

Mycobacterium indicus pranii induces potent dendritic cell activation, enhances dendritic cell survival, and promotes Th1/Th17 polarization potential in TLR2- and TLR9-dependent manner.
Keywords: cytokines, costimulatory molecules, apoptosis, T cell polarization, pattern recognition receptors
Abstract
MIP is a nonpathogenic, soil-borne predecessor of Mycobacterium avium. It has been reported previously that MIP possesses strong immunomodulatory properties and confers protection against experimental TB and tumor. DCs, by virtue of their unmatched antigen-presentation potential, play a critical role in activation of antitumor and antimycobacterial immune response. The effect of MIP on the behavior of DCs and the underlying mechanisms, however, have not been investigated so far. In the present study, we showed that MIP induces significant secretion of IL-6, IL-12p40, IL-10, and TNF-α by DCs and up-regulates the expression of costimulatory molecules CD40, CD80, and CD86. MIP(L) induced a significantly higher response compared with MIP(K). PI and Annexin V staining showed that MIP increases DC survival by inhibiting apoptosis. Consistently, higher expression of antiapoptotic proteins Bcl-2 and Bcl-xl was observed in MIP-stimulated DCs. Cytokines, produced by naïve T cells, cocultured with MIP-stimulated DCs, showed that MIP promotes Th1/Th17 polarization potential in DCs. Response to MIP was lost in MyD88−/−DCs, underscoring the critical role of TLRs in MIP-induced DC activation. Further studies revealed that TLR2 and TLR9 are involved in DC activation by MIP(L), whereas MIP(K) activates the DCs through TLR2. Our findings establish the DC activation by MIP, define the behavior of MIP-stimulated DCs, and highlight the role of TLRs in MIP-induced DC activation.
Introduction
DCs are the most potent APCs, strategically placed at the interface of the innate and adaptive immune system. Depending on the nature of microbial stimuli, DCs can promote a Th1, Th2, or Th17 type of immune response [1]. The Th1 type of immune response is critical for protection against intracellular pathogens and neoplastic diseases. In the case of M.tb infection, for example, the Th1 immune response leads to activation of infected macrophages and subsequent killing of intracellular bacilli [2]. M.tb-infected DCs can directly present the antigens to T cells, whereas uninfected DCs take up the apoptotic bodies of infected macrophages and cross-prime the T cells against M.tb antigens [3]. In a normal scenario, DCs lead to a robust immune response against mycobacteria, but virulent mycobacterial species prevent their clearance by inhibiting phagolysosomal fusion, which not only protects them against lytic enzymes but also hampers the presentation of their components on MHC molecules [4]. Additionally, mycobacteria-infected DCs produce IL-10, which acts in an autocrine manner to inhibit DC trafficking to the draining lymph node and to suppress IL-12 production by them [5].
DCs also play a critical role in the activation of the antitumor immune response. Tumor cells are actively phagocytosed by DCs through CD36 and the vitronectin receptor [6]. This is followed by activation of tumor-specific CD8+T cells and NK cells, which can lyse the tumor cells in an antigen-dependent and -independent manner, respectively [7, 8]. By virtue of their ability to activate a potent antitumor immune response, DCs are being investigated for cancer immunotherapy using different approaches, which include ex vivo loading with tumor antigens and genetic modifications [9]. Although DCs are highly efficient in inducing an antitumor immune response, in the absence of proper activation signals inside of the tumor mass, their antigen-presentation ability is compromised [10]. Growing tumors, on the other hand, actively suppress the maturation of DCs by producing immunosuppressive cytokines, such as TGF-β and IL-10. Immature DCs, in turn, promote T regulatory cells, which further dampen the antitumor immune response [11].
MIP is an atypical Mycobacterium with strong immunomodulatory properties and antigenic determinants shared with M.tb and Mycobacterium leprae [12, 13]. It is a potential vaccine candidate against TB, and we have previously evaluated the protective efficacy of MIP(L) and MIP(K) against experimental TB. Even though MIP is a nonpathogenic Mycobacterium, it is approved for human use only in the heat-killed form. The nonpathogenic nature of MIP has been authenticated by its recently published whole-genome sequence [14]. We previously observed that MIP(L) confers significantly higher protection against M.tb compared with MIP(K) and BCG [15, 16]. MIP promoted the Th1 type of immune response in animal models of TB, as indicated by IFN-γ production by immune cells from lung and lymphoid tissue. The Th1 type of immune response has also been shown to be crucial for protection against neoplastic diseases. Accordingly, MIP therapy was found to protect the mice against poorly immunogenic melanoma tumors [17]. Tumors in MIP-treated mice were infiltrated with a high number of DCs, and consistently, a higher antitumor immune response was observed in them. As DCs play a key role in the activation of an antitumor and antimycobacterial immune response, in the present study, we examined the effect of MIP on DC responses and delineated the underlying mechanisms. It was observed that MIP leads to potent DC activation, as indicated by the production of proinflammatory cytokines and up-regulation of costimulatory molecules. MIP(L) led to a higher response compared with MIP(K). Stimulation with MIP resulted in an increased DC survival. MIP-stimulated DCs induced Th1/Th17 polarization in naïve T cells. It was further observed that TLR2 and TLR9 but not TLR4 play a crucial role in DC activation by MIP.
MATERIALS AND METHODS
Ethics statement
All animal experiments described here were approved by the Institutional Animal Ethics Committee of the National Institute of Immunology (New Delhi, India; Approval No. 205/08/13) and were performed in accordance with the guidelines from the same.
Mice
Six- to 8-week-old inbred BALB/c, C57BL/6, and MyD88−/− mice were obtained from the animal facility of the National Institute of Immunology. TLR2−/− and TLR4−/− mice were kind gifts from Dr. Ruslan Medzhitov (Yale University School of Medicine, New Haven, CT, USA) and were maintained at the International Centre for Genetic Engineering and Biotechnology (New Delhi, India).
Mycobacteria
MIP was cultured in 7H9 medium with 0.05% Tween-80 and 0.1% glycerol and supplemented with 10% albumin-dextrose-catalase. Log-phase culture was harvested by centrifugation at 2000 g and washed in PBS-3. The count of MIP was determined on the basis of OD at 600 nm. MIP(K) was prepared by autoclaving bacterial suspension at 15 pounds/square inch for 15 min.
