

Abstract
Purpose
To characterize the toxicity, pharmacokinetics, and pharmacodynamics of selinexor, a selective inhibitor of nuclear export, when combined with fludarabine and cytarabine, in children with relapsed or refractory leukemia.
Patients and Methods
Eighteen patients with relapsed or refractory acute leukemia were enrolled in the SELHEM (Selinexor With Fludarabine and Cytarabine for Treatment of Refractory or Relapsed Leukemia or Myelodysplastic Syndrome) clinical trial (NCT02212561). Selinexor, initially at 30 mg/m2 per dose, was given orally on days 1, 3, 8, 10, 22, and 24 and was escalated according to a rolling-six design. Fludarabine 30 mg/m2 and cytarabine 2 g/m2 were administered on days 15 to 19. Pharmacokinetic and pharmacodynamic studies were performed on days 1 and 22. Response evaluations were performed on day 15 and at the completion of course 1.
Results
Among the 17 patients who were evaluable for toxicity, three were treated at 30 mg/m2, three at 40 mg/m2, six at 55 mg/m2, and five at 70 mg/m2. The most common grade 3 nonhematologic toxicity was asymptomatic hyponatremia. Two patients who were treated at 70 mg/m2 experienced reversible cerebellar toxicity, thereby defining the dose-limiting toxicity. Pharmacokinetic parameters demonstrated that plasma exposure was dose proportional. Fifteen of 16 patients demonstrated at least a twofold increase of XPO1 mRNA, indicating inhibition of the XPO1 protein. In this group of heavily pretreated, relapsed, and refractory patients, seven of 15 evaluable patients (47%) achieved complete response or complete response with incomplete count recovery.
Conclusion
Selinexor, in combination with fludarabine and cytarabine, is tolerable at doses up to 55 mg/m2 in pediatric patients with relapsed or refractory leukemia. All patients who received selinexor at ≥ 40 mg/m2 demonstrated XPO1 target inhibition. Response rates are promising and will be further explored in a phase II trial.
INTRODUCTION
Although survival rates are approximately 90% for children with acute lymphoblastic leukemia (ALL) and 60% for those with acute myeloid leukemia (AML), patients with relapsed or refractory disease have dismal outcomes.1-4 However, because only a minority of pediatric patients have known targetable lesions, novel and broadly active agents are urgently needed. Selinexor (KPT-330), a selective inhibitor of exportin 1 (XPO1), may represent such an agent.
XPO1 is the primary nuclear exporter of key tumor suppressor and growth regulatory proteins, including p53, p73, p21, p27, NPM1, FOXO, and I-ĸβ.5,6 Because the majority of these proteins require nuclear localization for their genome-surveying activities, increased nuclear export leads to decreased activity. Many malignancies, including AML, display elevated levels of XPO1.5,6 Higher levels of XPO1 are independently associated with a worse prognosis in adult patients with AML.7 These data have led to the development of selective inhibitor of nuclear export (SINE) compounds that covalently bind to the Cys528 residue of XPO1 and prevent binding of cargo proteins.6 In preclinical models, SINE compounds are active in a wide variety of malignancies and may act synergistically with traditional chemotherapy.8-16
Selinexor is an orally bioavailable, slowly reversible SINE compound that specifically blocks XPO1 and undergoes hepatic metabolism and excretion. It has been studied extensively in preclinical models of T-lineage ALL and AML.7,17-20 Selinexor causes apoptosis and differentiation in T-lineage ALL and AML cells lines, primary samples, and murine xenograft models, while sparing normal hematopoietic cells.17,18 Activity was evident in xenograft models of AML cells, with on-target effects demonstrated by reduction in XPO1 protein and nuclear accumulation of p53 and NPM1.17,20 In xenograft models, selinexor eliminated leukemia-initiating cells, as well as bulk tumor cells.20 A recent phase I study demonstrated that selinexor can be safely administered as a single agent to adult patients with advanced solid tumors.21 To our knowledge, this is the first report of selinexor in patients with hematologic malignancies, as well as the first report of selinexor given with conventional chemotherapy. We describe the safety, pharmacokinetics, pharmacodynamics, and activity of selinexor, in combination with fludarabine and cytarabine, in pediatric patients with relapsed acute leukemia.
