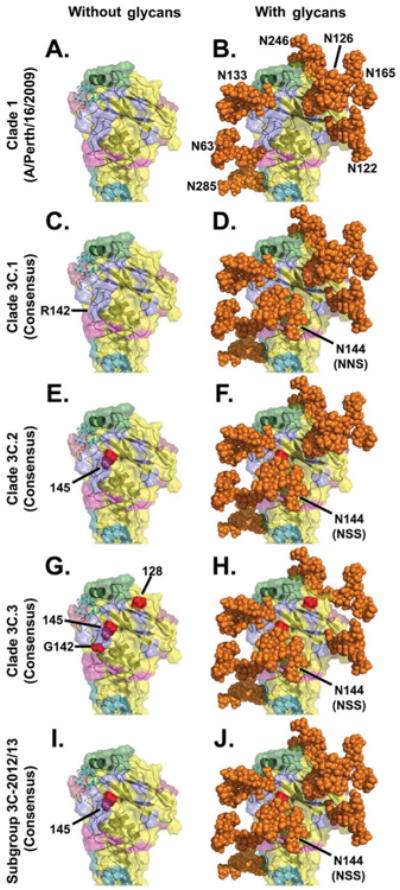
In all panels, the peptide backbone of the HA globular head is represented as a ribbon with a translucent solid surface using the A/Finland/486/2004 HA crystal structure, 2YP3, bound to a synthetic 2,6-sialic acid ligand (cyan stick structure) [40]. In panels on the right, all potential N-linked glycans in the H3 globular head were modelled using the AllosMod server [41] and rendered by PyMOL [45] as solid orange spheres.
Panels A and B: A representation of A/Perth/16/2009 (clade 1) illustrating previously defined H3 antigenic sites [60-63]: lavender (antigenic site A), green (antigenic site B), deep teal (antigenic site C), raspberry (antigenic site D), and light magenta (antigenic site E). A/Perth/16/2009 lacks the N144 glycosylation site that all subclade/subgroup 3C viruses have.
Panels C and D: A representation of 3C.1 viruses illustrating the H3 antigenic sites and the presence of the NNS sequon that may allow for glycosylation at N144, possibly at a reduced efficiency [55].
Panels E–J: Critical amino acid differences between the 3C.1 consensus and the indicated 3C subclade/subgroup consensuses are shown in red and labelled with H3 structural numbering.
Panel G: 3C.3 viruses have an R142G substitution, which removes most of the bulk of that protruding side chain; in this panel, the structure was modified to remove the bulky arginine (R) side chain (cf. the small red spheres for residue 142 with the larger lavender protrusion that extends downward in the other panels, as labelled in C).
Panel F: Some 3C.2 viruses have lost the N122 glycan.
Panel H: 3C.3 viruses lack the N126 glycan.
Panel J: 3C-2012/13 viruses are among the most heavily glycosylated.
Discussion
The H3N2 subtype predominated in Houston during the 2012/13 season, as was the case throughout the US for this epidemic [10]. While viruses from several known clades were co-circulating throughout the season, the majority of IAV H3s belonged to the 3C subclades, 3C.2 and 3C.3, as well as a new subgroup designated here as 3C-2012/13. Subgroup 3C-2012/13 represented an emerging group of viruses that differed primarily in nucleotide, but not amino acid, sequence from subclades 3C.2 and 3C.3, suggesting the possibility of convergent evolution of these drift variants in the human population.
Our data demonstrate that intrasubtypic reassortment created some of the co-circulating viruses in the 2012/13 epidemic. This finding is consistent with a recent study [12] and demonstrates that intrasubtypic reassortment is another evolutionary mechanism that antigenic drift variants can employ to gain fitness advantages and spread among the viral population. In some cases, intrasubtypic reassortment events among H3N2 segments can become fixed in the population as part of periodic selective sweeps and incorporated into the trunk of the phylogeny [49,50]. Intrasubtypic reassortment may also serve as a mechanism whereby drift variants can recover from a putative fitness loss (e.g. reduced receptor binding by HA) by acquiring epistatic changes through reassortment (e.g. in NA or NS) and/or the subsequent increase in amino acid replacement rates that reassortment triggers [49,50]. The NS gene segment encodes the NS1 protein, which is important in the evasion of the innate host immune response [51,52]; therefore, it is reasonable to hypothesize that the acquisition of a unique NS gene segment in most 3C-2012/13 viruses may offer a fitness advantage in the host when paired with the 3C-2012/13 HA segment. Experimental testing of these types of reassortants (e.g. 3C-2012/13 constellations 9 and 10 in Figure 3B) is required to test this hypothesis and to better understand how various segments interact with drifted HA sequences to create successful H3N2 strains.