BMDCs
BMDCs were derived by culturing mouse BM cells in the presence of GM-CSF, as described previously [18]. In brief, 4 × 106 BM cells were plated/well of a 6-well plate in RPMI media supplemented with 10% FBS and 1% penicillin-streptomycin (RPMI 10) in the presence of 20 ng/ml GM-CSF. Culture medium along with nonadherent cells was removed on Days 3 and 5, and fresh, 4.0 ml GM-CSF-supplemented media were added to each well. Immature DCs were harvested on Day 7 by gentle pipetting. After a wash in RPMI-10 medium, cells were used for subsequent experiments. Purity of DCs was determined on the basis of CD11c and MHCII expression.
Isolation of MIP genomic DNA
DNA was isolated from MIP by the phenol-chloroform extraction method. In brief, the MIP pellet was suspended in buffered solution with 2 mg/ml lysozyme and incubated at 37°C for 6 h. Bacilli were lysed with 1% SDS, and proteinase K treatment was given for 1 h at 60°C. DNA was extracted with phenol-chloroform-isoamyl alcohol, precipitated with the help of isopropanol, and washed with 75% ethanol. Extracted DNA was finally solubilized in autoclaved double-distilled water, and concentration was determined by use of NanoDrop 1000.
DNAse I treatment
MIP genomic DNA was treated with DNAse I (Thermo Scientific, Pittsburgh, PA, USA), per the instructions provided by the manufacturer. Treated DNA was analyzed by agarose gel electrophoresis to confirm its digestion. MK-DNA was also analyzed on the gel.
Stimulation of BMDCs with MIP
DCs were plated in 24-well plate (1.5 × 106 cells/well) and stimulated with MIP(L) and MIP(K) at indicated MOI. Culture supernatants were collected after 24 h and analyzed for different cytokines by ELISA. For kinetics study, MIP(L) and MIP(K) were used at an MOI of 10, and culture supernatants were collected at 12, 24, and 36 h. To evaluate the effect of MIP on DC survival, cells were stimulated at the indicated MOI and analyzed for Annexin V and PI staining after 24 h. To elucidate the role of TLR9, genomic DNA from MIP (20 µg/ml) was used, and culture supernatants were analyzed after 24 h.
Cytokine estimation with ELISA
Cytokine levels in the culture supernatants were determined, according to the instruction manual provided with ELISA kits, purchased from BD Biosciences (San Jose, CA, USA) or eBioscience (San Diego, CA, USA).
Annexin V and PI staining
DCs were analyzed for Annexin V (BD Biosciences) staining, following the instructions provided by the manufacturer. In brief, 1.0 × 106 cells were suspended in 100 µl Annexin staining buffer, and 5 µl FITC-Annexin V was added to the suspension. After 15 min, volume was made up to 500 µl with Annexin staining buffer. For PI staining, 5 µl 100 µg/ml PI solution was added to 500 µl DC suspension. Cells were analyzed immediately by flow cytometry.
Flow cytometry
To analyze the purity of BMDCs, freshly harvested DCs were fixed in 1% paraformaldehyde. After 2 washes with PBS-10, cells were incubated with FITC-conjugated anti-mouse CD11c and PE-conjugated anti-mouse MHCII mAb. To analyze the expression of costimulatory molecules, MIP-stimulated DCs were harvested after 24 h, fixed with 1% paraformaldehyde, washed with PBS-10, and incubated with PE-conjugated anti-mouse CD40, CD80, CD86, and MHCII mAb (purchased from BD Biosciences or eBioscience). After 1 h, cells were washed with ice-cold PBS and run on a BD LSR or BD Accuri C6 flow cytometer (both BD Biosciences). Data were analyzed with Cyflogic 1.2.1 or BD Accuri C6 software.
Immunoblotting
Cellular proteins were prepared with the help of M2 lysis buffer [19]. Protein concentration was determined with Bicinchoninic Acid (BCA) Protein Assay Kit (Pierce, Thermo Scientific, Rockford, IL, USA). Cellular protein (10–20 µg) was resolved on 12% polyacrylamide gel at 30 mA and transferred to a 0.2 µm polyvinylidene difluoride membrane at 120 mA for 2 h. The membrane was blocked overnight with 10% BSA at 4°C before probing with anti-Bcl-2 (1:1000), anti-Bcl-xl (1:2000), and anti-β-actin (1:10,000) antibodies (Cell Signaling Technologies, Danvers, MA, USA). After probing with primary and secondary antibodies, proteins of interest were revealed with the help of ECL substrate (Bio-Rad Laboratories, Hercules, CA, USA), according to the manufacturer’s instructions. Band intensities were quantified with the help of ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA).
Purification of CD4+T cells and CD8+T cells
CD4+T cells and CD8+T cells were purified from splenocytes of naïve BALB/c mice by use of T Lymphocyte Enrichment Set (BD Biosciences), according to instructions provided by the manufacturer. In brief, splenocytes were incubated with T lymphocyte enrichment biotinylated antibody cocktail on ice for 15 min. After giving a wash, cells were incubated with streptavidin-magnetic particles for 30 min. Tubes were placed in a cell separation magnet (BD Biosciences), and cell suspension was collected after 10 min. Purified cells were washed and resuspended in RPMI-10 medium.
Allogeneic-MLR
BMDCs (2.0 × 104 cells) were added/well in a 96-well, round-bottom plate and stimulated with MIP(L) and MIP(K) at an MOI of 10. After 24 h, DCs were irradiated with gamma-rays (25 Gy), and 1.0 × 105 allogeneic CD4+T cells or CD8+T cells or splenocytes were cocultured with DCs. Culture supernatants were collected after 5 days and analyzed for IL-5, IL-17, and IFN-γ by ELISA.
Statistical analysis
Statistical analyses were performed with the help of GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA, USA). Data were analyzed by 1-way ANOVA with Tukey’s multiple comparison test applied postanalysis. P < 0.05 was considered significant.
RESULTS
MIP induces production of proinflammatory cytokines by DCs
APCs respond to microbial stimuli by producing proinflammatory cytokines that help in the activation of other immune cells and their recruitment to the site of insult. To examine the DC response to MIP, mouse BMDCs were stimulated with MIP(K) and MIP(L). Purity of BMDCs, as assessed on the basis of CD11c and MHCII expression, was found to be in the range of 85–90% (Supplemental Fig. 1). It was observed that MIP(K) and MIP(L) led to production of TNF-α, IL-12p40, and IL-6 in a dose-dependent manner. Significantly high levels of these cytokines, however, were observed in response to MIP(L) (Fig. 1A–C). Production of IL-10, in response to MIP(L) and MIP(K), was also analyzed. IL-10 has immunosuppressive properties and controls the damaging effects of excessive proinflammatory cytokines [20]. It was observed that unstimulated and MIP(K)-stimulated DCs produced a comparable level of IL-10. MIP(L), however, led to the significant level of IL-10 secretion by DCs (Fig. 1D). Time-kinetics study of the DC response to MIP(K) and MIP(L) showed that IL-6 and TNF-α were produced at early time-points, whereas the level of IL-12p40 increased with time (Fig. 1E–H). Compared with MIP(K), MIP(L) induced the higher levels of IL-6, IL-12p40, TNF-α, and IL-10 at all of the time-points.