PATIENTS AND METHODS
Patients
Patients with relapsed or refractory AML, ALL, myelodysplastic syndrome, or mixed phenotype acute leukemia (MPAL) who were ≤ 24 years old were eligible. Patients with AML, myelodysplastic syndrome, or MPAL were eligible if they were experiencing a first or subsequent relapse, whereas patients with ALL were eligible if they were experiencing a second or subsequent relapse. Refractory disease was defined as persistent disease after at least two courses of induction chemotherapy. Patients were required to have adequate hepatic and renal function, no evidence of graft-versus-host disease, and no uncontrolled infections. Patients could not receive investigational agents within 30 days of enrollment or myelosuppressive therapy within 14 days. Because selinexor is inactivated by glucuronidation and by conjugation with glutathione, acetaminophen was prohibited on days of selinexor administration. The protocol was approved by each site’s institutional review board and registered at ClinicalTrials.gov (NCT02212561). Informed consent, and assent when appropriate, was obtained from all patients or their legal guardians.
Treatment Plan
Selinexor was given orally on days 1, 3, 8, 10, 22, and 24. Fludarabine (30 mg/m2) and cytarabine (2 g/m2) were given on days 15 through 19. Allowance was made to start fludarabine and cytarabine early for patients with progressive disease. Intrathecal (IT) chemotherapy was given before day 1. If CNS disease was present, IT therapy was given weekly until the cerebrospinal fluid became clear of leukemia. The starting dose of selinexor was 30 mg/m2 (dose level 1), with planned dose escalations to 40 mg/m2 (dose level 2), 55 mg/m2 (dose level 3), and 70 mg/m2 (dose level 4) in a rolling-six design.22 Patients who had acceptable toxicity and clinical benefit were eligible to receive a second cycle of therapy. Supportive care included mandatory administration of dexamethasone, olanzapine, and ondansetron; voriconazole or posaconazole; and vancomycin plus ciprofloxacin or monotherapy levofloxacin during periods of neutropenia.
Definitions of Dose-Limiting Toxicities and Maximum Tolerated Dose
Toxicity was graded according to the Common Terminology Criteria for Adverse Events (version 4). Dose-limiting toxicities (DLTs) were based on nonhematologic toxicities that occurred during cycle 1 and were deemed possibly attributable to selinexor. Any grade 5 events and any grade 4 nonhematologic events possibly related to selinexor were considered DLTs, with the exception of electrolyte abnormalities reversible with standard intervention. Failure to receive at least 5 doses of selinexor by day 35 because of toxicity was considered a DLT. The maximum tolerated dose (MTD) was defined as the highest dose level at which one or fewer of six patients experienced a DLT.
Pharmacokinetic Studies
Blood samples were collected for pharmacokinetic (PK) analysis on days 1 and 22. On day 1, samples were collected before the dose; at 30 minutes; and at 1, 2, 4, 8, 24, and 48 hours after the dose. Samples were also collected before the 22-day dose and at 1, 2, 4, and 8 hours after the dose.
Pharmacodynamic Studies
To assess inhibition of XPO1, blood samples were collected before the first dose of selinexor and at 4, 8, 24, and 48 hours after the dose. Cells isolated from these samples were evaluated for mRNA expression levels by quantitative reverse transcription-polymerase chain reaction (qRT-PCR) of XPO1, which is upregulated in response to XPO1 inactivation. See Appendix (online only) for methods used for gene expression analysis.9
Response Criteria and Definitions
Response to single-agent selinexor was assessed by bone marrow aspiration on day 15 of cycle 1 unless fludarabine and cytarabine were started early because of progressive disease. Response to combination therapy was initially assessed between days 28 and 35 and reassessed at the time of count recovery. Patients were considered evaluable for response if they received at least five doses of selinexor. However, those who received fewer than five doses because of progressive leukemia were considered nonresponders. Measurement of response and minimal residual disease (MRD) were made on the basis of blast percentage by flow cytometry. Complete remission (CR) was defined as bone marrow with < 5% blasts, absolute neutrophil count (ANC) with ≥ 500 cells/µL, platelets with ≥ 75,000 cells/µL without transfusions, and no evidence of extramedullary disease. CR with incomplete blood count recovery (CRi) was defined as < 5% blasts, no extramedullary disease, but ANC < 500 cells/µL or platelet count < 75,000 cells/µL. Partial response was defined as bone marrow with 5% to 25% blasts and a decrease of at least 50% in blast percentage. MRD was defined as negative if the level was < 0.1%.
RESULTS
Patient Characteristics
Four patients had primary refractory disease at the time of enrollment, seven were experiencing a first relapse, and seven were experiencing a second relapse (Table 1). All patients who were enrolled while experiencing a first relapse had relapsed within 1 year of initial remission. Fifteen patients had AML, two had MPAL, and one had early T-cell precursor ALL after an initial diagnosis of AML. Ten patients had undergone prior stem-cell transplantation (SCT).
Table 1.
