

Abstract
Cardiotoxicity is an important issue for breast cancer patients receiving anthracycline-trastuzumab therapy in the adjuvant setting. Studies show that 3–36% of patients receiving anthracyclines and/or trastuzumab experience chemotherapy related cardiac dysfunction (CRCD) and approximately 17% of patients must stop chemotherapy due to the consequences of CRCD. There is currently no standardized, clinically verified way to detect CRCD early, but common practices include serial echocardiography and troponin measurements, which can be timely, costly, and not always available in areas where health care resources are scarce. Furthermore, detection of CRCD, before there is any echocardiographic evidence of dysfunction or clinical symptoms present, would allow maximal benefit of chemotherapy and minimize cardiac complications. Creating a panel of serum biomarkers would allow for more specificity and sensitivity in the early detection of CRCD, which would be easy to implement and cost effective in places with limited health care. Based on a review of the literature, we propose creating a biomarker panel consisting of topoisomerase 2β, serum troponin T/I, myeloperoxidase, NT-proBNP, miR-208b, miR-34a, and miR-150 in breast cancer patients receiving anthracyclines and/or trastuzumab to detect CRCD before any signs of overt cardiotoxicity are apparent.
Keywords: Chemotherapy related cardiotoxicity, Trastuzumab, Anthracyclines, Breast cancer, Biomarkers
Introduction
Chemotherapy and radiation therapy have revolutionized cancer treatment but they can lead to potential adverse cardiovascular effects, termed “chemotherapy related cardiac dysfunction” (CRCD). CRCD can include one or more of the following: a reduction in left ventricular ejection fraction (LVEF), either global or specific in the interventricular septum, symptoms or signs associated with heart failure (HF), and/or a reduction in LVEF by less than 5% from baseline (in the presence of signs or symptoms of HF) to less than 55% or a reduction in LVEF greater than 10% from baseline (without signs or symptoms of HF) to less than 55% [1].
Cardiac dysfunction associated with chemotherapy in breast cancer can be acute, subacute or chronic [1]. Acute or subacute cardiotoxicity develops any time from the initiation of chemotherapy up to two weeks after the completion of therapy. It can be characterized by different types of arrhythmias, abnormalities in ventricular repolarization and QT intervals, acute coronary syndromes, or pericardial reaction and alteration in myocardial function. Chronic cardiotoxicity is classified as either early (within 1 year after completing treatment) or late (>1 year after completing treatment). There are two types of CRCD–type I and type II. Type I CRCD is actual myocardial damage and generally the damage is irreversible. Though the damage may stabilize, it appears to be permanent. Type II is reversible with a high likelihood of recovery in 2–4 months to, or near, baseline status. Type I is cumulative dose related and mostly associated with anthracyclines whereas type II is not dose related and mostly associated with trastuzumab. The primary difference to keep in mind is that type I results in structural myocardial damage and necrosis, while type II is a functional deficit that has been described as “stunning” (Table 1) [2].
Table 1.
Comparison of type I vs. type II CRCD.
| Types of CRCD [2] | ||
|---|---|---|
| Type 1 (Myocardial Damage) | Type II (Myocardial Dysfunction) | |
| Typical Agent | Anthracyclines (doxorubicin, daunorubicin, epirubicin, idarubicin, valrubicin) | HER2-targeted agents (trastuzumab) |
| Clinical Course | Irreversible-damage may stabilize but underlying damage appears to be permanent | Reversible-high likelihood of recovery in 2–4 months to near baseline cardiac status |
| Dose Effects | Cumulative, dose related | Not dose related |
| Mechanism | Free radical formation; oxidative stress and damage of cardiac muscle cells | Blocked ErbB2 signaling; loss of contractility/stunning or hibernation |
| Ultrastructural Changes | Vacuoles; myofibrillar disarray and dropout; necrosis (changes resolve over time) | No ultrastructural abnormalities |
| Echocardiography | Decreased EF, global decrease in wall motion | |
| Effect of Rechallenge | High probability of recurrent, progressive dysfunction. May result in heart failure and death | Increasing evidence for relative safety of rechallenge, but more data needed |
| Effect of Late Sequential Stress | High likelihood of sequential stress related cardiac dysfunction | Low likelihood of sequential stress related cardiac dysfunction |
| Risk Factors | IV high single dose, time of drug infusion<30mins, history of irradiation, prior use of anthracyclines, use of concomitant agents, female, old or young age, underlying CVD, increased time elapsed since therapy administration [3] | Age >50 yrs, underlying CVD, prior anthracycline treatment, use of concomitant agents [3] |
Cardiotoxicity is a particularly important issue given the large number of women with breast cancer receiving combination anthracycline-trastuzumab therapy in the adjuvant setting.
Based on the literature, the incidence of CRCD in patients receiving trastuzumab alone is 3–7%, anthracyclines before trastuzumab 5%, and anthracyclines alone is reportedly 4–36% (6% have clinically overt cardiotoxicity and 18% subclinical cardiotoxicity). And on average, 17% of patients receiving this treatment for the most aggressive forms of breast cancer have to stop therapy due to cardiac complications. However, cardiotoxicity is reversible in many patients, if chemotherapy is stopped immediately [3–5].
Patients receiving a cumulative doxorubicin dose of ≥ 500 mg/m2 have a significantly increased risk of developing cardiac toxicity, including cardiomyopathy and congestive heart failure. Doxorubicin is believed to cause dose-dependent cardiotoxicity through redox cycling and the generation of reactive oxygen species (ROS) [6]. The ROS hypothesis, however, has been tempered by a series of studies in which ROS scavenger treatment failed to prevent cardiac toxicity caused by doxorubicin [7,8]. Thus, the mechanism by which anthracyclines causes cardiotoxicity has remained elusive and to date there is no method available to predict whether a patient will develop heart damage as a result of an anthracycline-based therapy.
Several strategies have been proposed to reduce anthracycline-trastuzumab-induced cardiotoxicity, though none are studied in controlled clinical trials or standardized practice. Conventional strategies include evaluation of LVEF at the beginning of chemotherapy, after the administration of half the total anthracycline cumulative dose, and before every subsequent dose. During follow up, LVEF evaluation is recommended 3, 6 and 12 months after the end of treatment [9–11]. And a decline of LVEF by more than 10%, associated with an absolute LVEF less than 50%, is suggested as criteria for suspending chemotherapy. Using this approach reportedly, reduces the risk of developing heart failure to less than 5% [12,13]. Discontinuing potentially cardiotoxic therapy when cardiotoxicity arises and instituting heart failure medications early are both critical steps in addressing CRCD [3–5].
Issues with using the aforementioned approach however include the poor cost effectiveness, since not every patient requires such frequent, repeated echocardiographs [14]. Furthermore, echocardiography is neither sensitive nor specific to predict early development of cardiac dysfunction. In other words, it only allows for identification of cardiac damage after there is already dysfunction, which is not ideal for a screening tool since it will not allow for early intervention [15].
An alternate approach to the early diagnosis of CRCD is through the use of serum biomarkers that can detect myocardial damage before any cardiac dysfunction is apparent on echocardiography and before any clinical symptoms are present. This diagnostic modality is becoming preferable because of its ease of administration (a simple blood draw), cost effectiveness, and most importantly, the timing of when these biomarkers are aberrant. The trend towards minimally invasive medicine has resulted in the research of individually studied biomarkers. Now, based on a review of the literature, we have compiled these studies and are suggesting a panel of biomarkers be used in combination to create an innovative way to detect CRCD in breast cancer patients who are receiving chemotherapy.