Figure 1. Cytokine production by MIP-stimulated DCs. BMDCs were stimulated with MIP(K) and MIP(L) at MOIs of 5, 10, and 20. Culture supernatants from unstimulated or MIP-stimulated DCs were collected after 24 h and analyzed for TNF-α, IL-6, IL-12p40, and IL-10 by ELISA. Higher levels of these cytokines were produced in response to MIP(L). MIP(K) induced moderate levels of TNF-α, IL-6, IL-12p40, and IL-10 (A–D). For time-kinetics study, BMDCs were stimulated with MIP(K) and MIP(L) at an MOI of 10. Culture supernatants were collected after 12, 24, and 36 h and analyzed for different cytokines. Higher levels of TNF-α, IL-6, IL-12p40, and IL-10 were observed in response to MIP(L) at all time-points (E–H). Mean ± sem of 3 independent experiments is shown. **P < 0.01, and ***P < 0.001.

DCs up-regulate costimulatory molecules in response to MIP
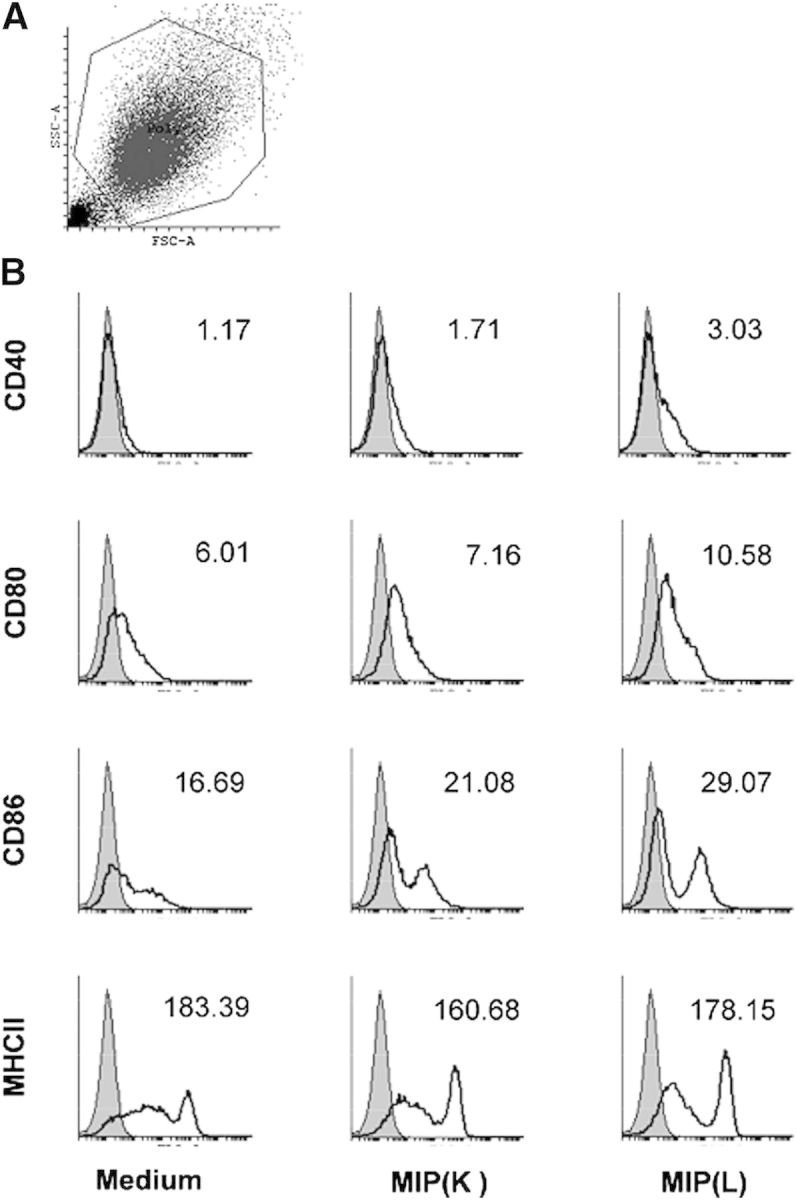
In addition to cytokine production, activated DCs up-regulate the expression of costimulatory molecules, which provide the necessary signals for T cell activation and acquisition of effector function [21]. The effect of MIP on the expression of costimulatory markers by DCs was examined henceforth. It was observed that DCs up-regulate the expression of CD40, CD80, and CD86 in response to MIP. Consistent with cytokine production, higher up-regulation of these markers was observed in response to MIP(L) (Fig. 2). However, up-regulation of CD40 on MIP-stimulated DCs was very low. To analyze whether MIP is inefficient in up-regulating CD40, BMDCs were stimulated with LPS. Expression of CD40 was moderately up-regulated in response to LPS, indicating that stronger stimulation is required for optimal CD40 expression (data not shown). DCs were further analyzed for the expression of the MHCII molecule. Comparable levels of MHCII expression were observed in unstimulated and MIP-stimulated DCs.
Figure 2. Expression of costimulatory markers by MIP-stimulated DCs. BMDCs were stimulated with MIP(K) and MIP(L) at an MOI of 10. After 24 h, cells were stained with PE-conjugated anti-mouse CD40, CD80, CD86, and MHCII mAb and analyzed by flow cytometry. The total population of DCs was gated on for analysis (A). SSC-A, Side-scatter-area; FSC-A, forward-scatter area. Higher expression of CD40, CD80, and CD86 was observed in MIP(L)-stimulated DCs. MIP(K) led to a moderate up-regulation of these molecules. Comparable levels of MHCII expression were observed in unstimulated and MIP-stimulated DCs (B). Shaded histograms, Unstimulated DCs; open histograms, MIP-stimulated DCs. Mean fluorescence intensity (1/1000) is mentioned with each histogram. Representative data of 2 independent experiments are shown.