Patient Characteristics and Treatment Outcomes

Toxicity and MTD
Grade 3 and 4 nonhematologic toxicities occurring during cycle 1 are presented in Table 2. The most common Grade 3 nonhematologic toxicity related to selinexor was asymptomatic hyponatremia (median sodium nadir, 128.5 meq/L; range, 123 to 132 meq/L), which was easily corrected by oral or intravenous supplementation in all patients. Asymptomatic hypokalemia was also common.
Table 2.
Toxicity Profile of Patients
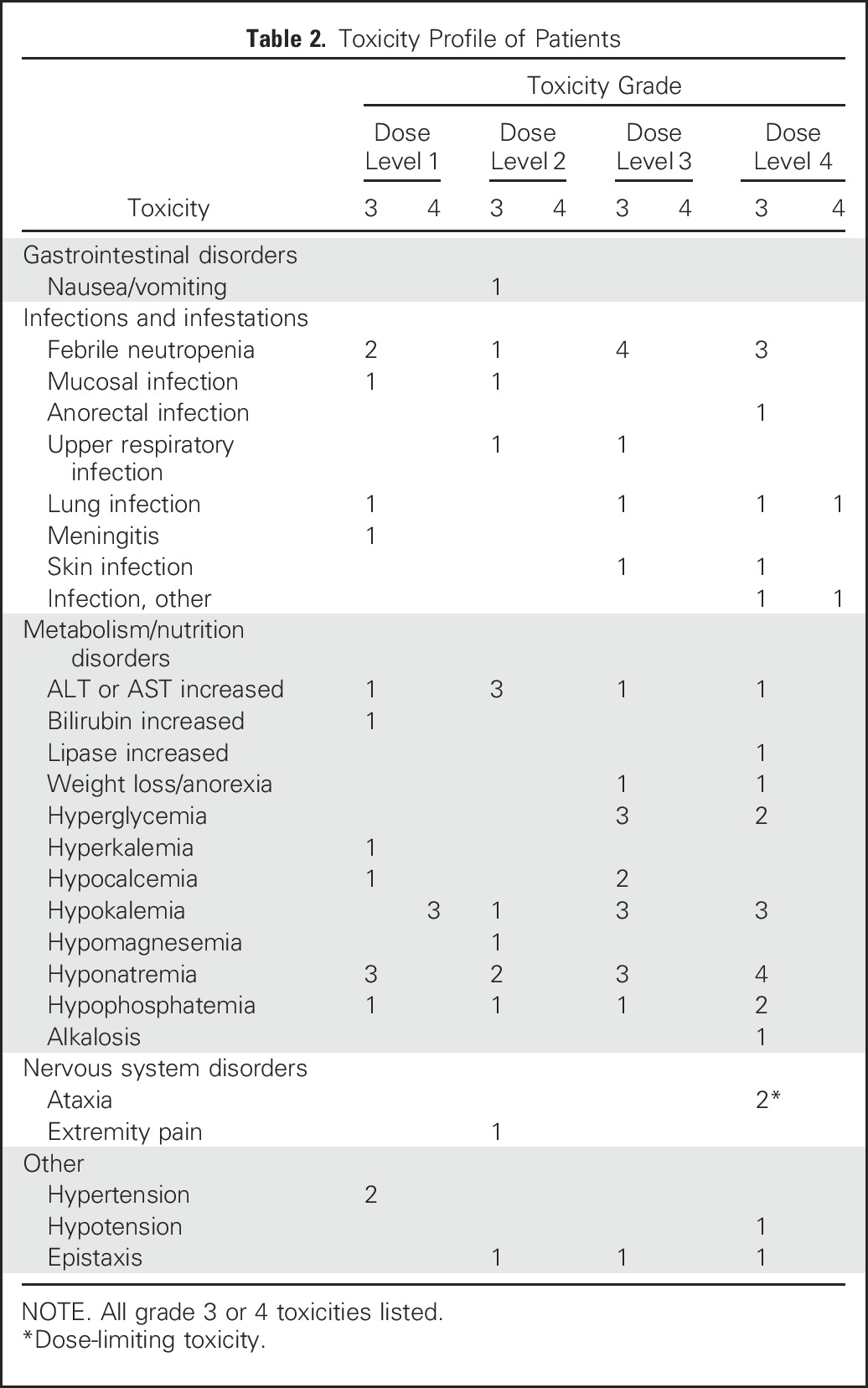
All 16 patients who received fludarabine and cytarabine after initial dosing of selinexor developed pancytopenia. Three patients with persistent disease were not evaluable for time to count recovery because they began alternative chemotherapy on day 37, 43, and 45, respectively. Two patients began SCT conditioning on day 43 and 53, respectively. One patient (No. 17), with a history of two SCTs before enrolling in this trial, developed concurrent disseminated Fusarium fungemia and enterococcus faecium bacteremia on day 36 and had no follow-up after day 45 because he transitioned to palliative care. One patient had prolonged neutropenia before enrollment, which persisted after protocol therapy. Of the remaining 9 patients, the median time of ANC recovery to ≥ 500 cell/µL was protocol day 49. This translates to a median of 35 days (range, 21 to 47) after the start of fludarabine and cytarabine. The patients did not receive granulocyte colony-stimulating factor.