Topoisomerase 2β
There are two isozymes of topoisomerase 2 (Top2) in mammalian cells – Top2α and Top2β. Top2α is only expressed in proliferating and tumor cells, and plays an important role in cell cycle events such as DNA replication, chromosome condensation/decondensation, and sister chromatid segregation. Doxorubicin, as well as other Top2-directed anticancer drugs such as etoposide, amsacrine, and mitoxantrone, targets both isozymes. The antitumor activity of doxorubicin is due to the formation of a topoisomerase 2-doxorubicin-DNA ternary complex and the high efficacy of doxorubicin is thought to be due to highly elevated expression of Top2α in cancer cells. Top2β however is present in all cells, but it is noteworthy that it is the only Top2 isozyme expressed in the adult heart (while Top2α is not) [6].
Top2β is the most physiologically relevant target of anthracycline compounds in heart tissue. Cardiomyocyte-specific deletion of Top2β protects cardiomyocytes from doxorubicin-induced DNA double-strand breaks and transcriptome changes that are responsible for defective mitochondrial biogenesis and ROS formation [6]. Thus, Lyu et al. demonstrated that anthracycline-induced DNA breaks and cell death in a cardiomyocyte cell line depended on the presence of Top2β. Building on this, Zhang et al. genetically engineered mice to lack Top2β in cardiomyocytes and in contrast to control mice, mutant mice did not have acute or chronic cardiac injury after doxorubicin exposure. Analysis of cardiac tissue from the doxorubicin-treated wild-type mice revealed the activation of DNA-damage–response pathways by suppression of transcription factors known to be critical for regulation of mitochondrial biogenesis. These changes were not present in cardiac tissue from the mutant mice treated with doxorubicin, a finding consistent with their protection against cardiotoxicity secondary to lack of Top2β [16,17]. Furthermore, studies have detailed that the cardioprotective compound, dexrazoxane, functions (at least in part) by reducing Top2β protein levels in heart tissue. Accordingly, a subject identified to be at high risk of anthracycline-induced cardiotoxicity (e.g., a subject having elevated Top2β expression) can be treated with an agent that reduces Top2β levels, such as dexrazoxane, to reduce the risk of anthracycline-induced cardiotoxicity [6,18,19]. Kersting et al. demonstrated that Top2β, usually considered a constitutively expressed protein, varied 3-, 18-, and 16-fold on the mRNA, protein and activity levels, respectively, among individuals tested. Interestingly, Top2β activity correlated significantly with the apoptotic response in peripheral leukocytes when exposed to doxorubicin, suggesting that higher Top2β activity most likely leads to a higher production of double-stranded DNA breaks, thus increasing the probability of p53-mediated apoptosis. Furthermore, Kersting et al. suggested that variability in Top2β expression in normal tissues could affect the severity of side-effects. Thus, assessment of Top2β expression level may be used to predict whether a subject is at risk for developing cardiotoxicity in response to anthracycline-based therapeutics [20]. Unfortunately, at the time of this review, there are no published human clinical trials on the utility of Top2β as a biomarker for CRCD. But, given its mechanism, in vitro and in vivo findings, and the effect of dexrazoxane, it stands to reason, that it would be a beneficial addition to a panel of biomarkers that could effectively predict CRCD.
Troponin T & Troponin I
Troponin T (TnT) and troponin I (TnI) are thin-filament contractile proteins present in high concentrations in the myocardium. The serum concentration of these proteins increases within 3–6 h after myocardial injury and remains elevated for 7–10 days, long before any functional impairment is detected. In the evaluation of myocardial necrosis, troponins, in contrast to CK-MB, are more cardiospecific. TnT is released rapidly after myocardial injury in direct proportion to the extent of injury and persists in the serum for several days, probably as a result of ongoing release from the heart [15].
Serum TnT levels are known to increase within 4–6 h after the onset of myocardial infarction and peak at about 24 h. This increase lasts for 10–14 days. In a study of patients with hematologic malignancies receiving anthracyclines, in 34.1% of patients, the TnT levels were found to be significantly higher after completion of therapy compared with baseline levels and values after the first cycle of therapy. After chemotherapy was completed, echocardiography demonstrated there was a decrease in the E/A ratio and an increase in the IRT (isovolumetric relaxation time) (both of which are predictive of diastolic dysfunction) in those patients whose TnT (42.9% of patients <44 years old; 71.4% of patients >44 years old) levels increased during therapy [21].
In a population of patients with hematologic malignancies receiving anthracyclines, Auner et al. showed that TnT was positive in 15% of patients and was observed on day +21.5 (median, range: +6 to +35) after the initiation of therapy. TnT positivity lasted at least three days in 63% of cycles and began after a median of two anthracycline doses. Follow up echocardiography revealed that TnT+ patients had a significant decrease in LVEF compared to TnT− patients (10% vs. 2% respectively) [22].
Lipshultz et al. studied pediatric patients with acute lymphoblastic leukemia receiving doxorubicin and 46% of them had a positive TnT during the course of their treatment and 38% had at least one positive TnT after doxorubicin was complete. Most patients were TnT+ between days 60–240 of doxorubicin treatment [23], however, prior studies by Lipshultz et al. in the same type of patient population demonstrated that they were TnT+ after initial doxorubicin dosing and/or through treatment and this was significantly correlated with LVs that had thinner walls and were more dilated 9 months later (as evidenced by echocardiography) [24]. This same group also demonstrated that low-level elevations of cardiac troponin T, presumably induced by doxorubicin, are associated with histologic evidence of myocardial injury and are clinically meaningful in spontaneously hypertensive rats (SHR) [25]. Herman et al. administered doxorubicin weekly to SHR and assessed cardiomyopathy and myocardial localization of TnT and showed increases in both the serum levels of TnT and myocardial lesions. The authors found that the increase in TnT levels was associated with the cumulative doxorubicin doses, and there was a positive correlation between the increase in serum TnT levels and cardiomyopathy scores based on microscopic examination of cardiac tissue. Similarly, another study showed that TnT is a useful marker in rabbits and serum TnT levels increased pathologically after a cumulative dose of 400 mg/m2 doxorubi cin [26].
Timing of serum collection may be particularly important and require further research to determine the optimum time, as demonstrated by Kremer et al. This study showed that measuring TnT in the first 24 hours of chemotherapy administration did not have a high sensitivity for the identification of patients with subsequent subclinical cardiotoxicity as only 1 patient of 7 with any detectable cardiac dysfunction on echocardiography was TnT+ [27]. Furthermore, only one TnT collection may not be sufficient. Another point to consider is that TnT is also expressed when skeletal muscle has been injured so it may not be as specific as TnI and several studies have documented the utility of TnI.
Patients receiving HDC (high dose chemotherapy) for various malignancies had TnI tested before and immediately after receiving HDC and in the hours following administration for every cycle of treatment. Increased TnI was detected in 32% of the study population [28]. In subsequent studies by Cardinale et al., which included patients with various malignancies undergoing high-dose anthracycline chemotherapy, 33% were TnI+ starting as early as the first cycle immediately after HDC was administered up to 72 h later. In the TnI+ group, a significant reduction in LVEF was observed at the first month of follow-up and thereafter. A strong relationship was also observed between the TnI maximal value detected after HDC and the LVEF maximal reduction during follow-up. There was also a significant correlation between the number of positive TnI assays per patient and the LVEF maximal deficit [29]. Sandri et al. later confirmed that after treatment with HDC, there was a significant decrease in LVEF in TnI+ patients, after the first month of treatment and then increasingly pronounced during the following months (from −6.8% change in LVEF after 1 month to −18.2% after 12 months) [30]. In this study, they used very low TnI concentrations cutoffs (<0.1 μg/L) in order to detect very small myocardial injuries. These findings are in accordance with Missov et al. who described TnI increases during the course of anthracycline treatment in patients with hematological malignancies. It was the first study that correlated the presence of a TnI increase with the long-term development of cardiac dysfunction, even asymptomatic dysfunction [31]. Ky et al. evaluated levels of TnI+ and subsequent cardiotoxicity (based on echocardiography) and showed that for each increase in standard deviation in TnI+, there was a nearly 40% increased risk of subsequent cardiotoxicity. Those patients at the 90th percentile of change of TnI+ had the greatest predicted risk of cardiotoxicity (34.2% at 15 months), with little difference in the predicted risk between the 10th and 50th percentile (23.6% at 15 months after treatment) [32].