MIP stimulation leads to enhanced survival of DCs
Enhanced DC survival has been shown to result in heightened T cell response. Survival of DCs is increased by components of the immune system, such as RANKL, and microbial molecules, such as LPS. Given the induction of higher antimycobacterial and the antitumor immune response by MIP, we examined the effect of MIP on the survival of DCs. Interestingly, it was observed that MIP stimulation results in an enhanced DC survival. Nearly 28% of unstimulated DCs were PI positive, whereas the level of PI-positive cells in MIP(K)- and MIP(L)-stimulated DCs was ∼10% and ∼6%, respectively (Fig. 3A). Both MIP(K) and MIP(L) increased the DC survival in a dose-dependent manner, but MIP(L) led to higher survival compared with MIP(K) (Fig. 3B). Annexin V staining showed that MIP enhanced the DC survival by inhibiting apoptosis. Involvement of antiapoptotic proteins in MIP-induced DC survival was also analyzed. Consistent with their enhanced survival, higher levels of antiapoptotic proteins were expressed by MIP(L)-stimulated DCs. Furthermore, MIP(L) increased the expression of both Bcl-2 and Bcl-xl, whereas MIP(K) induced the expression of Bcl-xl only (Fig. 3C).
Figure 3. Effect of MIP on DC survival. BMDCs were stimulated with MIP(K) and MIP(L) at an MOI of 10 and analyzed for Annexin V and PI staining after 24 h. Lower levels of Annexin V and PI-positive cells were observed in MIP(K)- and MIP(L)-stimulated DCs (A). Both MIP(K) and MIP(L) enhanced DC survival in a dose-dependent manner when used at MOIs of 5, 10, and 20 (B). Mean ± sem of 3 independent experiments is shown. **P < 0.01; ns, not significant. Expression of antiapoptotic proteins Bcl-2 and Bcl-xl was analyzed in unstimulated and MIP-stimulated BMDCs by Western blotting. MIP(K) induced the expression of Bcl-xl, whereas MIP(L) enhanced the expression of both Bcl-2 and Bcl-xl (C). Fold expression to unstimulated DCs is indicated with respective bands. Representative data of 2 independent experiments are shown.

MIP-stimulated DCs promote Th1/Th17 polarization in naïve T cells
Th1 polarization of the immune response in MIP-treated animals has been reported previously [16]. To analyze the involvement of DCs in MIP-induced polarization of naïve T cells, allogeneic splenocytes, CD4+T cells and CD8+T cells were cocultured with MIP(L)- or MIP(K)-stimulated DCs, and culture supernatants were analyzed for IL-5, IL-17, and IFN-γ. It was observed that neither of the splenocytes, CD4+T and CD8+T cells, produced significant levels of IL-5 in the coculture assay (Fig. 4A). Likewise, splenocytes and CD8+T cells did not produce significant levels of IL-17 in the coculture system. A significant level of IL-17, however, was produced by CD4+T cells cocultured with MIP(L)-stimulated DCs (Fig. 4B). As expected, splenocytes, CD4+T cells, as well as CD8+T cells produced the significant levels of IFN-γ when cocultured with MIP(L)-stimulated DCs (Fig. 4C). A moderate level of IFN-γ was induced by MIP(K)-stimulated DCs. These results showed that MIP-stimulated DCs induce Th1 and Th17 polarization in naïve T cells.
Figure 4. Polarization of naïve T cells by MIP-stimulated DCs. Allogeneic splenocytes, CD4+T cells, and CD8+T cells were cocultured with MIP(K)- and MIP(L)-stimulated, γ-irradiated DCs in a 96-well round-bottom plate. Culture supernatants were collected after 5 days and analyzed for IL-5, IL-17, and IFN-γ. Neither of the splenocytes, CD4+T cells and CD8+T cells, produced significant levels of IL-5 in the coculture assay (A). MIP(L)-stimulated but not MIP(K)-stimulated DCs induced the significant production of IL-17 by CD4+T cells. Splenocytes or CD8+T cells did not produce significant levels of IL-17 in the coculture (B). MIP(L)-stimulated DCs induced significantly higher production of IFN-γ by splenocytes, CD4+T cells and CD8+T cells. Higher levels of IFN-γ were also produced by splenocytes, CD4+T cells and CD8+T cells, cultured with MIP(K)-stimulated DCs compared with those cultured with unstimulated DCs (C). Mean ± sem of 3 independent experiments is shown. **P < 0.01, and ***P < 0.001.

Production of IL-12p70 by DCs plays the most decisive role in Th1 polarization of naïve T cells. Likewise, IL-23 has been shown to promote Th17 polarization. As MIP induced the Th1 and Th17 polarization potential in DCs, MIP-stimulated DCs were analyzed for IL-12p70 and IL-23 production. It was observed that MIP(L)-stimulated DCs produced a significantly higher level of IL-12p70 compared with unstimulated DCs (Fig. 5A). MIP(K), however, was not effective in inducing IL-12p70 production by DCs. Surprisingly, DCs did not produce IL-23 in response to MIP(K) or MIP(L) (Fig. 5B). LPS-stimulated DCs produced both IL-12p70 and IL-23.
Figure 5. Production of TH-polarizing cytokines by MIP-stimulated DCs. BMDCs were stimulated with MIP(K) and MIP(L) at an MOI of 10, and culture supernatants were analyzed for IL-12-p70 and IL-23 by ELISA. A significant amount of IL-12p70 was produced by MIP(L)-stimulated DCs (A). Comparable amounts of IL-23 were produced by unstimulated and MIP-stimulated DCs (B). LPS, TLR4 ligand (10 ng/ml). Mean ± sem of 2 independent experiments is shown. *P < 0.05.

MIP activates DCs in a MyD88-dependent manner
MyD88 is a common adapter protein used by all TLRs (except TLR3) for intracellular passage of signals initiated with the recognition of microbial molecules. To examine the involvement of MyD88 in MIP-mediated DC activation, BMDCs from WT and MyD88−/− mice were analyzed for their response to MIP. It was observed that the response to MIP(K) and MIP(L) was lost in MyD88−/− DCs. MIP(L)-stimulated MyD88−/− DCs produced significantly reduced levels of IL-6 and IL-12p40 compared with MIP(L)-stimulated WT DCs (Fig. 6A and B). However, levels of IL-6 and IL-12p40, produced by MIP(K)-stimulated MyD88−/− DCs were comparable with those produced by unstimulated DCs.