Two patients (No. 16 and No. 18; Table 1) experienced reversible cerebellar toxicity after receiving selinexor at 70 mg/m2. The first patient (No. 16) was a 5-year-old male with refractory AML who had not undergone prior SCT. On day 23, after receiving four doses of selinexor and 5 days of fludarabine and cytarabine (without IT therapy), he had severe body pain, followed the next day by irritability, aphasia, and lower extremity weakness. Lumbar puncture showed normal opening pressure and no evidence of infection. Magnetic resonance imaging (MRI) showed diffuse cerebellar restricted diffusion with patchy restriction in the medulla and pons (Fig 1). He was empirically treated with intravenous immunoglobulin for 5 days, improved significantly over the next 3 weeks, and had resolution of symptoms after 6 weeks. A repeated MRI scan 10 days after the initial imaging showed nearly complete resolution of diffusion abnormalities. The second patient (No. 18) was a 19-month-old male with relapsed AML who had received prior SCT with busulfan, cyclophosphamide, and fludarabine conditioning. No CNS disease was detected at relapse. On day 13, after receiving four doses of selinexor and before receiving fludarabine or cytarabine, he became unable to hold his trunk or head straight and demonstrated erratic use of his hands. He had normal muscular tone and strength, but truncal instability when seated upright. An MRI scan revealed restricted diffusion and fluid-attenuated inversion recovery signal changes throughout the cerebellum (Fig 1). Lumbar puncture was deferred because of cerebellar edema seen on MRI. Because the clinical and imaging findings were consistent with cerebellar toxicity, protocol therapy was discontinued. His ataxia showed improvement over the course of 2 weeks. Neither patient had received radiation therapy, and neither had unusual difficulties with hyponatremia. No CNS toxicity was observed in any patients who received selinexor at doses ≤ 55 mg/m2, thus establishing 55 mg/m2 as the MTD.
Fig 1.

Axial T2-weighted magnetic resonance imaging scan of (A and B) the two patients with dose-limiting toxicity at 70 mg/m2. Both patients show diffuse cerebellar edema and hyperintensity (white arrows), sparing the brainstem (blue arrows), deep cerebellar nuclei, and cerebrum.
Selinexor Pharmacodynamics and Pharmacokinetics
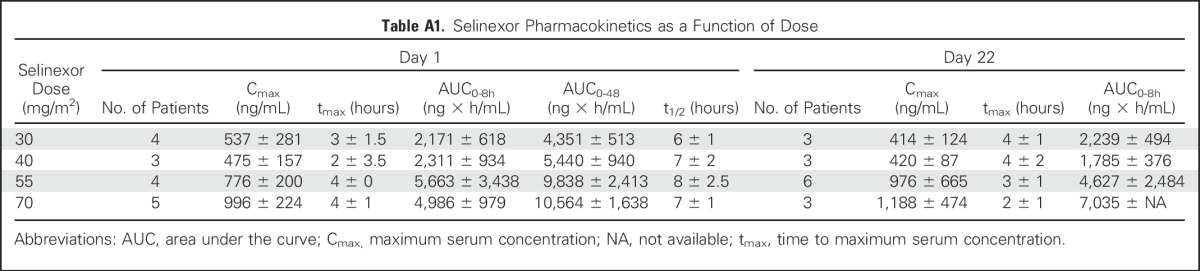
Plasma selinexor concentrations were assessed after the initial dose and after the day 22 dose. Mean plasma selinexor concentration showed little separation between the 30- and 40-mg/m2 dose levels, with an increase in exposure evident at the 55- and 70-mg/m2 dose levels (Fig 2). The mean half-life of selinexor at 40, 55, and 70 mg/m2 was 7.1, 7.6, and 7.2 hours, respectively. PK parameters measured on days 1 and 22 were comparable (Fig 2 and Appendix Table A1, online only). To assess the effects of selinexor on XPO1 protein, we used qRT-PCR to measure XPO1 expression, which is upregulated at the RNA level in response to XPO1 protein inactivation.9 Of the 16 patients assessed, 15 demonstrated at least twofold induction of XPO1 mRNA (Fig 3). The only patient who did not achieve this level was receiving a dose of 30 mg/m2. Increased expression persisted for at least 48 hours.