Combined, the studies on troponins can be summarized as follows: troponins are able to predict at least 3 months in advance, clinically significant left ventricular dysfunction [21,29]; early increases in troponin also predict the degree and severity of future left ventricular dysfunction [29,33,34]. Persistence of positive troponins one month after chemotherapy is completed, is related to 85% probability of major cardiac events within the first year of follow up [33,35,36]; persistently negative troponins can identify with a predictive negative value of 99%, patients with the lowest cardiotoxicity risk, who will most likely never encounter cardiac complications (at least for the first year after completing chemotherapy). However, troponins come with some negatives – peak values occur at different intervals after HDC, so several samples are required to detect it. Also it is not possible to define the time point at which a negative troponin value reaches 100% specificity for no further troponin release.
Myeloperoxidase
Myeloperoxidase (MPO) is an enzyme secreted by polymorphonuclear leukocytes that has atherogenic and pro-oxidant effects on cardiac tissues by causing lipid peroxidation (a marker of oxidative stress), scavenging of nitric oxide, and inhibition of nitric oxide synthase [32]. In patients with acute coronary syndromes, elevated levels of MPO are predictive of adverse outcomes. Similarly, studies in heart failure also suggest that MPO is related to increased risk of more severe disease [37]. Because oxidative stress is posited to be central to the mechanism of anthracycline cardiotoxicity [38], it is plausible that elevated MPO levels after anthracycline exposure are associated with subsequent cardiac dysfunction and recent studies have demonstrated this [39].
Ky et al.’s findings suggest that MPO can be used in combination with TnI to identify patients that are at a substantially increased risk for cardiotoxicity from combination anthracycline therapy. Those at the 10th and 50th percentile in change of MPO values between visit 2 and baseline did not have a significant difference in their predicted cardiotoxicity rate (26.5% at 15 months after treatment). However those with a change in MPO in the 90th percentile, had a predicted rate of cardiotoxicity of 36.1% at 15 months. They also identified an association between an increase in MPO at 3 months and first signs of cardiotoxicity [32]. Putt et al. followed breast cancer patients treated with doxorubicin and trastuzumab for 15 months and those with MPO levels at the 75th and 90th percentiles were associated with increases in subsequent cardiotoxicity risk with a relative risk of 1.1–1.6 [40]. These two studies, though very promising, are the only ones with an in depth look at the progression of MPO throughout the treatment course of patients, so data is limited. However, the findings of Ky and Putt et al. are strong enough to suggest that MPO would be beneficial to add to a diagnostic biomarker panel.
NT-proBNP
The B-type natriuretic peptide is synthesized in cardiac myocytes as a 134 amino acid pre-pro hormone, which is subsequently cleaved to yield a 108 amino acid pro-peptide that is stored in secretory granules within the myocyte. In response to the appropriate stimulus, pro-BNP is proteolytically cleaved into the biologically active, mature BNP and the inactive N-terminal (NT)-proBNP, which is released into the blood stream. Although BNP is produced in both atrial tissue and the right ventricular myocardium, highest concentrations of BNP have been found in the left ventricle myocardium. BNP and NT-proBNP are secreted by the heart in response to ventricular wall distention and therefore are used as markers of ventricular dysfunction [41]. NT-proBNP is raised in both symptomatic and asymptomatic patients with left ventricular dysfunction and increased blood concentrations are associated with a pathophysiological model of overload cardiomyopathy [41,42]. According to Lipshultz et al., increases in NT-proBNP detected in the first 90 days of anthracycline (doxorubicin) treatment were significantly associated with changes in the left ventricle thickness-to-dimension ratio 4 years later, while increased levels detected during the last 90 days of treatment were not significantly related to any echocardiographic outcome 4 years later [43]. Gimeno et al. showed that a NT-proBNP level above 900 pg/mL predicts a high risk of death from any cause in patients treated for non-Hodgkins lymphoma with CHOP(Cyclophosphamide, Doxorubicin, Oncovin, Prednisolone) and suggested that measurement of NT-proBNP levels should be considered for risk assessment in patients with non-Hodgkins lymphoma prior to chemotherapy [44]. Sandri et al. showed that patients with NT-proBNP levels that were persistently elevated 72 h after the end of HDC administration, and not those with transiently elevated levels, developed some form of cardiac impairment during the 12 months of observation after HDC administration [45]. De Iuliis et al. showed that NT-proBNP levels, in patients treated with anthracyclines (with or without trastuzumab), were significantly increased at 3 months, 6 months, and 1 year after treatment was completed and this was before any echocardiographic evidence of LVEF decrease became apparent. They concluded that NT-proBNP detection was able to support an early diagnosis of CRCD in the absence of LVEF decrease and is more sensitive than echocardiography for early cardiac changes [46]. A marker like this would be a valuable addition to a panel of markers in which the goal is to detect CRCD prior to there being any echocardiographic or clinical evidence of the condition.
MicroRNA
MicroRNAs (miRNA) are a class of endogenous, naturally occurring, small non-protein-coding RNA molecules which are approximately 22 nucleotides long. They are partially complementary to one or more messenger RNA molecules and their main function is to downregulate gene expression at the posttranscriptional level in both physiological and disease conditions. More than 1000 miRNAs have been identified in humans [47]. Circulating miRNAs have been detected in whole blood, peripheral blood mononuclear cells, platelets, serum, plasma, and other body fluids (saliva, urine, tears, breast milk) [48] and can be detected with high sensitivity and specificity using real-time PCR, deep sequencing, and microarray [49]. Circulating miRNAs display remarkable stability under harsh conditions, including endogenous RNase activity, multiple freeze-thaw cycles, boiling, extreme pH levels, and long-term storage at room temperature, which renders them extremely suitable and preferable for use as blood-based biomarkers [49,50]. Also, because of their tissue specific expression profiles, miRNAs are being profiled for use as biomarkers [51–54]. Over 200 miRNAs have been detected in the heart; miRNAs such as miR-1, let-7, miR-133, miR-126-3p, miR-30c, and miR-26a were found to be predominant in the cardiac muscles while miR-145, let-7, miR-125b, miR-125a, miR-23, and miR-143 are dominant in arterial smooth muscles. More importantly though, miRNAs have been implicated in various cardiovascular diseases (STEMI, NSTEMI, CHF, CAD, DCM) [47,48,51,55]. Traditional biomarkers, such as troponins detect cardiomyocyte damage, which puts them at a disadvantage to detect conditions in which there is no cardiomyocyte damage (such as type II CRCD) or other clinical syndromes such as drug induced arrhythmias, valvular disease, and contractile dysfunction and recent studies have demonstrated that miRNAs have the disease and condition specificity to make this distinction [56]. miRNA biomarker research is an extremely new field that has a significant amount of potential because of the above listed reasons. However since it is a blossoming field, human clinical studies are limited and supporting data for their inclusion in a biomarker panel, comes primarily from animal models. But given the findings in these animal models, the selected miRNAs appear to be highly promising.