Figure 6. Role of MyD88 in MIP-induced DC activation. DCs derived from MyD88−/− and WT mice were stimulated with MIP(K) or MIP(L) at an MOI of 10, and culture supernatants were analyzed for IL-6 and IL-12p40. Response to MIP(K) and MIP(L) was lost in MyD88−/− DCs, as evident by a drastically reduced production of IL-6 and IL-12p40 by these cells compared with WT cells (A and B). Pam, Pam3CSK4 (TLR2 agonist; 10 ng/ml). Mean ± sem of 3 independent experiments is shown. **P < 0.01, and ***P < 0.001. Role of MyD88 in MIP-induced DC survival was also analyzed. A significant increase in the level of PI-positive cells was observed in MIP(K)-stimulated MyD88−/− DCs compared with MIP(K)-stimulated WT DCs. DC survival efficacy of MIP(L) was also reduced in MyD88−/− DCs compared with WT DCs (C). Representative data of 2 independent experiments are shown.

It has been previously reported that microbial stimuli promote DC survival in a TLR-dependent manner. As TLR signaling occurs through MyD88, the role of MyD88 in a MIP-mediated increase in DC survival was also analyzed. It was observed that the DC survival efficacy of MIP was drastically reduced in MyD88−/− DCs (Fig. 6C). The level of PI-positive cells in MIP(K)-stimulated MyD88−/− DCs was significantly high compared with that in MIP(K)-stimulated WT DCs. Likewise, nearly 50% increase in the level of PI-positive cells was observed in MIP(L)-stimulated MyD88−/− DCs compared with MIP(L)-stimulated WT DCs. These results showed that MyD88 plays an important role in MIP-induced DC survival.
TLR2 and TLR9 are involved in MIP-induced DC activation
To further delineate the role of different TLRs in DC activation by MIP, DCs derived from TLR knockout mouse strains were analyzed for their response to MIP. It was observed that MIP-stimulated WT and TLR4−/− DCs produced comparable levels of IL-6 (Fig. 7A), suggesting a minimal role of TLR4 in DC activation by MIP. A drastically reduced level of IL-6, however, was produced by MIP(L)-stimulated TLR2−/− DCs compared with MIP(L)-stimulated WT DCs (Fig. 7A). The level of IL-6 produced by MIP(K)-stimulated TLR2−/− DCs was also comparable with that produced by unstimulated DCs. These results provided evidence for a major role of TLR2 in MIP-induced DC activation.
Figure 7. Involvement of TLRs in DC activation by MIP. BMDCs from TLR2−/−, TLR4−/−, and WT mice were stimulated with MIP(K) and MIP(L) at an MOI of 10. A substantially reduced level of IL-6 was produced by MIP(K)- and MIP(L)-stimulated TLR2−/− DCs compared with WT DCs. WT and TLR4−/− DCs produced comparable levels of IL-6 upon stimulation with MIP (A). Mean ± sem of 3 independent experiments is shown. To elucidate the role of TLR9 in DC activation by MIP(L) and MIP(K), BMDCs were stimulated with native ML-DNA or MK-DNA. ML-DNA induced a significant level of IL-6 production by WT DCs. MK-DNA did not lead to IL-6 production by BMDCs. Loss of response to ML-DNA, as indicated by a drastically reduced level of IL-6, was observed in MyD88−/− DCs (B). A significantly reduced level of IL-6 was produced by DNAse I-treated, ML-DNA-stimulated DCs compared with ML-DNA-stimulated DCs (C). Agarose gel electrophoresis of DNA showed that ML-DNA was degraded with DNAse I treatment. Degradation of MK-DNA was also confirmed by agarose gel electrophoresis (D). ML-DNA induced a significantly reduced level of IL-6 in the presence of chloroquine, confirming the role of TLR9 in the recognition of ML-DNA (E). LPS, TLR4 agonist (10 ng/ml); Pam3CSK4, TLR2 agonist (10 ng/ml); CpG, CpG oligonucleotide (TLR9 agonist; 2 µg/ml). Mean ± sem of 3 independent experiments is shown. ***P < 0.001.

To elucidate the role of TLR9 in MIP-induced DC activation, BMDCs were stimulated with MIP genomic DNA. TLR9 recognizes the CpG motifs present in the microbial genomic DNA. It was observed that ML-DNA led to a significantly higher level of IL-6 production by DCs (Fig. 7B). MK-DNA, however, did not result in IL-6 production by BMDCs. Furthermore, the response to ML-DNA was lost in the MyD88−/− DCs, indicating that TLR9 is involved in the recognition of MIP genomic DNA (Fig. 7B). To rule out the possibility of DC activation as a result of unknown heat-labile contaminants in DNA preparation, ML-DNA was treated with DNAse I and subsequently used for DC stimulation. Degradation of DNAse I-treated ML-DNA was confirmed by agarose gel electrophoresis (Fig. 7D). MK-DNA was also found to be degraded into small fragments, indicating that DNA is degraded during the process of autoclaving. A significantly reduced level of IL-6 was produced by DNAse I-treated ML-DNA-stimulated DCs, showing that DNA is the active component in the MIP genomic DNA preparation (Fig. 7C). The role of TLR9 in the recognition of ML-DNA was further ascertained with the help of chloroquine, an inhibitor of the intracellular TLRs. It was observed that IL-6 production by ML-DNA-stimulated DCs was drastically reduced in the presence of chloroquine (Fig. 7E). These results showed that TLR9 is involved in DC activation by MIP(L) but not by MIP(K).
DISCUSSION
DCs and macrophages are the two major cell types that interact with mycobacteria in the host body. Being the most potent APCs, DCs play a decisive role in antimycobacterial and the antitumor immune response. Based on the nature of the encountered pathogens, DCs acquire a differing ability to migrate to draining lymph nodes, produce proinflammatory cytokines, up-regulate costimulatory molecules and present the processed antigens to T cells [22]. The DC response varies even with different Mycobacterium species that can be partly attributed to their specific cell-wall composition [23]. Our results show that MIP leads to the production of proinflammatory cytokines TNF-α, IL-12p40, and IL-6 and up-regulation of costimulatory molecules CD80 and CD86 by DCs. Stimulation with MIP(L) induced a significantly higher response compared with MIP(K). Potent DC activation in response to MIP suggests the presence of immunostimulatory arabinose-capped lipoarabinomannan in the cell wall of MIP. We did not observe a difference in MHCII expression by unstimulated and MIP-stimulated DCs. Down-regulation of MHCII expression on APCs is peculiar to mycobacteria. BCG has been shown to inhibit the expression of MHCII by IL-10-dependent inhibition of Cathepsin S [24].