Fig 2.

Mean plasma selinexor concentration in pediatric patients at day 1 showed little separation between the 30 and 40 mg/m2 dose levels, with an increase in exposure evident at the 55 and 70 mg/m2 dose levels. Mean plasma selinexor concentration in pediatric patients after the day-22 dose showed peak levels and dynamics similar to the day-1 levels.
Fig 3.

XPO1 mRNA expression levels by dose. Inhibition of XPO1 was assessed by quantitative reverse transcription-polymer chain reaction of XPO1, which is upregulated at the RNA level in response to XPO1 protein inactivation.
Leukemia Response
Bone marrow aspirations and biopsies were performed on day 15 of therapy to assess for early activity of single-agent selinexor, which was given on days 1, 3, 8, and 10. Two patients (No. 2 and No. 16) were not evaluable for single-agent selinexor response because fludarabine and cytarabine were started early because of progressive leukemia. At the early response assessment on day 15, two patients with AML, including one who had suffered a second relapse after SCT (No. 5) and one with primary refractory leukemia (No. 15), had CR with undetectable MRD (Table 1).
Although initial response to combination therapy was assessed between days 28 and 35, patients were predictably hypocellular at this time. Therefore, evaluations of responses to combination (selinexor, fludarabine, and cytarabine) therapy were again performed when patients showed signs of hematopoietic recovery. Fifteen patients were evaluable for response to combination therapy. Two patients (No. 16 and No. 18) were not evaluable because they experienced DLTs before completion of cycle 1 and one patient (No. 17) because the family chose to pursue palliative care secondary to infectious complications before being evaluable for response. After the completion of one course of combination therapy, 7 of 15 evaluable patients (47%) achieved CR or CRi, including five who had no detectable MRD. All patients with CR/CRi subsequently underwent SCT (Table 1). Six of these seven patients were alive and in remission at a median time of 9.5 months (range, 3 to 12 months) from enrollment.
We incidentally observed that morphologic examination of the day-15 bone marrow aspirations showed clear evidence of myeloid differentiation in patients No. 5 and No. 14, despite the high day-15 level of MRD in patient No. 14 (Fig 4). The three patients (No. 5, No. 14, and No. 15) who demonstrated morphologic differentiation or flow cytometric complete response to single-agent selinexor at day 15 achieved a final response of CR or CRi, underwent SCT, and remain alive and in remission.
Fig 4.

Bone marrow aspirates of patient No. 5: (A) before therapy, showing myeloblasts along with neutrophils, myelocytes, and promyelocytes; and (B) at day 15, showing differentiation evidenced by the absence of blasts, with an increase in neutrophils and myelocytes. Bone marrow aspirates of patient No. 14: (C) before therapy, showing monoblasts and occasional neutrophils; and (D) at day 15, showing a decrease in monoblasts with a corresponding increase in immature promonocytes.
DISCUSSION
We report here the results of a phase I study to assess the safety, pharmacodynamics, pharmacokinetics, and activity of selinexor, the only inhibitor of nuclear export currently in clinical trials. Because preclinical3,18-20 and phase I21 data suggest that the DLTs of selinexor are anorexia, weight loss, and fatigue, we chose to give selinexor in combination with fludarabine and cytarabine, a commonly used regimen in which myelosuppression is the limiting toxicity. The treatment course included 2 weeks of single-agent selinexor, which allowed us to examine the safety and activity of selinexor alone in this pediatric population. Exposure to selinexor as a prephase may increase nuclear localization of many tumor suppressor proteins, which may augment the effectiveness of cytotoxic chemotherapy.14
Although data from a phase I trial of selinexor in adults with advanced solid tumors indicate that anorexia, weight loss, and fatigue are common, these adverse effects were rarely observed in this study.21 The occurrence of hyponatremia and the need for sodium supplementation in nearly all patients highlight the need for frequent monitoring of serum electrolytes among patients treated with selinexor. The DLT, observed at 70 mg/m2, was acute, severe, slowly reversible cerebellar toxicity. Cerebellar toxicity was also reported in one adult patient who received selinexor at a dose of 85 mg/m2 in the recently reported phase I trial.21 In our study, we cannot definitively attribute the cerebellar toxicity observed in the first patient to selinexor because this patient had also received fludarabine and cytarabine, which are known to cause neurotoxicity.23,24 However, a second patient developed cerebellar toxicity characterized by similar symptoms and nearly identical MRI changes after receiving single-agent selinexor, suggesting that both occurrences represent dose-dependent toxicities.