Importantly, our data show that the emerging 3C.3 and 3C-2012/13 subclades/subgroups represented the majority of the virus strains sequenced in this study, and the representative samples from these clades that were tested in HI assays reacted poorly with antisera raised against the vaccine strains used for the 2012/13 season (egg- and cell-grown A/Victoria/361/2011) and the 2013/14 season (A/Texas/50/2012) which are both in the 3C.1 subclade. While use of oseltamivir in the HA assay reduced the HA titre enough that HI assays could not be performed in the presence of oseltamivir, more recent data on vaccine effectiveness and vaccine strain updates have corroborated our findings that the 2012/13 H3N2 vaccine strain was generally poorly matched for the majority of circulating 2012/13 H3N2 strains [26,47,53].
Overall, our data demonstrate that a few simultaneously evolving amino acid substitutions played a central role in immune escape by the 2012/13 epidemic viruses. Our data indicate that the H3N2 viruses escape the human immune response through a combination of specific residue changes (e.g. N145S, H3 structural numbering) and the impact these changes have on N-linked glycosylation. Residue 145 is located on a loop of the H3 that is partly involved in receptor binding (Figure 4), and it is one of seven residues shown to play a central role in the antigenic drift of H3 viruses in humans [54]. Viruses in 3C.2, 3C.3, and 3C-2012/13 have the S145 substitution, while the clade 3C.1 viruses have N145. This residue is part of an N-linked glycosylation sequon, and the N145S change is likely to increase glycosylation of N144 [55].
Some 3C.2 viruses acquired an S124N substitution, which removes the N122 glycosylation site; they are antigenically similar to previously circulating viruses in clades 1, 5, and 3C.1 and are therefore likely to die out. However, the new 3C.2a subclade that emerged from 3C.2 (Figure 1A) and expanded in the 2014/15 season [47] has lost glycosylation at 144 (N144S) and added a new putative glycosylation site at N158.
The 3C.3 viruses acquired accessory changes in or near antigenic site A, including T128A, which removes the N126 glycosylation site, and R142G; these substitutions are likely to be responsible for these viruses' antigenic difference from 3C.1 and 3C.2 viruses. The alterations of N-linked glycosylation sequons among the various clades (Figure 2, Figure 4) are likely to contribute to more complex structural differences in the HA due to the gain or loss of glycan shields [56-58]. In particular, it appears that glycans at HA residues N126 and N144 are the major differences among the 3C subclades/subgroups (Figure 2).
Furthermore, because 3C.3-like viruses and their descendants currently circulate globally (data not shown), vaccines based on A/Texas/50/2012 are likely to offer suboptimal protection. This hypothesis is supported by the moderate vaccine effectiveness for the 2012/13 season in the US (47% for IAV), where the H3N2 subtype predominated [13]. The data presented here indicate that an ideal vaccine candidate would probably be derived from subclade 3C.3a or 3C2a. The WHO influenza vaccine selection committee recently recommended an A/Switzerland/9715293/2013-like strain, which belongs to the 3C.3a subgroup of the 3C.3 viruses (Figure 1A), for the 2015 southern hemisphere season [59]. In addition to N145S, T128A and R142G substitutions, the 3C.3a viruses have acquired additional changes (A138S, F159S) that are likely to be antigenically important.
Conclusion
This study provides insights into the dynamics of the 2012/13 influenza epidemic by demonstrating that multiple H3N2 strains co-circulated during the epidemic, that different antigenic drift variants evolved concurrently and that intrasubtypic reassortment occurred frequently, suggesting that reassortment plays a role in the evolution of seasonal influenza viruses. The emergent 3C.3 and 3C-2012/13 viruses, which had substitutions impacting N-linked glycosylation at major antigenic sites, predominated, and we predict that they will come to define the new trunk of the phylogeny. Finally, our data show that the accumulation of relatively few HA mutations can convey large selective advantages, sometimes through epistatic interactions, and support the new 2015 southern hemisphere vaccine strain recommendation.