An interesting observation was that in addition to proinflammatory cytokines, MIP(L) induced a moderate level of IL-10, which is an immunosuppressive cytokine involved in controlling the damage to host tissue that could be caused by an overtly active immune response [20]. Indeed, IL-10 and IL-12 have contradicting effects on the immune system. Production of IL-10 and IL-12 by DCs has also been reported in response to other mycobacteria, including BCG. In one such study, the ratio of IL-10 to IL-12-p40, produced by BCG-stimulated DCs, was much higher than that observed with MIP(L)-stimulated DCs [25]. An excessive amount of IL-10, produced in response to pathogenic mycobacteria, impairs the antigen-presenting ability of DCs, resulting in a suboptimal adaptive-immune response [25].
With the help of TLR2-expressing human embryonic kidney 293 cells, we have shown earlier that MIP(K) possesses a significantly reduced TLR2 agonist activity compared with MIP(L) [26]. Interestingly, TLR2 agonist activity of MIP(K) was restored with its sonication, indicating that TLR2 ligands are masked during the process of autoclaving. Data presented here further show that MIP genomic DNA gets degraded during the autoclaving process, precluding the TLR9 engagement by MIP(K). Degradation or inaccessibility of immunogenic molecules in MIP(K) is therefore responsible for its reduced immunostimulatory nature. Reduced DC activation by MIP(K), however, raises the questions about its immunotherapeutic properties. However, it should be recalled here that MIP(K) induced the DC activation in a dose-dependent manner, which indicates that DCs would produce a substantial level of proinflammatory cytokines and up-regulate the costimulatory molecules at higher MOIs of MIP(K). That MIP(K) is efficacious at a higher dose is also evident from the fact that for human use, the 1st dose of MIP consists of 1.0 × 109 heat-killed bacilli (given i.d.), and subsequent doses consist of 5.0 × 108 bacilli [27]. Furthermore, we have previously analyzed the macrophage response to MIP. It was observed that macrophages produce significant levels of TNF-α, IL-12, and IL-6 in response to MIP(K) [26], suggesting that immunotherapeutic effects of MIP(K) are partly mediated by DCs and macrophages.
The lifespan of DCs serves as a key factor to regulate the intensity of the adaptive-immune response [28]. Inhibition of DC apoptosis with the caspase inhibitor has been shown to result in heightened T cell responses [29]. Components of the immune system, such as RANKL, and microbial stimuli, such as TLR ligands, can increase the survival and lifespan of DCs [30, 31]. We observed enhanced survival of DCs upon stimulation with MIP. In chronic conditions, such as TB, where the immune system is not effective enough to protect against active disease, an increase in DC survival will have favorable effects by increasing potential T cell–DC interactions, resulting in a heightened immune response. This could be one of the mechanisms responsible for a higher antimycobacterial and antitumor response observed in MIP-treated mice. The role of antiapoptotic proteins Bcl-2 and Bcl-xl has been implicated in enhanced DC survival in response to microbial stimuli [31]. Consistent with previous reports, we have also observed an increased expression of Bcl-2 and Bcl-xl in MIP-stimulated DCs.
Depending on the nature of microbial stimuli, DCs can promote a Th1, Th2, or Th17 immune response. The Th1 immune response is indispensable for protection against intracellular pathogens and cancer [32, 33]. Th1 signature cytokine, IFN-γ plays a critical role in macrophage activation and elimination of mycobacteria or lysis of tumor cells. Our results showed that MIP induces the Th1 and Th17 polarization potential in DCs. Both CD4+T cells and CD8+T cells produced substantial levels of IFN-γ in the presence of MIP-stimulated DCs, indicating that MIP promotes classical- as well as cross-presentation of antigens by DCs. Engagement of intracellular TLRs results in enhanced antigen cross-presentation. We observed TLR9 engagement by live MIP DNA and consistent with this, MIP(L)- stimulated DCs induced higher IFN-γ secretion by CD8+ T cells. Furthermore, a high Th1 polarization potential of MIP(L)-stimulated DCs correlates with the higher anti-TB efficacy of MIP(L) compared with MIP(K). A higher immunotherapeutic efficacy of MIP compared with BCG is also explicable from these findings, as BCG-stimulated DCs have been shown to induce IL-10-producing T cells with no Th1 or Th2 bias [34]. CD4+T cells also produced a significant level of IL-17 when cocultured with MIP(L)-stimulated DCs. IL-17 has been shown to play an important role in protection against M.tb in initial stages of infection. The inability of CD4+T cells or CD8+T cells to produce IL-5 in the presence of MIP-stimulated DCs was again in agreement with the beneficial effects of MIP against tumor and TB. IL-5 is a Th2 signature cytokine and is thought to be involved in the pathology of TB. Growing tumors also promote a Th2 type of immune response and take advantage of its homeostatic and tissue-regenerative properties.
Cytokines produced by DCs direct the polarization of naïve T cells. IL-12p70 and IL-23 promote Th1 and Th17 polarization, respectively. A significant amount of IL-12p70 secretion in response to MIP(L) was consistent with a higher Th1-polarization potential of MIP(L)-stimulated DCs. Surprisingly, DCs did not produce a significant level of IL-23 in response to MIP(K) or MIP(L). In fact, it has been noted that IL-23 is not absolutely necessary for Th17 polarization of T cells [35]. A significant level of IL-17 is produced by immune cells from the mediastinal lymph nodes of BCG-immunized and M.tb-challenged IL-12−/−/IL-23−/− mice. Clearly, there exists an IL-23-independent mechanism for IL-17 production in response to BCG, which could be responsible for the Th17 polarization of naïve T cells by MIP-stimulated DCs.
TLRs are the most widely characterized pattern recognition receptors expressed by a variety of immune cells and play a central role in innate and adaptive immunity. MyD88 is a common adapter molecule used by different TLRs. MyD88−/− mouse strains display a profound loss in resistance to M.tb, which is attributed to impaired production of Th1 cytokines and NO [36]. MyD88−/− DCs have been shown to exhibit reduced production of TNF-α, IL-12p40, and IL-6 in response to BCG [37]. An observed loss of response to MIP in MyD88−/− DCs indicated that TLRs play a critical role in MIP-mediated DC activation. Furthermore, MIP-mediated DC survival was drastically reduced in MyD88−/− DCs compared with WT cells. Interestingly, DC survival efficacy of MIP(L) was not completely abrogated in DCs derived from MyD88−/− mice, suggesting that some other mechanisms are also involved in DC survival in response to MIP(L).