Plasma concentration levels and measured half-lives in pediatric patients are similar to levels achieved in adults.21 There were no significant differences in PK levels between day 1 and day 22 in our study, consistent with data in adults.21 We did not observe differences in the PK levels between patients with or without DLT. Induction of XPO1 mRNA expression, which was observed in all patients treated at ≥ 40 mg/m2 and which was sustained for at least 48 hours, demonstrates on-target inhibition of XPO1 protein.9 Although the ideal length of inhibition is not known, the PK and pharmacodynamic data suggest that the convenient dosing schedule used in this trial is sufficient to achieve target inhibition for at least 96 hours during the weeks of selinexor dosing.
The complete responses observed in this study suggest activity of the combined therapeutic regimen of selinexor, fludarabine, and cytarabine. Two recent large trials evaluating regimens for refractory or first relapse of pediatric AML showed complete response rates of 48% and 64%.3,25 The higher-risk nature of our patient population and the small number of patients in the phase I portion of this study preclude direct comparisons with the response rates reported from these studies. The characteristics of our patient cohort more closely resemble those of patients enrolled in other early-phase trials for children with relapsed AML, such as a phase I trial of gemtuzumab ozogamicin and a phase II trial of clofarabine, in which response rates of 28% and 26%, respectively, were reported.26,27 Notably, two patients in our study became MRD negative after receiving only selinexor, indicating that there is a patient population that might benefit from single-agent selinexor therapy. In addition, three of the seven patients who attained CR or CRi were in second relapse. Interestingly, the patient in our study with AML harboring the high-risk translocation t(6;9), which causes a DEK-NUP214 chimeric protein, had an MRD-negative complete response to single-agent selinexor. The full role of this fusion protein in AML remains unclear, but NUP214 is a member of the nuclear pore complex and is the nucleoporin with the highest affinity for XPO1.28-30 The relationship between selinexor-mediated XPO1 inhibition and NUP214 fusions warrants further study. In addition to correlating response to selinexor regimens with cytogenetic or molecular leukemia subtypes, specific possibilities for analysis of predictive biomarkers include assessment of p53 mutational status, ex vivo treatment with selinexor followed by assays of apoptosis, or quantitation of XPO1 transcript expression or XPO1 protein levels.5,7,20
In summary, to our knowledge, this is the first report of the use of selinexor in pediatric patients, as well as the first report of selinexor in patients with hematologic malignancies. We found that six doses of selinexor, given at 55 mg/m2/dose, was well tolerated in combination with fludarabine and cytarabine in children with relapsed and refractory acute leukemia. The dose-limiting reversible cerebellar toxicity was only observed at a dose level of 70 mg/m2. Complete responses observed in this high-risk population, after single-agent and combination therapy, suggest that selinexor may have a role in the treatment of pediatric acute leukemia. Taken together, this phase I study of selinexor, along with promising preclinical studies demonstrating efficacy in high-risk leukemia and on leukemia-initiating cells,20 justifies further studies to evaluate the safety and efficacy of selinexor-based therapy in children with relapsed and refractory acute leukemia. In this regard, preclinical studies have shown that selinexor has synergistic activity when combined with agents such as DNA methyltransferase inhibitors16 or MDM2 inhibitors,7 suggesting that future trials include such combinations. We are currently planning a phase II study to further characterize the combination of selinexor, fludarabine, and cytarabine.
ACKNOWLEDGMENT
We thank Karyopharm Therapeutics, Newton, MA, for providing selinexor, for performing pharmacokinetic and pharmacodynamic testing, and for critical review of the manuscript. We also thank Stanley Pounds for providing input on the initial study design and offering editorial advice. We also thank Kathy Jackson and Heidi Clough for data collection and management.
Appendix
XPO1 Gene Expression Analysis Methods
Total RNA was isolated from whole-blood PAXgene samples (cat#762165; BD Biosciences, Sparks, MD) according to the manufacturer’s protocol. cDNA was synthesized using high-capacity cDNA Reverse Transcription Kit (cat#4368813; Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s protocol. XPO1 mRNA levels were measured by quantitative polymerase chain reaction (QPCR) using predesigned TaqMan Gene Expression Assay (cat#Hs00185645; ThermoFisher). Each QPCR consisted of the following components: 0.5μL of XPO1 TaqMan assay, 0.5μl of GAPDH Control (cat#4326317E; Thermo Fisher Scientific), 5μL of Fast Advanced Master Mix ( cat#4444963; Thermo Fisher Scientific), and 2μL water. QPCR was performed on the Viia7 System (Life Technologies, Carlsbad, CA), and results were analyzed using Excel (Microsoft, Redmond, WA).