Acknowledgments
We thank the members of the Joint Technology Center at the J. Craig Venter Institute (JCVI) for performing the Illumina sequencing and Susmita Shrivastava in the JCVI Bioinformatics Group for annotating and submitting the genomes to NCBI. We also greatly appreciate the excellent technical assistance provided by Anju Subba and Haley Hochstein, past members of the JCVI Virology Group. We are very grateful to CBER/FDA for providing the sheep antisera used in the HI experiments and to Robert McHugh for developing the OrionPlot genome constellation visualization software. We thank Dr. Kathryn Stockbauer for her critique of our manuscript and gratefully acknowledge the authors and the originating and submitting laboratories of the sequences from GISAID's EpiFlu database that were used in the phylogenetic and genome constellation analyses. The data for this manuscript and its preparation was generated while DEW was employed at JCVI. The opinions expressed in this article are the authors' own and do not reflect the views of the Centers for Disease Control, the Department of Health and Human Services, or the United States government. This project was funded with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under contract number HHSN272200900007C (Genome Sequencing Centers for Infectious Diseases) and grant number U19AI110819 (Genomic Centers for Infectious Diseases).
Funding for the studies was obtained by DEW and JMM.
Footnotes
Conflict of interest: None declared.
Authors' contributions: KMS, SAS, RJO, SRD, DEW, and JMM conceived and designed the study. RJO and JMM conducted clinical diagnostics. XL and RAH performed the sequencing, TBS performed the sequencing data analysis and computational genome assemblies, and NF performed the finishing analysis to assess the final sequence quality. KMS and SAS performed the phylogenetic and genome constellation analyses. KMS and HLH performed the laboratory experiments. AT modelled the H3 N-linked glycosylation, and AT and KMS created the final structural figures. KMS, SAS, SRD, DEW, and JMM wrote the manuscript, and all authors reviewed and approved the final version.
Contributor Information
D E Wentworth, Email: dwentworth@cdc.gov.
J M Musser, Email: jmmusser@houstonmethodist.org.
References
- 1.World Health Organization (WHO) Fact Sheet No 211. Geneva: WHO; Mar, 2014. Influenza (Seasonal) Available from: http://www.who.int/mediacentre/factsheets/fs211/en/ [Google Scholar]
- 2.Wiley DC, Skehel JJ. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu Rev Biochem. 1987;56(1):365–94. doi: 10.1146/annurev.bi.56.070187.002053. http://dx.doi.org/10.1146/annurev.bi.56.070187.002053. [DOI] [PubMed] [Google Scholar]
- 3.Hay AJ, Gregory V, Douglas AR, Lin YP. The evolution of human influenza viruses. Philos Trans R Soc Lond B Biol Sci. 2001;356(1416):1861–70. doi: 10.1098/rstb.2001.0999. http://dx.doi.org/10.1098/rstb.2001.0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stöhr K, Bucher D, Colgate T, Wood J. Influenza virus surveillance, vaccine strain selection, and manufacture. Methods Mol Biol. 2012;865:147–62. doi: 10.1007/978-1-61779-621-0_9. http://dx.doi.org/10.1007/978-1-61779-621-0_9. [DOI] [PubMed] [Google Scholar]
- 5.Ampofo WK, Al Busaidy S, Cox NJ, Giovanni M, Hay A, Huang S, et al. WHO Writing Group. Strengthening the influenza vaccine virus selection and development process: outcome of the 2nd WHO Informal Consultation for Improving Influenza Vaccine Virus Selection held at the Centre International de Conférences (CICG) Geneva, Switzerland, 7 to 9 December 2011. Vaccine. 2013;31(32):3209–21. doi: 10.1016/j.vaccine.2013.05.049. http://dx.doi.org/10.1016/j.vaccine.2013.05.049. [DOI] [PubMed] [Google Scholar]