Further characterization of TLRs involved in DC activation by MIP highlighted the major role of TLR2. Mycobacteria possess different molecules with a varying TLR2 engagement property. Lipoarabinomannans, lipoteichoic acid, and trehalose dimycolate have been shown to be major TLR2 ligands in different mycobacterial species. Involvement of TLR2 has also been previously observed in macrophage activation by MIP(K) and MIP(L) [26]. Interestingly, our findings showed that TLR9 is involved in DC activation by MIP(L) but not by MIP(K). TLR9 is activated by CpG motifs present in the microbial genomic DNA and promotes a Th1 type of immune response. Lack of TLR9 engagement property in MIP(K) indicated that MIP genomic DNA is degraded during the process of autoclaving, which was confirmed by the agarose gel electrophoresis of MK-DNA.
Our findings provide the insights into DC activation by MIP, delineate the involvement of TLRs in MIP-induced DC activation, and implicate these cells in the immunomodulatory properties exhibited by MIP. Ever since it was selected in the 1970s for its ability to induce a cell-mediated immune response against M. leprae, MIP has come a long way and has been approved as an adjunct to chemotherapy for treatment of non small cell lung cancer recently. Overall, our studies showed that MIP(L) induced a higher DC activation compared with MIP(K). MIP(L) has also been observed to confer higher protection against experimental TB, and studies in the animal model have shown that MIP(L) does not cause any untoward reaction [15]. Based on the present and previous observations, we suggest further evaluation of MIP(L) for immunotherapy of TB and cancer.
AUTHORSHIP
P.K., V.J., and S.M. performed the experiments. P.K., G.D., and S.B. designed the study. P.K. and S.B. prepared the manuscript.
ACKNOWLEDGMENTS
This work was supported by core research grant from the National Institute of Immunology, New Delhi, India.
Glossary
- −/−
deficient
- BCG
bacillus Calmette-Guérin
- BM
bone marrow
- BMDC
bone-marrow-derived dendritic cell
- DC
dendritic cell
- M.tb
Mycobacterium tuberculosis
- MIP
Mycobacterium indicus pranii
- MIP(K)
heat-killed Mycobacterium indicus pranii
- MIP(L)
live Mycobacterium indicus pranii
- MK-DNA
autoclaved Mycobacterium indicus pranii genomic DNA
- ML-DNA
live Mycobacterium indicus pranii genomic DNA
- MOI
multiplicity of infection
- Pam3CSK4
palmitoyl-3-cysteine-serine-lysine-4
- PBS-3
PBS with 3% FBS
- PI
propidium iodide
- RANKL
receptor activator for NF-κB ligand
- TB
tuberculosis
- WT
wild-type
Footnotes
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
DISCLOSURES
The authors declare no commercial or financial conflict of interest.
REFERENCES
- 1.Maldonado-López R., Moser M. (2001) Dendritic cell subsets and the regulation of Th1/Th2 responses. Semin. Immunol. 13, 275–282. [DOI] [PubMed] [Google Scholar]
- 2.Hernández-Pando R., Orozcoe H., Sampieri A., Pavón L., Velasquillo C., Larriva-Sahd J., Alcocer J. M., Madrid M. V. (1996) Correlation between the kinetics of Th1, Th2 cells and pathology in a murine model of experimental pulmonary tuberculosis. Immunology 89, 26–33. [PMC free article] [PubMed] [Google Scholar]
- 3.Winau F., Weber S., Sad S., de Diego J., Hoops S. L., Breiden B., Sandhoff K., Brinkmann V., Kaufmann S. H., Schaible U. E. (2006) Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity 24, 105–117. [DOI] [PubMed] [Google Scholar]
- 4.Deretic V., Singh S., Master S., Harris J., Roberts E., Kyei G., Davis A., de Haro S., Naylor J., Lee H. H., Vergne I. (2006) Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell. Microbiol. 8, 719–727. [DOI] [PubMed] [Google Scholar]
- 5.Geijtenbeek T. B., Van Vliet S. J., Koppel E. A., Sanchez-Hernandez M., Vandenbroucke-Grauls C. M., Appelmelk B., Van Kooyk Y. (2003) Mycobacteria target DC-SIGN to suppress dendritic cell function. J. Exp. Med. 197, 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nouri-Shirazi M., Banchereau J., Bell D., Burkeholder S., Kraus E. T., Davoust J., Palucka K. A. (2000) Dendritic cells capture killed tumor cells and present their antigens to elicit tumor-specific immune responses. J. Immunol. 165, 3797–3803. [DOI] [PubMed] [Google Scholar]
- 7.Whiteside T. L., Vujanovic N. L., Herberman R. B. (1998) Natural killer cells and tumor therapy. Curr. Top. Microbiol. Immunol. 230, 221–244. [DOI] [PubMed] [Google Scholar]
- 8.Klebanoff C. A., Gattinoni L., Restifo N. P. (2006) CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol. Rev. 211, 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palucka K., Banchereau J. (2012) Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 12, 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang L., Carbone D. P. (2004) Tumor-host immune interactions and dendritic cell dysfunction. Adv. Cancer Res. 92, 13–27. [DOI] [PubMed] [Google Scholar]
- 11.Hirohashi Y., Sato N. (2011) Tumor-associated dendritic cells: molecular mechanisms to suppress antitumor immunity. Immunotherapy 3, 945–947. [DOI] [PubMed] [Google Scholar]
- 12.Purswani S., Talwar G. P., Vohra R., Pal R., Panda A. K., Lohiya N. K., Gupta J. C. (2011) Mycobacterium indicus pranii is a potent immunomodulator for a recombinant vaccine against human chorionic gonadotropin. J. Reprod. Immunol. 91, 24–30. [DOI] [PubMed] [Google Scholar]
- 13.Yadava A., Mukherjee R. (1993) An immunodominant 30-kDa antigen of a candidate anti-leprosy vaccine, Mycobacterium w, shares T and B cell determinants with M. leprae and M. tuberculosis. Med. Microbiol. Immunol. (Berl.) 182, 243–253. [DOI] [PubMed] [Google Scholar]
- 14.Saini V., Raghuvanshi S., Khurana J. P., Ahmed N., Hasnain S. E., Tyagi A. K., Tyagi A. K. (2012) Massive gene acquisitions in Mycobacterium indicus pranii provide a perspective on mycobacterial evolution. Nucleic Acids Res. 40, 10832–10850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta A., Ahmad F. J., Ahmad F., Gupta U. D., Natarajan M., Katoch V. M., Bhaskar S. (2012) Protective efficacy of Mycobacterium indicus pranii against tuberculosis and underlying local lung immune responses in guinea pig model. Vaccine 30, 6198–6209. [DOI] [PubMed] [Google Scholar]