miR-208b
The miR-208 family is compiled of two transcripts-miR-208a and miR-208b. miR-208a is encoded by an intron of its host gene, Myh6 (which encodes protein α-MHC) and both miR-208a and α-MHC are cardiac specific and concurrently expressed during development, suggesting that their expression levels are controlled by a common regulatory element [57]. miR-208b, which shares a high level of sequence similarity with miR-208a, is located within an intron of the gene Myh7 (which encodes β-MHC). Unlike miR-208a, expression of miR-208b does not appear to be coupled to β-MHC as deletion of miR-208b does not alter β-MHC expression [58]. miR-208a is however required for expression Myh7b and β-MHC. Activation of Myh7b by miR-208a is constitutive, whereas activation of β-MHC also requires stress signals or absence of thyroid hormone [57]. In summary, this network of miRNAs within myosin genes, regulates myosin expression, fiber type gene expression, muscle performance and the function of myosin genes thus it extends far beyond the mere expression of myosin proteins because miRNA 208 governs a feedback loop that results in impaired contractility when dysregulated. Because of this, miRNA 208b has been implicated in conditions such as acute coronary syndrome and myocardial infarction [59,60]. Vacchi-Suzzi et al. treated rats with doxorubicin for 6 weeks and achieved cardiotoxicity by week 2 (evaluated by histopathology and genomic cardiomyopathy indicators). Up-regulation of miR-208b was detected in rat hearts starting at 2 weeks of treatment and significantly increased over the 6 weeks of treatment. There was also a dose dependent relationship between doxorubicin and miR-208b expression [61]. Using an established mouse model of doxorubicin cardiotoxicity [62], Desai et al. showed a 2.3-fold increase in miR-208b in mice hearts when given a cumulative dose of 18 mg/kg doxorubicin and 8.2-fold increase after exposure to 24 mg/kg cumulative dose of doxorubicin [63]. In their mouse model, cardiotoxicity was evident based on histopathological findings with a cumulative dose of 24 mg/kg and higher. Therefore, miR-208b was increased prior to and at the time when pathological findings of cardiotoxicity were present. It must be noted, that miR-208a does not have the same findings in the literature. It has not been shown to have an association with doxorubicin association cardiotoxicity, which further highlights their specific roles in gene regulation [52,64].
miR-34a
miR-34a is one of three members of the miR-34 family, which is encoded by its own transcript, whereas miR-34b and miR-34c share a common primary transcript. miR-34a is a direct transcriptional target of p53 because the promoter region of miR-34a contains an established p53 binding site. Its downstream effects are on apoptosis, inhibition of proliferation, cell cycle arrest, inhibition on migration, inhibition on invasion, and inhibition of differentiation – all oncogenic processes [65]. Increased plasma levels of miR-34a in patients after acute myocardial infarction has been suggested as a predictive marker of future heart failure [66]. Desai et al. (using the established mouse model of doxorubicin cardiotoxicity discussed above) found there was upregulation of miR-34a before doxorubicin-induced cardiac tissue injury occurred [63]. miR-34a was the only miRNA in this study that was significantly up-regulated in the mouse heart at all cumulative doxorubicin doses. However, histologically visual cardiac lesions were only noted at a cumulative dose of 24 mg/kg, suggesting that the upregulation of myocardial miR-34a is indicative of early stages of myocardial injury, prior to the onset of overt cardiotoxicity.
miR-150
miR-150 plays an essential regulatory role in normal hematopoiesis, as it is mainly expressed in B-cells, T-cells and natural killer cells [67]. The exact role miR-150 plays in the heart however, is still poorly understood. miR-150 is thought to protect the heart against ischemic stress by decreasing cell death in response to injury, in part, through its repression of egr2 and p2×7r (apoptotic genes) [68]. A recent study in a mouse model of myocardial infarction reported that overexpression of miR-150 protects the heart from ischemic injury by inhibiting inflammatory monocyte migration [69]. According to the miRNA-target databases, miR-150 is predicted to regulate the expression of 3 genes associated with left ventricle remodeling: ADRβ1 (β1 adrenergic receptor) [70], C-reactive protein [71], and tumor necrosis factor receptor–associated factor 2 [50]. Previously, miR-150 has been shown to be downregulated in patients with acute myocardial infarction, atrial fibrillation, and dilated and ischemic cardiomyopathy [72,73] as well as in various mouse models of heart failure, myocardial infarction, and cardiac hypertrophy [72,74–77]. It appears that low levels of miR-150 are associated with hypertrophy and left ventricle rupture after acute myocardial infarction. Given that inflammation is a critical component of the development of left ventricle remodeling after myocardial infarction and miR-150 is expressed by monocytes, miR-150 may inhibit cardiac structural and functional remodeling during and following ischemic injury [73]. Similarly, doxorubicin treatment in mouse models has shown comparable trends with regard to miR-150 expression. Desai et al. showed that there was downregulation of miR-150 even before doxorubicin-induced cardiac tissue injury occurred. At a cumulative dose of 12 mg/kg doxorubicin, miR-150 was significantly down-regulated, with a continued decline in expression at higher cumulative doses (i.e. there was a dose dependent decline) [56]. miR-150 is another promising miRNA which has demonstrated changes in expression prior to overt cardiotoxicity being present, which may aid in early detection of CRCD. miRNAs are advantageous over the traditional biomarkers for several reasons - they are released early after damage is done and are stable in circulation, are detected with high specificity and sensitivity, and offer a less invasive alternative to perform population-wide screening. However, the major disadvantage of using miRNAs as biomarkers is their arduous isolation and detection procedures. Compared to the ELISA-based detection of other markers like troponins, the PCR-based methods for detection of circulating miRNAs are highly time consuming and costly. In addition, the current technology employed to quantify miRNA requires optimization and needs to be uniform between laboratories [77]. While miRNAs may not be ideal from a logistics and cost perspective at this time, their biomarker applications will only grow as research and interest in the field expands.
Discussion
Generally, detecting CRCD only occurs with periodic echocardiography, at which point there is already a functional deficit, whether or not it is clinically apparent. There are however, some biomarkers which have been shown to be aberrant, prior to any detectable functional or clinical deficit in cardiac function. These include topoisomerase 2 β, myeloperoxidase, troponin T/I, and NT-proBNP [15,29,32,40,78–80] (Figure 1 and Table 2). The use of miRNA in CRCD detection is relatively unexplored, therefore this review presents a unique and exciting opportunity to create an extremely innovative panel to study in clinical trials and potentially use in clinical practice. Based on an extensive literature review we suggest three microRNAs that would be of benefit in CRCD detection based on findings from in vivo studies: miR-208b, miR-34a, and miR-150 (Figure 1 and Table 2). Given how new miRNA research is, there were no human clinical trials which studied miRNAs as biomarkers to predict CRCD. However animal models have been used and have been highlighted (Table 2). These serum markers and/or microRNAs, examined individually may not be sufficient to predict CRCD reliably, however taken together, would be highly sensitive and specific to detect it. They would be extremely beneficial considering they are minimally invasive and cost effective (when compared to serial echocardiography), something which is particularly relevant in rural areas where health care access is a concern. Furthermore, many of these biomarkers demonstrate dose dependent relationships with CRCD and are effective at predicting CRCD before there is any evidence of overt cardiotoxicity.
Figure 1.
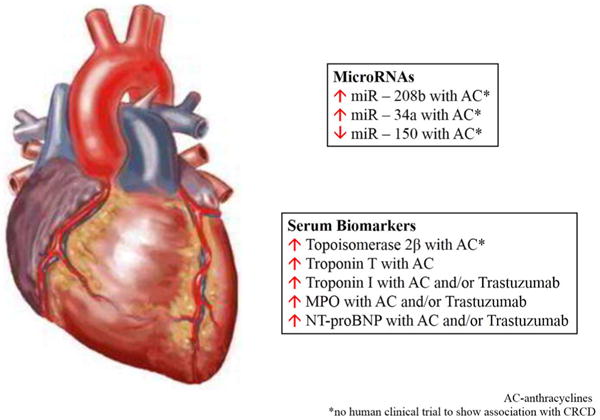
In vivo studies: miR-208b, miR-34a, and miR-150.
Table 2.