- 6.Ampofo WK, Baylor N, Cobey S, Cox NJ, Daves S, Edwards S, et al. Improving influenza vaccine virus selection: report of a WHO informal consultation held at WHO headquarters, Geneva, Switzerland, 14-16 June 2010. Influenza Other Respir Viruses. 2012;6(2):142–52. doi: 10.1111/j.1750-2659.2011.00277.x. http://dx.doi.org/10.1111/j.1750-2659.2011.00277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus AD, et al. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305(5682):371–6. doi: 10.1126/science.1097211. http://dx.doi.org/10.1126/science.1097211. [DOI] [PubMed] [Google Scholar]
- 8.Das SR, Hensley SE, Ince WL, Brooke CB, Subba A, Delboy MG, et al. Defining influenza A virus hemagglutinin antigenic drift by sequential monoclonal antibody selection. Cell Host Microbe. 2013;13(3):314–23. doi: 10.1016/j.chom.2013.02.008. http://dx.doi.org/10.1016/j.chom.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.World Health Organization Regional Office for Europe (WHO/Europe) Copenhagen: WHO/Europe; 2013. Summary of the 2012-2013 influenza season in the WHO European Region. Available from: http://www.euro.who.int/en/health-topics/communicable-diseases/influenza/publications/2013/summary-of-the-20122013-influenza-season-in-the-who-european-region. [Google Scholar]
- 10.Review of the 2012-2013 winter influenza season, northern hemisphere. Wkly Epidemiol Rec. 2013;88(22):225–32. [PubMed] [Google Scholar]
- 11.Cockburn WC, Delon PJ, Ferreira W. Origin and progress of the 1968-69 Hong Kong influenza epidemic. Bull World Health Organ. 1969;41(3):345–8. [PMC free article] [PubMed] [Google Scholar]
- 12.Westgeest KB, Russell CA, Lin X, Spronken MI, Bestebroer TM, Bahl J, et al. Genomewide analysis of reassortment and evolution of human influenza A(H3N2) viruses circulating between 1968 and 2011. J Virol. 2014;88(5):2844–57. doi: 10.1128/JVI.02163-13. http://dx.doi.org/10.1128/JVI.02163-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Centers for Disease Control and Prevention (CDC) Interim adjusted estimates of seasonal influenza vaccine effectiveness - United States, February 2013. MMWR Morb Mortal Wkly Rep. 2013;62(7):119–23. [PMC free article] [PubMed] [Google Scholar]
- 14.Bragstad K, Emborg H, Fischer TK, Voldstedlund M, Gubbels S, Andersen B, et al. Low vaccine effectiveness against influenza A(H3N2) virus among elderly people in Denmark in 2012/13--a rapid epidemiological and virological assessment. Euro Surveill. 2013;18(6):11–7. [PubMed] [Google Scholar]
- 15.Kelvin DJ, Farooqui A. Extremely low vaccine effectiveness against influenza H3N2 in the elderly during the 2012/2013 flu season. J Infect Dev Ctries. 2013;7(3):299–301. doi: 10.3855/jidc.3544. http://dx.doi.org/10.3855/jidc.3544. [DOI] [PubMed] [Google Scholar]
- 16.McMenamin J, Andrews N, Robertson C, Fleming D, Durnall H, von Wissmann B, et al. Effectiveness of seasonal 2012/13 vaccine in preventing laboratory-confirmed influenza infection in primary care in the United Kingdom: mid-season analysis 2012/13. Euro Surveill. 2013;18(5):1–5. doi: 10.2807/ese.18.05.20393-en. [DOI] [PubMed] [Google Scholar]
- 17.Skowronski DJN, De Serres G, Dickinson J, Winter A, Mahmud S, Sabaiduc S, et al. Interim estimates of influenza vaccine effectiveness in 2012/13 from Canada's sentinel surveillance network, January 2013. Euro Surveill. 2013;18(5):1–10. doi: 10.2807/ese.18.05.20394-en. [DOI] [PubMed] [Google Scholar]