Table A1.
Selinexor Pharmacokinetics as a Function of Dose
Footnotes
Presented in part at the American Society of Hematology 2015 annual meeting and the American Society of Pediatric Hematology/Oncology 2016 annual meeting.
Authors’ disclosures of potential conflicts of interest are found in the article online at www.jco.org.
Clinical trial information: NCT02212561.
AUTHOR CONTRIBUTIONS
Conception and design: John K. Choi, Ching-Hon Pui, Jeffrey E. Rubnitz
Collection and assembly of data: Thomas B. Alexander, Norman J. Lacayo, John K. Choi, Raul C. Ribeiro, Jeffrey E. Rubnitz
Data analysis and interpretation: Thomas B. Alexander, Norman J. Lacayo, Ching-Hon Pui, Jeffrey E. Rubnitz
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Phase I Study of Selinexor, a Selective Inhibitor of Nuclear Export, in Combination With Fludarabine and Cytarabine, in Pediatric Relapsed or Refractory Acute Leukemia
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Thomas B. Alexander
Stock or Other Ownership: Covenant Surgical Partners (I)
Norman J. Lacayo
No relationship to disclose
John K. Choi
No relationship to disclose
Raul C. Ribeiro
No relationship to disclose
Ching-Hon Pui
No relationship to disclose
Jeffrey E. Rubnitz
No relationship to disclose
REFERENCES
- 1.Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373:1541–1552. doi: 10.1056/NEJMra1400972. [DOI] [PubMed] [Google Scholar]
- 2.Zwaan CM, Kolb EA, Reinhardt D, et al. Collaborative efforts driving progress in pediatric acute myeloid leukemia. J Clin Oncol. 2015;33:2949–2962. doi: 10.1200/JCO.2015.62.8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaspers GJ, Zimmermann M, Reinhardt D, et al. Improved outcome in pediatric relapsed acute myeloid leukemia: Results of a randomized trial on liposomal daunorubicin by the International BFM Study Group. J Clin Oncol. 2013;31:599–607. doi: 10.1200/JCO.2012.43.7384. [DOI] [PubMed] [Google Scholar]
- 4.Pui CH, Yang JJ, Hunger SP, et al. Childhood acute lymphoblastic leukemia: Progress through collaboration. J Clin Oncol. 2015;33:2938–2948. doi: 10.1200/JCO.2014.59.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishizawa J, Kojima K, Hail N, Jr, et al. Expression, function, and targeting of the nuclear exporter chromosome region maintenance 1 (CRM1) protein. Pharmacol Ther. 2015;153:25–35. doi: 10.1016/j.pharmthera.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Senapedis WT, Baloglu E, Landesman Y. Clinical translation of nuclear export inhibitors in cancer. Semin Cancer Biol. 2014;27:74–86. doi: 10.1016/j.semcancer.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 7.Kojima K, Kornblau SM, Ruvolo V, et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood. 2013;121:4166–4174. doi: 10.1182/blood-2012-08-447581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lapalombella R, Sun Q, Williams K, et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood. 2012;120:4621–4634. doi: 10.1182/blood-2012-05-429506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tai YT, Landesman Y, Acharya C, et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: Molecular mechanisms and therapeutic implications. Leukemia. 2014;28:155–165. doi: 10.1038/leu.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green AL, Ramkissoon SH, McCauley D, et al. Preclinical antitumor efficacy of selective exportin 1 inhibitors in glioblastoma. Neuro Oncol. 2015;17:697–707. doi: 10.1093/neuonc/nou303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun H, Hattori N, Chien W, et al. KPT-330 has antitumour activity against non-small cell lung cancer. Br J Cancer. 2014;111:281–291. doi: 10.1038/bjc.2014.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kazim S, Malafa MP, Coppola D, et al. Selective nuclear export inhibitor KPT-330 enhances the antitumor activity of gemcitabine in human pancreatic cancer. Mol Cancer Ther. 2015;14:1570–1581. doi: 10.1158/1535-7163.MCT-15-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walker CJ, Oaks JJ, Santhanam R, et al. Preclinical and clinical efficacy of XPO1/CRM1 inhibition by the karyopherin inhibitor KPT-330 in Ph+ leukemias. Blood. 2013;122:3034–3044. doi: 10.1182/blood-2013-04-495374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turner JG, Dawson J, Cubitt CL, et al. Inhibition of CRM1-dependent nuclear export sensitizes malignant cells to cytotoxic and targeted agents. Semin Cancer Biol. 2014;27:62–73. doi: 10.1016/j.semcancer.