

NKG2D ligand expression, NK cells, and cytokine expression by NK cells could play a role in the lupus nephritic process.
Keywords: MICA, Rae, MRL/MpJ, MRL/lpr, T-bet, Eomes
Abstract
NK cells are a major component of the immune system, and alterations in their activity are correlated with various autoimmune diseases. In the present work, we observed an increased expression of the NKG2D ligand MICA in SLE patients’ kidneys but not healthy subjects. We also show glomerulus-specific expression of the NKG2D ligands Rae-1 and Mult-1 in various murine SLE models, which correlated with a higher number of glomerular-infiltrating NK cells. As the role of NK cells in the immunopathogenesis of SLE is poorly understood, we explored NK cell differentiation and activity in tissues and organs in SLE-prone murine models by use of diseased and prediseased MRL/MpJ and MRL/lpr mice. We report here that phenotypically iNK cells accumulate only in the spleen but not in BM or kidneys of diseased mice. Infiltrating NK cells in kidneys undergoing a lupus nephritic process showed a more mature, activated phenotype compared with kidney, as well as peripheral NK cells from prediseased mice, as determined by IFN-γ and STAT5 analysis. These findings and the presence of glomerulus-specific NKG2D ligands in lupus-prone mice identify a role for NK cells and NKG2D ligands in the lupus nephritic process, which could aid in understanding their role in human SLE.
Introduction
NK cells are large granular lymphocytes that have a major role in the innate immune system as important in vivo regulators of immune and nonimmune cells [1–3]. NK cells participate in various autoimmune diseases, such as type 1 diabetes and rheumatoid arthritis, and in SLE, a chronic autoimmune disease of unknown origin that can affect various organs, including the lungs, blood, kidneys, and nervous system [4, 5]. Lupus nephritis, 1 of the SLE hallmarks, is characterized by immune complex formation, followed by the accumulation of macrophages, T cells, and B cells in the interstitial infiltrates that amplify the local response and correlate with the severity of the glomerular lesions [6–8].
The role of NK cells in the development of SLE pathogenesis remains unclear and is widely debated. Recent reports from several groups show a significantly lower proportion and total numbers of NK cells in SLE patient blood compared with controls, especially in lupus nephritis patients [9–11]. There also is evidence of reduced cytotoxicity and impaired differentiation of peripheral NK cells from SLE patients and from mouse SLE models, such as MRL/lpr [9, 12, 13].
The role of NKG2D, 1 of the main NK-activating receptors in SLE, is also debated [14, 15]. NKG2D is expressed on all NK cells and on subsets of NKT, CD8, and CD4, and γδ T cells [16]. NKG2D is activated by NKG2D ligands, a “stress-induced” family of MHC-I-like proteins, which in mice, are Rae-1α/β/γ/ε/δ, MULT-1, and H60 and in humans, MICA, MICB, ULBP1-ULBP4, and retinoic acid early transcript 1G protein. The only information available to date concerning NKG2D and its ligands in SLE is a report of a larger proportion of CD4+NKG2D+ cells in PBMCs of SLE patients and higher levels of the human NKG2D MICA ligand in juvenile-onset SLE patient serum, suggesting NKG2D ligand up-regulation in SLE pathogenesis [17, 18]. There are few data regarding the role of NKG2D and its ligands in SLE pathogenesis and etiology or on the functional characterization of NK cells in a SLE environment, specifically in the target organs of this disease.
We used immunohistochemical procedures to analyze NKG2D ligand involvement in nephritic lupus development and observed expression of the human NKG2D ligand MICA in kidneys of SLE patients. We also show glomerulus-specific expression of NKG2D ligands in MRL/MpJ, MRL/lpr, and NZBxNZW(F1) strains compared with healthy C57BL/6J and BALB/c mice. NKG2D ligand expression increased with disease progression and correlated with greater NK cell infiltration in the glomeruli of affected mice, suggesting a role for NKG2D ligands in lupus nephritis development. As NKG2D ligands can activate NK cells, we characterized NK cells phenotypically and functionally in a SLE-like environment in BM, in the periphery (spleen), and in the kidney in MRL/MpJ and MRL/lpr SLE mouse models. In our SLE models, the NK cell population showed down-regulation of key NK cell markers, accumulation of iNK cells, and decreased functional capacity in the periphery but not in kidneys of diseased MRL/MpJ and MRL/lpr mice, where mainly mature and functional NK cells are detected. These findings indicate that NKG2D ligand expression and NK cells could play a role in the lupus nephritic process.