- 18.Valenciano M, Kissling E. I-MOVE Case-Control Study Team. Early estimates of seasonal influenza vaccine effectiveness in Europe: results from the I-MOVE multicentre case-control study, 2012/13. Euro Surveill. 2013;18(7):3. [PubMed] [Google Scholar]
- 19.Nelson MI, Tan Y, Ghedin E, Wentworth DE, St George K, Edelman L, et al. Phylogeography of the spring and fall waves of the H1N1/09 pandemic influenza virus in the United States. J Virol. 2011;85(2):828–34. doi: 10.1128/JVI.01762-10. http://dx.doi.org/10.1128/JVI.01762-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chambers BS, Li Y, Hodinka RL, Hensley SE. Recent H3N2 influenza virus clinical isolates rapidly acquire hemagglutinin or neuraminidase mutations when propagated for antigenic analyses. J Virol. 2014;88(18):10986–9. doi: 10.1128/JVI.01077-14. http://dx.doi.org/10.1128/JVI.01077-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris County, Texas. Washington: United States Census Bureau; [Accessed: 7 Oct 2014]. State and county QuickFacts. Available from: http://quickfacts.census.gov/qfd/states/48/48201.html. [Google Scholar]
- 22.Emerson MO, Bratter J, Howell J, Jeanty PW, Cline M. Houston: Rice University; 2012. Houston region grows more racially/ethnically diverse, with small declines in segregation. A joint report analyzing census data from 1990, 2000 and 2010. Available from: http://kinder.rice.edu/uploadedFiles/Urban_Research_Center/Media/Houston%20Region%20Grows%20More%20Ethnically%20Diverse%202-13.pdf. [Google Scholar]
- 23.Zhou B, Wentworth DE. Influenza A virus molecular virology techniques. Methods Mol Biol. 2012;865:175–92. doi: 10.1007/978-1-61779-621-0_11. http://dx.doi.org/10.1007/978-1-61779-621-0_11. [DOI] [PubMed] [Google Scholar]
- 24.Zhou B, Donnelly ME, Scholes DT, St George K, Hatta M, Kawaoka Y, et al. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J Virol. 2009;83(19):10309–13. doi: 10.1128/JVI.01109-09. http://dx.doi.org/10.1128/JVI.01109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cox NJ. Seasonal influenza and zoonotic influenza Information for the vaccine and related biological products advisory committee meeting. Silver Spring, Maryland: Feb 28, 2012. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/BloodVaccinesandOtherBiologics/VaccinesandRelatedBiologicalProductsAdvisoryCommittee/UCM300419.pptx. [Google Scholar]
- 26.McCauley J, Daniels R, Lin YP, Xiang Z, Gregory V, Whittaker L, et al. Report prepared for the WHO annual consultation on the composition of influenza vaccine for the Southern Hemisphere 2013. 2012 Available from: http://www.crick.ac.uk/media/221897/interim_report_september_2012_2.pdf.
- 27.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–66. doi: 10.1093/nar/gkf436. http://dx.doi.org/10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008;9(4):286–98. doi: 10.1093/bib/bbn013. http://dx.doi.org/10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 29.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80. doi: 10.1093/molbev/mst010. http://dx.doi.org/10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bazinet AL, Cummings MP. The Lattice Project: a grid research and production environment combining multiple grid computing models. In: Weber MHW, editor. Distributed & Grid Computing - Science Made Transparent for Everyone Principles, Applications and Supporting Communities. Marburg, Germany: 2008. pp. 2–13. Rechenkraft.net. [Google Scholar]
- 31.Zwickl DJ. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion [Thesis] Austin: The University of Texas; 2006. Available from: http://repositories.lib.utexas.edu/handle/2152/2666. [Google Scholar]