A biomaker panel to detect CRCD - a summary of the literature.
| Summary of Literature | |||
|---|---|---|---|
| Biomarker | Mechanism | Main Findings | Ref |
| Top2β | dsDNA breaks lead to cardiomyocyte death, increases ROS, causes mitochondrial dysfunction | -In vitro studies demonstrate that cardiomyocyte specific deletion of Top2β protects cells from doxorubicin-induced DNA double-strand breaks. | [6,16–20] |
| - In vivo studies of genetically engineered mice with cardiomyocyte Top2β deletion are resistant to doxorubicin induced cardiotoxicity. | |||
| -Dexrazoxane functions by reducing Top2β protein levels in heart tissue and has been shown to be cardioprotective in breast cancer patients receiving doxorubicin. | |||
| -The expression level of Top2β is intra- and inter-individually variable and may determine the apoptotic response to doxorubicin and other anthracyclines. | |||
| TnT | Released after cardiomyocyte damage starting 3-6 hours after injury | -Levels can be elevated anywhere from immediately after the first chemotherapy dose to 240 days after therapy is completed, while echocardiographic evidence of CRCD presents when therapy is completed to 9 months after it is completed. | [21–24] |
| -Levels during first 90 days after anthracyclines can predict CRCD at 4 year follow up. | |||
| TnI | Released after cardiomyocyte damage starting 3–6 h after injury | -In patients with TnI >0.5ng/mL, 33%, 27%, and 25% of increases occur right after, 12 hours, and 24 hours after anthracycline dose and predicts LVEF decrease 1 month later. | [28–30, 32, 33, 43,81–83] |
| -anthracycline treated patients with TnI >0.5ng/mL have a significant reduction in LVEF for 3–7 months compared to those with TnI <0.5ng/mL who show a transient decrease in LVEF at 3 months, followed by a complete recovery at 7 months. | |||
| -TnI >0.08ng/mL persisting 1 month after anthracycline treatment is completed is associated with 84% risk of CRCD compared to 37% when elevation is transient. | |||
| -When anthracyclines and trastuzumab are combined, TnI elevation early after anthracyclines and at 3 months is an independent predictor of cardiotoxicity with 17.6 times increased risk. | |||
| - Absolute value of TnI may not be as valuable as the change in value from baseline to subsequent visits. Patients at the 90th percentile of change in TnI+ had the greatest predicted risk of cardiotoxicity- 34.2% at 15 months after anthracycline treatment. | |||
| MPO | Secreted by PMN leukocytes, prooxidant | -Increase from baseline to 3 months (HR 1.34) is associated with increased risk of CRCD following anthracycline, taxane, and trastuzumab treatment. | [32,40] |
| -Increases in MPO beyond 3 months remain a predictor of cardiotoxicity risk over the duration of doxorubicin/trastuzumab. | |||
| -A change in MPO in the 90th percentile, has a predicted rate of cardiotoxicity of 36.1% at 15 months after anthracycline treatment. | |||
| NT-proBNP | Released in response to elevation in LV filling pressure and wall stress | - NT-proBNP levels during the first 90 days after anthracycline therapy predict cardiotoxicity at 4 years of follow up | [43–46,84] |
| -Patients with elevated NT-proBNP have higher risks of cardiac toxicity, HF progression, and death. | |||
| -Persistently elevated NT-proBNP at 72 h (after anthracycline administration) is associated with systolic/diastolic dysfunction at 12 months follow up. | |||
| - NT-proBNP detection is able to support an early diagnosis of CRCD in the absence of LVEF decrease and is more sensitive than echocardiography for early cardiac changes. | |||
| miR-208b | Regulates cardiac myocyte MHC expression, fiber type gene expression, muscle performance | -Rats and mice treated with doxorubicin show an increase in miR-208b in a dose dependent fashion starting when and sometimes before signs of cardiotoxicity are present. | [61–63] |
| miR-34a | Cell cycle arrest, proapoptotic | - In mouse models, increased levels of miR-34a are detected before doxorubicin-induced cardiac injury occurs; upregulation of miR-34a occurs at all cumulative doses of doxorubicin. | [63] |
| miR-150 | Inhibits inflammatory monocyte migration to the heart, represses apoptotic genes, regulates LV remodeling | - In mouse models, decreased levels of miR-150 are detected before doxorubicin-induced cardiac injury occurs. | [63] |
It is evident from reviewing the literature that there is a paucity of data on biomarker panels for the detection of CRCD, especially when it concerns type II (trastuzumab related) CRCD [77–80] and there are currently no published human clinical trials exploring the utility of microRNAs in this regard. There needs to be greater emphasis on research into biomarker panels for detecting CRCD as this information is particularly salient to physicians and patients in areas with limited healthcare access. It is especially relevant to patients in rural, lower socioeconomic communities, who may not be have access to serial echocardiography as a means to diagnose CRCD. A biomarker panel may in fact be better than serial echocardiography, because the information gathered from a biomarker panel allows oncologists to adjust chemotherapeutic regimens and/or allows cardiologists to start heart failure medications earlier in their treatment regimen. In other words, appropriate intervention can be taken before any cardiac damage has occurred. Overall, an effective biomarker panel, has the potential to improve morbidity and mortality for many patients on chemotherapy.
In the future, hopefully multiple institutions will study the effectiveness of this particular biomarker panel in clinical trials of their own. Collaborative efforts amongst institutions will demonstrate which combination of markers will be most sensitive and specific for CRCD.
Conclusion
Anthracyclines and trastuzumab, though they have revolutionized contemporary oncology and breast cancer treatment, are associated with an increased risk of short and long-term cardiac events. There are no longitudinal studies to say that there is ever complete resolution of chemotherapy induced cardiac dysfunction. Furthermore, there is no standardized, easily implemented, cost-effective method of detecting and managing CRCD. This is particularly problematic in areas with limited health care available. In order for this to occur, there needs to be stronger collaboration between cardiologists and oncologists to improve the care of oncology patients receiving cardiotoxic therapy. Compared to the current standards for diagnosing CRCD, the biomarker panel we have suggested would be cost effective and easy to implement, but more importantly, would aid in earlier diagnosis, risk assessment, and CRCD progression monitoring that would ultimately improve patient care and outcomes.
Acknowledgments
This work was supported by National Institutes of Health Grants HL109015, HL071556, and HL105649 (to JIS).