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hing ZA, Mantel R, Beckwith KA, et al. Selinexor is effective in acquired resistance to ibrutinib and synergizes with ibrutinib in chronic lymphocytic leukemia. Blood. 2015;125:3128–3132. doi: 10.1182/blood-2015-01-621391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranganathan P, Yu X, Santhanam R, et al. Decitabine priming enhances the antileukemic effects of exportin 1 (XPO1) selective inhibitor selinexor in acute myeloid leukemia. Blood. 2015;125:2689–2692. doi: 10.1182/blood-2014-10-607648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ranganathan P, Yu X, Na C, et al. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood. 2012;120:1765–1773. doi: 10.1182/blood-2012-04-423160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Etchin J, Sanda T, Mansour MR, et al. KPT-330 inhibitor of CRM1 (XPO1)-mediated nuclear export has selective anti-leukaemic activity in preclinical models of T-cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Br J Haematol. 2013;161:117–127. doi: 10.1111/bjh.12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Etchin J, Sun Q, Kentsis A, et al. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia. 2013;27:66–74. doi: 10.1038/leu.2012.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Etchin J, Montero J, Berezovskaya A, et al. Activity of a selective inhibitor of nuclear export, selinexor (KPT-330), against AML-initiating cells engrafted into immunosuppressed NSG mice. Leukemia. 2016;30:190–199. doi: 10.1038/leu.2015.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. doi: 10.1200/JCO.2015.65.3949. Abdul Razak AR, Mau-Soerensen M, Gabrail NY, et al: First-in-class, first-in-human phase I study of selinexor, a selective inhibitor of nuclear export, in patients with advanced solid tumors. J Clin Oncol 34:4142-4150, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skolnik JM, Barrett JS, Jayaraman B, et al. Shortening the timeline of pediatric phase I trials: The rolling six design. J Clin Oncol. 2008;26:190–195. doi: 10.1200/JCO.2007.12.7712. [DOI] [PubMed] [Google Scholar]
- 23.Beitinjaneh A, McKinney AM, Cao Q, et al. Toxic leukoencephalopathy following fludarabine-associated hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2011;17:300–308. doi: 10.1016/j.bbmt.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 24.Helton KJ, Patay Z, Triplett BM. Fludarabine-induced severe necrotizing leukoencephalopathy in pediatric hematopoietic cell transplantation. Bone Marrow Transplant. 2013;48:729–731. doi: 10.1038/bmt.2012.196. [DOI] [PubMed] [Google Scholar]
- 25.Cooper TM, Franklin J, Gerbing RB, et al. AAML03P1, a pilot study of the safety of gemtuzumab ozogamicin in combination with chemotherapy for newly diagnosed childhood acute myeloid leukemia: A report from the Children’s Oncology Group. Cancer. 2012;118:761–769. doi: 10.1002/cncr.26190. [DOI] [PubMed] [Google Scholar]
- 26.Arceci RJ, Sande J, Lange B, et al. Safety and efficacy of gemtuzumab ozogamicin in pediatric patients with advanced CD33+ acute myeloid leukemia. Blood. 2005;106:1183–1188. doi: 10.1182/blood-2004-10-3821. [DOI] [PubMed] [Google Scholar]
- 27.Jeha S, Razzouk B, Rytting M, et al. Phase II study of clofarabine in pediatric patients with refractory or relapsed acute myeloid leukemia. J Clin Oncol. 2009;27:4392–4397. doi: 10.1200/JCO.2008.18.8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sandahl JD, Coenen EA, Forestier E, et al. t(6;9)(p22;q34)/DEK-NUP214-rearranged pediatric myeloid leukemia: An international study of 62 patients. Haematologica. 2014;99:865–872. doi: 10.3324/haematol.2013.098517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tarlock K, Alonzo TA, Moraleda PP, et al. Acute myeloid leukaemia (AML) with t(6;9)(p23;q34) is associated with poor outcome in childhood AML regardless of FLT3-ITD status: A report from the Children’s Oncology Group. Br J Haematol. 2014;166:254–259. doi: 10.1111/bjh.12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Port SA, Monecke T, Dickmanns A, et al. Structural and functional characterization of CRM1-Nup214 interactions reveals multiple fg-binding sites involved in nuclear export. Cell Reports. 2015;13:690–702. doi: 10.1016/j.celrep.2015.09.042. [DOI] [PubMed] [Google Scholar]