- 32.Sukumaran J, Holder MT. DendroPy: a Python library for phylogenetic computing. Bioinformatics. 2010;26(12):1569–71. doi: 10.1093/bioinformatics/btq228. http://dx.doi.org/10.1093/bioinformatics/btq228. [DOI] [PubMed] [Google Scholar]
- 33.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772. doi: 10.1038/nmeth.2109. http://dx.doi.org/10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704. doi: 10.1080/10635150390235520. http://dx.doi.org/10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 35.Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21(9):2104–5. doi: 10.1093/bioinformatics/bti263. http://dx.doi.org/10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 36.Li K, Venter E, Yooseph S, Stockwell TB, Eckerle LD, Denison MR, et al. ANDES: Statistical tools for the ANalyses of DEep Sequencing. BMC Res Notes. 2010;3(1):199. doi: 10.1186/1756-0500-3-199. http://dx.doi.org/10.1186/1756-0500-3-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schobel-McHugh R. GitHub, Inc; 2014. OrionPlot. Available from: https://github.com/sschobel/orion-plot/tree/master/OrionPlot/dist/alpha. [Google Scholar]
- 38.Dormitzer PR, Suphaphiphat P, Gibson DG, Wentworth DE, Stockwell TB, Algire MA, et al. Synthetic generation of influenza vaccine viruses for rapid response to pandemics. Sci Transl Med. 2013;5(185):185ra68. doi: 10.1126/scitranslmed.3006368. http://dx.doi.org/10.1126/scitranslmed.3006368. [DOI] [PubMed] [Google Scholar]
- 39.World Health Organization (WHO) Geneva: WHO; 2011. Manual for the Laboratory Diagnosis and Virological Surveillance of Influenza. Available from: http://www.who.int/influenza/gisrs_laboratory/manual_diagnosis_surveillance_influenza/en/ [Google Scholar]
- 40.Lin YP, Xiong X, Wharton SA, Martin SR, Coombs PJ, Vachieri SG, et al. Evolution of the receptor binding properties of the influenza A(H3N2) hemagglutinin. Proc Natl Acad Sci U S A. 2012;109(52):21474–9. doi: 10.1073/pnas.1218841110. http://dx.doi.org/10.1073/pnas.1218841110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guttman M, Weinkam P, Sali A, Lee KK. All-atom ensemble modeling to analyze small-angle x-ray scattering of glycosylated proteins. Structure. 2013;21(3):321–31. doi: 10.1016/j.str.2013.02.004. http://dx.doi.org/10.1016/j.str.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guvench O, Greene SN, Kamath G, Brady JW, Venable RM, Pastor RW, et al. Additive empirical force field for hexopyranose monosaccharides. J Comput Chem. 2008;29(15):2543–64. doi: 10.1002/jcc.21004. http://dx.doi.org/10.1002/jcc.21004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guvench O, Hatcher ER, Venable RM, Pastor RW, Mackerell AD., Jr CHARMM Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J Chem Theory Comput. 2009;5(9):2353–70. doi: 10.1021/ct900242e. http://dx.doi.org/10.1021/ct900242e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. http://dx.doi.org/10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 45.Schrodinger L. The PyMOL Molecular Graphics System, Version 1.7.1.7. 2010 [Google Scholar]
- 46.Tovchigrechko A. Sydney: Atlassian; 2015. Modeling and visualization of influenza hemagglutinin glycosylation. Available from: https://bitbucket.org/andreyto/flu_ha_glyc. [Google Scholar]
- 47.McCauley J, Daniels R, Lin YP, Xiang Z, Gregory V, Whittaker L, et al. Report prepared for the WHO annual consultation on the composition of influenza vaccine for the Southern Hemisphere 2015. 2014 Available from: http://www.crick.ac.uk/media/221823/nimr-vcm-report-sep-14-web.pdf.