Abbreviations
- ADRβ1
β1 Adrenergic Receptor
- BNP
Brain Natriuretic Peptide
- CAD
Coronary Artery Disease
- CHF
Congestive Heart Failure
- CKMB
Creatine Kinase Myocardial b Fraction
- CRCD
Chemotherapy Related Cardiac Dysfunction
- DCM
Diabetic Cardiomyopathy
- EF
Ejection Fraction
- egr2
Early Growth Response 2 Gene
- HDC
High Dose Chemotherapy
- HF
Heart Failure
- IRT
Isovolumetric Relaxation Time
- LVEF
Left Ventricle Ejection Fraction
- MHC
Myosin Heavy Chain
- miR
MicroRNAMPO: Myeloperoxidase
- Myh6/7
Myosin Heavy Chain 6/7 Gene
- NSTEMI
Non ST Segment Elevation Myocardial Infarction
- p2×7r
purinoceptor 7 gene
- ROS
Reactive Oxygen Species
- SHR
Spontaneous Hypertensive Rats
- STEMI
ST Segment Elevation Myocardial Infarction
- TnI/TnT
Troponin I/T
- Top2
Topoisomerase 2
Footnotes
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contributions
K Srikanthan – Wrote and edited the manuscript
RK-Wrote the manuscript
MT-Edited the manuscript
ET-Edited the manuscript
HV-Wrote the manuscript
NP-Edited the manuscript
JIS-Edited the manuscript
K Sodhi-Edited the manuscript
References
- 1.Florescu M, Cinteza M, Vinereanu D. Chemotherapy-induced Cardiotoxicity. Maedica (Buchar) 2013;8:59–67. [PMC free article] [PubMed] [Google Scholar]
- 2.Ewer MS, Lippman SM. Type II chemotherapy-related cardiac dysfunction: time to recognize a new entity. J Clin Oncol. 2005;13:2900–2902. doi: 10.1200/JCO.2005.05.827. [DOI] [PubMed] [Google Scholar]
- 3.Bovelli D, Plataniotis G, Roila F, ESMO Guidelines Working Group Cardiotoxicity of chemotherapeutic agents and radiotherapy-related heart disease: ESMO Clinical Practice Guidelines. Ann Oncol. 2010;21(Suppl 5):277–282. doi: 10.1093/annonc/mdq200. [DOI] [PubMed] [Google Scholar]
- 4.Christenson ES, James T, Agrawal V, Park BH. Use of biomarkers for the assessment of chemotherapy-induced cardiac toxicity. Clin Biochem. 2015;48:223–235. doi: 10.1016/j.clinbiochem.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seidman A, Hudis C, Pierri MK, Shak S, Paton V, et al. Cardiac dysfunction in the trastuzumab clinical trials experience. J Clin Oncol. 2002;5:1215–1221. doi: 10.1200/JCO.2002.20.5.1215. [DOI] [PubMed] [Google Scholar]
- 6.Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, et al. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007;18:8839–8846. doi: 10.1158/0008-5472.CAN-07-1649. [DOI] [PubMed] [Google Scholar]
- 7.Myers C, Bonow R, Palmeri S, Jenkins J, Corden B, et al. A randomized controlled trial assessing the prevention of doxorubicin cardiomyopathy by N-acetylcysteine. Semin Oncol. 1983;10:53–55. [PubMed] [Google Scholar]
- 8.Martin E, Thougaard AV, Grauslund M, Jensen PB, Bjorkling F, et al. Evaluation of the topoisomerase II-inactive bisdioxopiperazine ICRF-161 as a protectant against doxorubicin-induced cardiomyopathy. Toxicology. 2009;255:72–79. doi: 10.1016/j.tox.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 9.Ritchie JL, Bateman TM, Bonow RO, Crawford MH, Gibbons RJ, et al. Guidelines for clinical use of cardiac radionuclide imaging: A report of the American College of Cardiology/American Heart Association Task Force on assessment of diagnostic and therapeutic cardiovascular procedures (Committee on Radionuclide Imaging).–developed in collaboration with the American Society of Nuclear Cardiology. J Nucl Cardiol. 1995;2:172–192. doi: 10.1016/s1071-3581(06)80030-9. [DOI] [PubMed] [Google Scholar]
- 10.Lipshultz SE, Sanders SP, Goorin AM, Krischer JP, Sallan SE, et al. Monitoring for anthracycline cardiotoxicity. Pediatrics. 1994;93:433–437. [PubMed] [Google Scholar]
- 11.Steinherz LJ, Graham T, Hurwitz R, Sondheimer HM, Schwartz RG, et al. Guidelines for cardiac monitoring of children during and after anthracycline therapy: report of the Cardiology Committee of the Childrens Cancer Study Group. Pediatrics. 1992;89:942–949. [PubMed] [Google Scholar]
- 12.Schwartz RG, McKenzie WB, Alexander J, Sager P, D‘Souza A, et al. Congestive heart failure and left ventricular dysfunction complicating doxorubicin therapy. Seven-year experience using serial radionuclide angiocardiography. Am J Med. 1987;82:1109–1118. doi: 10.1016/0002-9343(87)90212-9. [DOI] [PubMed] [Google Scholar]
- 13.Mitani I, Jain D, Joska TM, Burtness B, Zaret BL. Doxorubicin cardiotoxicity: prevention of congestive heart failure with serial cardiac function monitoring with equilibrium radionuclide angiocardiography in the current era. J Nucl Cardiol. 2003;10:132–139. doi: 10.1067/mnc.2003.7. [DOI] [PubMed] [Google Scholar]
- 14.Sabel MS, Levine EG, Hurd T, Schwartz GN, Zielinski R, et al. Is MUGA scan necessary in patients with low-risk breast cancer before doxorubicin-based adjuvant therapy? Multiple gated acquisition. Am J Clin Oncol. 2001;4:425–428. doi: 10.1097/00000421-200108000-00027. [DOI] [PubMed] [Google Scholar]
- 15.Dolci A, Dominici R, Cardinale D, Sandri MT, Panteghini M. Biochemical markers for prediction of chemotherapy-induced cardiotoxicity: systematic review of the literature and recommendations for use. Am J Clin Pathol. 2008;130:688–695. doi: 10.1309/AJCPB66LRIIVMQDR. [DOI] [PubMed] [Google Scholar]
- 16.Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 17.Vejpongsa P, Yeh ET. Topoisomerase 2beta: a promising molecular target for primary prevention of anthracycline-induced cardiotoxicity. Clin Pharmacol Ther. 2014;95:45–52. doi: 10.1038/clpt.2013.201. [DOI] [PubMed] [Google Scholar]
- 18.Swain SM, Whaley FS, Gerber MC, Ewer MS, Bianchine JR, et al. Delayed administration of dexrazoxane provides cardioprotection for patients with advanced breast cancer treated with doxorubicin-containing therapy. J Clin Oncol. 1997;15:1333–1340. doi: 10.1200/JCO.1997.15.4.1333. [DOI] [PubMed] [Google Scholar]
- 19.Swain SM, Whaley FS, Gerber MC, Weisberg S, York M, et al. Cardioprotection with dexrazoxane for doxorubicin-containing therapy in advanced breast cancer. J Clin Oncol. 1997;15:1318–1332. doi: 10.1200/JCO.1997.15.4.1318. [DOI] [PubMed] [Google Scholar]
- 20.Kersting G, Tzvetkov MV, Huse K, Kulle B, Hafner V, et al. Topoisomerase II beta expression level correlates with doxorubicin-induced apoptosis in peripheral blood cells. Naunyn Schmiedebergs Arch Pharmacol. 2006;374:21–30. doi: 10.1007/s00210-006-0091-0. [DOI] [PubMed] [Google Scholar]
- 21.Kilickap S, Barista I, Akgul E, Aytemir K, Aksoyek S, et al. cTnT can be a useful marker for early detection of anthracycline cardiotoxicity. Ann Oncol. 2005;16:798–804. doi: 10.1093/annonc/mdi152. [DOI] [PubMed] [Google Scholar]
- 22.Auner HW, Tinchon C, Linkesch W, Tiran A, Quehenberger F, et al. Prolonged monitoring of troponin T for the detection of anthracycline cardiotoxicity in adults with hematological malignancies. Ann Hematol. 2003;82:218–222. doi: 10.1007/s00277-003-0615-3. [DOI] [PubMed] [Google Scholar]
- 23.Lipshultz SE, Rifai N, Dalton VM, Levy DE, Silverman LB, et al. The effect of dexrazoxane on myocardial injury in doxorubicin-treated children with acute lymphoblastic leukemia. N Engl J Med. 2004;351:145–153. doi: 10.1056/NEJMoa035153. [DOI] [PubMed] [Google Scholar]
- 24.Lipshultz SE, Rifai N, Sallan SE, Lipsitz SR, Dalton V, et al. Predictive value of cardiac troponin T in pediatric patients at risk for myocardial injury. Circulation. 1997;96:2641–2648. doi: 10.1161/01.cir.96.8.2641. [DOI] [PubMed] [Google Scholar]