- 16.Gupta A., Geetha N., Mani J., Upadhyay P., Katoch V. M., Natrajan M., Gupta U. D., Bhaskar S. (2009) Immunogenicity and protective efficacy of “Mycobacterium w” against Mycobacterium tuberculosis in mice immunized with live versus heat-killed M. w by the aerosol or parenteral route. Infect. Immun. 77, 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmad F., Mani J., Kumar P., Haridas S., Upadhyay P., Bhaskar S. (2011) Activation of anti-tumor immune response and reduction of regulatory T cells with Mycobacterium indicus pranii (MIP) therapy in tumor bearing mice. PLoS ONE 6, e25424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inaba K., Inaba M., Romani N., Aya H., Deguchi M., Ikehara S., Muramatsu S., Steinman R. M. (1992) Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 176, 1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Ayoubi A. M., Zheng H., Liu Y., Bai T., Eblen S. T. (2012) Mitogen-activated protein kinase phosphorylation of splicing factor 45 (SPF45) regulates SPF45 alternative splicing site utilization, proliferation, and cell adhesion. Mol. Cell. Biol. 32, 2880–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Garra A., Vieira P. L., Vieira P., Goldfeld A. E. (2004) IL-10-producing and naturally occurring CD4+ Tregs: limiting collateral damage. J. Clin. Invest. 114, 1372–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Medzhitov R., Janeway C. A. Jr. (1997) Innate immunity: impact on the adaptive immune response. Curr. Opin. Immunol. 9, 4–9. [DOI] [PubMed] [Google Scholar]
- 22.Mellman I., Steinman R. M. (2001) Dendritic cells: specialized and regulated antigen processing machines. Cell 106, 255–258. [DOI] [PubMed] [Google Scholar]
- 23.Cheadle E. J., O’Donnell D., Selby P. J., Jackson A. M. (2005) Closely related mycobacterial strains demonstrate contrasting levels of efficacy as antitumor vaccines and are processed for major histocompatibility complex class I presentation by multiple routes in dendritic cells. Infect. Immun. 73, 784–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sendide K., Deghmane A. E., Pechkovsky D., Av-Gay Y., Talal A., Hmama Z. (2005) Mycobacterium bovis BCG attenuates surface expression of mature class II molecules through IL-10-dependent inhibition of cathepsin S. J. Immunol. 175, 5324–5332. [DOI] [PubMed] [Google Scholar]
- 25.Demangel C., Bertolino P., Britton W. J. (2002) Autocrine IL-10 impairs dendritic cell (DC)-derived immune responses to mycobacterial infection by suppressing DC trafficking to draining lymph nodes and local IL-12 production. Eur. J. Immunol. 32, 994–1002. [DOI] [PubMed] [Google Scholar]
- 26.Kumar P., Tyagi R., Das G., Bhaskar S. (2014) Mycobacterium indicus pranii and Mycobacterium bovis BCG lead to differential macrophage activation in Toll-like receptor-dependent manner. Immunology 143, 258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma P., Kar H. K., Misra R. S., Mukherjee A., Kaur H., Mukherjee R., Rani R. (1999) Induction of lepromin positivity following immuno-chemotherapy with Mycobacterium w vaccine and multidrug therapy and its impact on bacteriological clearance in multibacillary leprosy: report on a hospital-based clinical trial with the candidate antileprosy vaccine. Int. J. Lepr. Other Mycobact. Dis. 67, 259–269. [PubMed] [Google Scholar]
- 28.Chen M., Wang Y. H., Wang Y., Huang L., Sandoval H., Liu Y. J., Wang J. (2006) Dendritic cell apoptosis in the maintenance of immune tolerance. Science 311, 1160–1164. [DOI] [PubMed] [Google Scholar]
- 29.Chen M., Huang L., Wang J. (2007) Deficiency of Bim in dendritic cells contributes to overactivation of lymphocytes and autoimmunity. Blood 109, 4360–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong B. R., Josien R., Lee S. Y., Sauter B., Li H. L., Steinman R. M., Choi Y. (1997) TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J. Exp. Med. 186, 2075–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hou W. S., Van Parijs L. (2004) A Bcl-2-dependent molecular timer regulates the lifespan and immunogenicity of dendritic cells. Nat. Immunol. 5, 583–589. [DOI] [PubMed] [Google Scholar]
- 32.Salgame P. (2005) Host innate and Th1 responses and the bacterial factors that control Mycobacterium tuberculosis infection. Curr. Opin. Immunol. 17, 374–380. [DOI] [PubMed] [Google Scholar]
- 33.Haabeth O. A., Lorvik K. B., Hammarström C., Donaldson I. M., Haraldsen G., Bogen B., Corthay A. (2011) Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat. Commun. 2, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madura Larsen J., Benn C. S., Fillie Y., van der Kleij D., Aaby P., Yazdanbakhsh M. (2007) BCG stimulated dendritic cells induce an interleukin-10 producing T-cell population with no T helper 1 or T helper 2 bias in vitro. Immunology 121, 276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wozniak T. M., Ryan A. A., Britton W. J. (2006) Interleukin-23 restores immunity to Mycobacterium tuberculosis infection in IL-12p40-deficient mice and is not required for the development of IL-17-secreting T cell responses. J. Immunol. 177, 8684–8692. [DOI] [PubMed] [Google Scholar]
- 36.Scanga C. A., Bafica A., Feng C. G., Cheever A. W., Hieny S., Sher A. (2004) MyD88-deficient mice display a profound loss in resistance to Mycobacterium tuberculosis associated with partially impaired Th1 cytokine and nitric oxide synthase 2 expression. Infect. Immun. 72, 2400–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicolle D. M., Pichon X., Bouchot A., Maillet I., Erard F., Akira S., Ryffel B., Quesniaux V. F. (2004) Chronic pneumonia despite adaptive immune response to Mycobacterium bovis BCG in MyD88-deficient mice. Lab. Invest. 84, 1305–1321. [DOI] [PubMed] [Google Scholar]