- 48.Lin YP, Gregory V, Collins P, Kloess J, Wharton S, Cattle N, et al. Neuraminidase receptor binding variants of human influenza A(H3N2) viruses resulting from substitution of aspartic acid 151 in the catalytic site: a role in virus attachment? J Virol. 2010;84(13):6769–81. doi: 10.1128/JVI.00458-10. http://dx.doi.org/10.1128/JVI.00458-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neverov AD, Lezhnina KV, Kondrashov AS, Bazykin GA. Intrasubtype reassortments cause adaptive amino acid replacements in H3N2 influenza genes. PLoS Genet. 2014;10(1):e1004037. doi: 10.1371/journal.pgen.1004037. http://dx.doi.org/10.1371/journal.pgen.1004037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rambaut A, Pybus OG, Nelson MI, Viboud C, Taubenberger JK, Holmes EC. The genomic and epidemiological dynamics of human influenza A virus. Nature. 2008;453(7195):615–9. doi: 10.1038/nature06945. http://dx.doi.org/10.1038/nature06945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hale BG, Randall RE, Ortín J, Jackson D. The multifunctional NS1 protein of influenza A viruses. J Gen Virol. 2008;89(Pt 10):2359–76. doi: 10.1099/vir.0.2008/004606-0. http://dx.doi.org/10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- 52.Haye K, Burmakina S, Moran T, García-Sastre A, Fernandez-Sesma A. The NS1 protein of a human influenza virus inhibits type I interferon production and the induction of antiviral responses in primary human dendritic and respiratory epithelial cells. J Virol. 2009;83(13):6849–62. doi: 10.1128/JVI.02323-08. http://dx.doi.org/10.1128/JVI.02323-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McLean HQ, Thompson MG, Sundaram ME, Kieke BA, Gaglani M, Murthy K, et al. Influenza vaccine effectiveness in the United States during 2012-2013: variable protection by age and virus type. J Infect Dis. 2015;211(10):1529–40. doi: 10.1093/infdis/jiu647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koel BF, Burke DF, Bestebroer TM, van der Vliet S, Zondag GC, Vervaet G, et al. Substitutions near the receptor binding site determine major antigenic change during influenza virus evolution. Science. 2013;342(6161):976–9. doi: 10.1126/science.1244730. http://dx.doi.org/10.1126/science.1244730. [DOI] [PubMed] [Google Scholar]
- 55.Shakin-Eshleman SH, Spitalnik SL, Kasturi L. The amino acid at the X position of an Asn-X-Ser sequon is an important determinant of N-linked core-glycosylation efficiency. J Biol Chem. 1996;271(11):6363–6. doi: 10.1074/jbc.271.11.6363. http://dx.doi.org/10.1074/jbc.271.11.6363. [DOI] [PubMed] [Google Scholar]
- 56.Wanzeck K, Boyd KL, McCullers JA. Glycan shielding of the influenza virus hemagglutinin contributes to immunopathology in mice. Am J Respir Crit Care Med. 2011;183(6):767–73. doi: 10.1164/rccm.201007-1184OC. http://dx.doi.org/10.1164/rccm.201007-1184OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arinaminpathy N, Grenfell B. Dynamics of glycoprotein charge in the evolutionary history of human influenza. PLoS One. 2010;5(12):e15674. doi: 10.1371/journal.pone.0015674. http://dx.doi.org/10.1371/journal.pone.0015674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kobayashi Y, Suzuki Y. Evidence for N-glycan shielding of antigenic sites during evolution of human influenza A virus hemagglutinin. J Virol. 2012;86(7):3446–51. doi: 10.1128/JVI.06147-11. http://dx.doi.org/10.1128/JVI.06147-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.World Health Organization (WHO) Geneva: WHO; 2014. Recommended composition of influenza virus vaccines for use in the 2015 southern hemisphere influenza season. Available from: http://www.who.int/influenza/vaccines/virus/recommendations/2015_south/en/ [PubMed] [Google Scholar]
- 60.Daniels RS, Douglas AR, Skehel JJ, Wiley DC. Analyses of the antigenicity of influenza haemagglutinin at the pH optimum for virus-mediated membrane fusion. J Gen Virol. 1983;64(Pt 8):1657–62. doi: 10.1099/0022-1317-64-8-1657. http://dx.doi.org/10.1099/0022-1317-64-8-1657. [DOI] [PubMed] [Google Scholar]
- 61.Popova L, Smith K, West AH, Wilson PC, James JA, Thompson LF, et al. Immunodominance of antigenic site B over site A of hemagglutinin of recent H3N2 influenza viruses. PLoS One. 2012;7(7):e41895. doi: 10.1371/journal.pone.0041895. http://dx.doi.org/10.1371/journal.pone.0041895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stray SJ, Pittman LB. Subtype- and antigenic site-specific differences in biophysical influences on evolution of influenza virus hemagglutinin. Virol J. 2012;9(1):91. doi: 10.1186/1743-422X-9-91. http://dx.doi.org/10.1186/1743-422X-9-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature. 1981;289(5796):373–8. doi: 10.1038/289373a0. http://dx.doi.org/10.1038/289373a0. [DOI] [PubMed] [Google Scholar]