- 25.Herman EH, Zhang J, Lipshultz SE, Rifai N, Chadwick D, et al. Correlation between serum levels of cardiac troponin-T and the severity of the chronic cardiomyopathy induced by doxorubicin. J Clin Oncol. 1999;17:2237–2243. doi: 10.1200/JCO.1999.17.7.2237. [DOI] [PubMed] [Google Scholar]
- 26.Adamcová M, Gersl V, Hrdina R, Melka M, Mazurová Y, et al. Cardiac troponin T as a marker of myocardial damage caused by antineoplastic drugs in rabbits. J Cancer Res Clin Oncol. 1999;125:268–274. doi: 10.1007/s004320050273. [DOI] [PubMed] [Google Scholar]
- 27.Kremer LC, Bastiaansen BA, Offringa M, Lam J, van Straalen JP, et al. Troponin T in the first 24 hours after the administration of chemotherapy and the detection of myocardial damage in children. Eur J Cancer. 2002;38:686–689. doi: 10.1016/s0959-8049(01)00431-2. [DOI] [PubMed] [Google Scholar]
- 28.Cardinale D, Sandri MT, Martinoni A, Tricca A, Civelli M, et al. Left ventricular dysfunction predicted by early troponin I release after high-dose chemotherapy. J Am Coll Cardiol. 2000;36:517–522. doi: 10.1016/s0735-1097(00)00748-8. [DOI] [PubMed] [Google Scholar]
- 29.Cardinale D, Sandri MT, Martinoni A, Borghini E, Civelli M, et al. Myocardial injury revealed by plasma troponin I in breast cancer treated with high-dose chemotherapy. Ann Oncol. 2002;13:710–715. doi: 10.1093/annonc/mdf170. [DOI] [PubMed] [Google Scholar]
- 30.Sandri MT, Cardinale D, Zorzino L, Passerini R, Lentati P, et al. Minor increases in plasma troponin I predict decreased left ventricular ejection fraction after high-dose chemotherapy. Clin Chem. 2003;49:248–252. doi: 10.1373/49.2.248. [DOI] [PubMed] [Google Scholar]
- 31.Missov E, Calzolari C, Davy JM, Leclercq F, Rossi M, et al. Cardiac troponin I in patients with hematologic malignancies. Coron Artery Dis. 1997;8:537–541. [PubMed] [Google Scholar]
- 32.Ky B, Putt M, Sawaya H, French B, Januzzi JL, Jr, et al. Early increases in multiple biomarkers predict subsequent cardiotoxicity in patients with breast cancer treated with doxorubicin, taxanes, and trastuzumab. J Am Coll Cardiol. 2014;63:809–816. doi: 10.1016/j.jacc.2013.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cardinale D, Sandri MT, Colombo A, Colombo N, Boeri M, et al. Prognostic value of troponin I in cardiac risk stratification of cancer patients undergoing high-dose chemotherapy. Circulation. 2004;109:2749–2754. doi: 10.1161/01.CIR.0000130926.51766.CC. [DOI] [PubMed] [Google Scholar]
- 34.La Vecchia L, Mezzena G, Zanolla L, Paccanaro M, Varotto L, et al. Cardiac troponin I as diagnostic and prognostic marker in severe heart failure. J Heart Lung Transplant. 2000;19:644–652. doi: 10.1016/s1053-2498(00)00120-0. [DOI] [PubMed] [Google Scholar]
- 35.Cardinale D, Colombo A, Sandri MT, Lamantia G, Colombo N, et al. Prevention of high-dose chemotherapy-induced cardiotoxicity in high-risk patients by angiotensin-converting enzyme inhibition. Circulation. 2006;114:2474–2481. doi: 10.1161/CIRCULATIONAHA.106.635144. [DOI] [PubMed] [Google Scholar]
- 36.Cardinale D, Lamantia G, Cipolla CM. Troponin I and cardiovascular risk stratification in patients with testicular cancer. J Clin Oncol. 2006;24:3508. doi: 10.1200/JCO.2006.06.7876. [DOI] [PubMed] [Google Scholar]
- 37.Baldus S, Heeschen C, Meinertz T, Zeiher AM, Eiserich JP, et al. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108:1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- 38.Tang WH, Tong W, Troughton RW, Martin MG, Shrestha K, et al. Prognostic value and echocardiographic determinants of plasma myeloperoxidase levels in chronic heart failure. J Am Coll Cardiol. 2007;49:2364–2370. doi: 10.1016/j.jacc.2007.02.053. [DOI] [PubMed] [Google Scholar]
- 39.Lipshultz SE, Cohen H, Colan SD, Herman EH. The relevance of information generated by in vitro experimental models to clinical doxorubicin cardiotoxicity. Leuk Lymphoma. 2006;47:1454–1458. doi: 10.1080/10428190600800231. [DOI] [PubMed] [Google Scholar]
- 40.Putt M, Hahn VS, Januzzi JL, Sawaya H, Sebag IA, et al. Longitudinal Changes in Multiple Biomarkers Are Associated with Cardiotoxicity in Breast Cancer Patients Treated with Doxorubicin, Taxanes, and Trastuzumab. Clin Chem. 2015;61:1164–1172. doi: 10.1373/clinchem.2015.241232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bay M, Kirk V, Parner J, Hassager C, Nielsen H, et al. NT-proBNP: a new diagnostic screening tool to differentiate between patients with normal and reduced left ventricular systolic function. Heart. 2003;89:150–154. doi: 10.1136/heart.89.2.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sparano JA, Brown DL, Wolff AC. Predicting cancer therapy-induced cardiotoxicity: the role of troponins and other markers. Drug Saf. 2002;25:301–311. doi: 10.2165/00002018-200225050-00001. [DOI] [PubMed] [Google Scholar]
- 43.Lipshultz SE, Miller TL, Scully RE, Lipsitz SR, Rifai N, et al. Changes in cardiac biomarkers during doxorubicin treatment of pediatric patients with high-risk acute lymphoblastic leukemia: associations with long-term echocardiographic outcomes. J Clin Oncol. 2012;30:1042–1049. doi: 10.1200/JCO.2010.30.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gimeno E, Gómez M, González JR, Comín J, Alvarez-Larrán A, et al. NT-proBNP: a cardiac biomarker to assess prognosis in non-Hodgkin lymphoma. Leuk Res. 2011;35:715–720. doi: 10.1016/j.leukres.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 45.Sandri MT, Salvatici M, Cardinale D, Zorzino L, Passerini R, et al. N-terminal pro-B-type natriuretic peptide after high-dose chemotherapy: a marker predictive of cardiac dysfunction? Clin Chem. 2005;51:1405–1410. doi: 10.1373/clinchem.2005.050153. [DOI] [PubMed] [Google Scholar]
- 46.De Iuliis F, Salerno G, Taglieri L, De Biase L, Lanza R, et al. Serum biomarkers evaluation to predict chemotherapy-induced cardiotoxicity in breast cancer patients. Tumour Biol. 2016;37:3379–3387. doi: 10.1007/s13277-015-4183-7. [DOI] [PubMed] [Google Scholar]
- 47.Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011;469:336–342. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bronze-da-Rocha E. MicroRNAs expression profiles in cardiovascular diseases. Biomed Res Int. 2014;2014:985408. doi: 10.1155/2014/985408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weber JA, Baxter DH, Zhang S, Huang DY, Huang KH, et al. The microRNA spectrum in 12 body fluids. Clin Chem. 2010;56:1733–1741. doi: 10.1373/clinchem.2010.147405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Donners MM, Beckers L, Lievens D, Munnix I, Heemskerk J, et al. The CD40-TRAF6 axis is the key regulator of the CD40/CD40L system in neointima formation and arterial remodeling. Blood. 2008;111:4596–4604. doi: 10.1182/blood-2007-05-088906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McManus DD, Ambros V. Circulating MicroRNAs in cardiovascular disease. Circulation. 2011;124:1908–1910. doi: 10.1161/CIRCULATIONAHA.111.062117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nishimura Y, Kondo C, Morikawa Y, Tonomura Y, Torii M, et al. Plasma miR-208 as a useful biomarker for drug-induced cardiotoxicity in rats. J Appl Toxicol. 2015;35:173–180. doi: 10.1002/jat.3044. [DOI] [PubMed] [Google Scholar]
- 53.Corsten MF, Dennert R, Jochems S, Kuznetsova T, Devaux Y, et al. Circulating MicroRNA-208b and MicroRNA-499 reflect myocardial damage in cardiovascular disease. Circ Cardiovasc Genet. 2010;3:499–506. doi: 10.1161/CIRCGENETICS.110.957415. [DOI] [PubMed] [Google Scholar]
- 54.Sandhu H, Maddock H. Molecular basis of cancer-therapy-induced cardiotoxicity: introducing microRNA biomarkers for early assessment of subclinical myocardial injury. Clin Sci (Lond) 2014;6:377–400. doi: 10.1042/CS20120620. [DOI] [PubMed] [Google Scholar]
- 55.Kondkar AA, Abu-Amero KK. Utility of circulating microRNAs as clinical biomarkers for cardiovascular diseases. Biomed Res Int. 2015;2015:821823. doi: 10.1155/2015/821823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oury C, Servais L, Bouznad N, Hego A, Nchimi A, et al. MicroRNAs in Valvular Heart Diseases: Potential Role as Markers and Actors of Valvular and Cardiac Remodeling. Int J Mol Sci. 2016;17 doi: 10.3390/ijms17071120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Rooij E, Olson EN. MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest. 2007;117:2369–2376. doi: 10.1172/JCI33099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malizia AP, Wang DZ. MicroRNAs in cardiomyocyte development. Wiley Interdiscip Rev Syst Biol Med. 2011;3:183–190. doi: 10.1002/wsbm.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Romaine SP, Tomaszewski M, Condorelli G, Samani NJ. MicroRNAs in cardiovascular disease: an introduction for clinicians. Heart. 2015;101:921–928. doi: 10.1136/heartjnl-2013-305402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schulte C, Zeller T. MicroRNA-based diagnostics and therapy in cardiovascular disease-Summing up the facts. Cardiovasc Diagn Ther. 2015;5:17–36. doi: 10.3978/j.issn.2223-3652.2014.12.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vacchi-Suzzi C, Bauer Y, Berridge BR, Bongiovanni S, Gerrish K, et al. Perturbation of microRNAs in rat heart during chronic doxorubicin treatment. PLoS One. 2012;7:e40395. doi: 10.1371/journal.pone.0040395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Desai VG, Herman EH, Moland CL, Branham WS, Lewis SM, et al. Development of doxorubicin-induced chronic cardiotoxicity in the B6C3F1 mouse model. Toxicol Appl Pharmacol. 2013;266:109–121. doi: 10.1016/j.taap.2012.10.025. [DOI] [PubMed] [Google Scholar]
- 63.Desai VG, C Kwekel J, Vijay V, Moland CL, Herman EH, et al. Early biomarkers of doxorubicin-induced heart injury in a mouse model. Toxicol Appl Pharmacol. 2014;281:221–229. doi: 10.1016/j.taap.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 64.Oliveira-Carvalho V, Ferreira LR, Bocchi EA. Circulating mir-208a fails as a biomarker of doxorubicin-induced cardiotoxicity in breast cancer patients. J Appl Toxicol. 2015;35:1071–1072. doi: 10.1002/jat.3185. [DOI] [PubMed] [Google Scholar]
- 65.Misso G, Di Martino MT, De Rosa G, Farooqi AA, Lombardi A, et al. Mir-34: a new weapon against cancer? Mol Ther Nucleic Acids. 2014;3:e194. doi: 10.1038/mtna.2014.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matsumoto S, Sakata Y, Suna S, Nakatani D, Usami M, et al. Circulating p53-responsive microRNAs are predictive indicators of heart failure after acute myocardial infarction. Circ Res. 2013;113:322–326. doi: 10.1161/CIRCRESAHA.113.301209. [DOI] [PubMed] [Google Scholar]
- 67.Wang F, Ren X, Zhang X. Role of microRNA-150 in solid tumors. Oncol Lett. 2015;10:11–16. doi: 10.3892/ol.2015.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang Y, Wang Y, Park KM, Hu Q, Teoh JP, et al. MicroRNA-150 protects the mouse heart from ischaemic injury by regulating cell death. Cardiovasc Res. 2015;106:387–397. doi: 10.1093/cvr/cvv121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu Z, Ye P, Wang S, Wu J, Sun Y, et al. MicroRNA-150 protects the heart from injury by inhibiting monocyte accumulation in a mouse model of acute myocardial infarction. Circ Cardiovasc Genet. 2015;8:11–20. doi: 10.1161/CIRCGENETICS.114.000598. [DOI] [PubMed] [Google Scholar]
- 70.Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, et al. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307:205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- 71.Ørn S, Manhenke C, Ueland T, Damås JK, Mollnes TE, et al. C-reactive protein, infarct size, microvascular obstruction, and left-ventricular remodelling following acute myocardial infarction. Eur Heart J. 2009;30:1080–1106. doi: 10.1093/eurheartj/ehp070. [DOI] [PubMed] [Google Scholar]
- 72.Topkara VK, Mann DL. Role of microRNAs in cardiac remodeling and heart failure. Cardiovasc Drugs Ther. 2011;25:171–182. doi: 10.1007/s10557-011-6289-5. [DOI] [PubMed] [Google Scholar]
- 73.Devaux Y, Vausort M, McCann GP, Zangrando J, Kelly D, et al. MicroRNA-150: a novel marker of left ventricular remodeling after acute myocardial infarction. Circ Cardiovasc Genet. 2013;6:290–298. doi: 10.1161/CIRCGENETICS.113.000077. [DOI] [PubMed] [Google Scholar]
- 74.Duan Y, Zhou B, Su H, Liu Y, Du C. miR-150 regulates high glucose-induced cardiomyocyte hypertrophy by targeting the transcriptional co-activator p300. Exp Cell Res. 2013;319:173–184. doi: 10.1016/j.yexcr.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 75.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA. 2006;103:18255–18260. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Divakaran V, Mann DL. The emerging role of microRNAs in cardiac remodeling and heart failure. Circ Res. 2008;103:1072–1083. doi: 10.1161/CIRCRESAHA.108.183087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. 2007;100:416–424. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 78.Hahn VS, Lenihan DJ, Ky B. Cancer therapy-induced cardiotoxicity: basic mechanisms and potential cardioprotective therapies. J Am Heart Assoc. 2014;3:e000665. doi: 10.1161/JAHA.113.000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stevens PL, Lenihan DJ. Cardiotoxicity due to Chemotherapy: the Role of Biomarkers. Curr Cardiol Rep. 2015;17:603. doi: 10.1007/s11886-015-0603-y. [DOI] [PubMed] [Google Scholar]
- 80.Meinardi MT, van Veldhuisen DJ, Gietema JA, Dolsma WV, Boomsma F, et al. Prospective evaluation of early cardiac damage induced by epirubicin-containing adjuvant chemotherapy and locoregional radiotherapy in breast cancer patients. J Clin Oncol. 2001;19:2746–2753. doi: 10.1200/JCO.2001.19.10.2746. [DOI] [PubMed] [Google Scholar]
- 81.Cardinale D, Colombo A, Torrisi R, Sandri MT, Civelli M, et al. Trastuzumab-induced cardiotoxicity: clinical and prognostic implications of troponin I evaluation. J Clin Oncol. 2010;28:3910–3916. doi: 10.1200/JCO.2009.27.3615. [DOI] [PubMed] [Google Scholar]
- 82.Sawaya H, Sebag IA, Plana JC, Januzzi JL, Ky B, et al. Early detection and prediction of cardiotoxicity in chemotherapy-treated patients. Am J Cardiol. 2011;107:1375–1380. doi: 10.1016/j.amjcard.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sawaya H, Sebag IA, Plana JC, Januzzi JL, Ky B, et al. Assessment of echocardiography and biomarkers for the extended prediction of cardiotoxicity in patients treated with anthracyclines, taxanes, and trastuzumab. Circ Cardiovasc Imaging. 2012;5:596–603. doi: 10.1161/CIRCIMAGING.112.973321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sherief LM, Kamal AG, Khalek EA, Kamal NM, Soliman AA, et al. Biomarkers and early detection of late onset anthracycline-induced cardiotoxicity in children. Hematology. 2012;17:151–156. doi: 10.1179/102453312X13376952196412. [DOI] [PubMed] [Google Scholar]