MATERIALS AND METHODS
Mice
Female C57BL/6J mice MRL/MpJ and MRL/lpr (The Jackson Laboratory, Bar Harbor, ME, USA) were maintained in the CNB Animal Facility (Madrid, Spain) in pathogen-free conditions. All animal studies were approved by the CNB Ethics Committee for Animal Experimentation (Ref. 11022) in compliance with national and European Union legislation.
Human kidney samples
Formalin-fixed paraffin sections from 11 patients with lupus nephritis (with active and/or chronic lesions) and 6 sections of human kidneys without disease used as healthy controls (healthy parenchyma of radical nephrectomies) were obtained from FJD Biobank (Biobanco-FJD, IIS-FJD, Madrid, Spain). Patient details are given (Table 1). The study was approved by the Ethical Committee of the FJD IIS-FJD and conducted following institutional guidelines.
TABLE 1.
Lupus nephritis patients’ details
| Patient | Age/sex | Current status | Kidney biopsy | Clinical history | Disease-activity parameters | Treatment |
|---|---|---|---|---|---|---|
| 1 | 44/F | Moderately active/inactive lupus | Lupus nephritis. Stage III (acute/chronic) | SLE diagnosed with focal nephropathy, ANA+, anti-DNA−, anti-Ro−, anti-Sm−, anti-La− | Proteinuria: 359 mg/24 h; lymphocytes: 2500/mm3 (<1500); complement: C3, 89 (90–170); C4, 16 (12–36); ANA: 1/320 (>1/80); α-DNA: −; α-SSA: − | Dezacor/Deflazacor |
| 2 | 21/F | Moderately active lupus | Lupus nephritis (focal proliferative). Stage III (acute) | SLE diagnosed with proliferative lupic nephritis, ANA+, anti-DNA+, anti-Ro+, anti-La+ | Proteinuria: 153 mg/24 h; lymphocytes: 1100/mm3 (<1500); complement: C3, 49 (90–170); C4, 3 (12–36); ANA: 1/1280 (>1/80); α-DNA: 433 UI/ml (>20); α-SSA: 240 U/ml (>10) | Micophenolate mofetil, Prednisone, Dolquine |
| 3 | 34/F | Active lupus | Lupus nephritis. Stage IV–global (acute) | SLE diagnosed with joint, pulmonary, and skin edemas, leukocytosis | Proteinuria: >500 mg/dl; leukocyturia: lymphocytes, 2700/ mm3 (<1500) | None |
| 4 | 48/F | Active lupus | Lupus nephritis. Stage IV–global (chronic) | SLE diagnosed with joint pain and vasculitis, ANA+, anti-DNA−, anti-Ro−, anti-La− | Proteinuria: 1739 mg/24 h; C3: 78 (90–170); C4: 33 (1236); ANA: 1/160 (>180); creatinine: 2.5 mg/dl (0.6–1.3) | Dolquine |
| 5 | 20/F | Active lupus | Lupus nephritis. Stage IV–segmental (acute) | SLE diagnosed with skin injury, joint pain, and serositis, ANA+, anti-DNA+, anti-Ro−, anti-La− | Proteinuria: 1750 mg/24 h; lymphocytes: 2900/ mm3 (<1500) complement: C3, 71 (90–170); C4, 11 (12–36); ANA: 1/640 (>1/80); α-DNA: 124 UI/ml (>20); α-SSA: − | Hydroxy-chloroquine, Azathioprine, Prednisone, Dolquine |
| 6 | 78/M | Active lupus | Lupus nephritis. Stage IV–global (acute) | Tubulointerstitial nephritis | Proteinuria: 6800 mg/24 h; lymphocytes: 2400/mm3 (<1500); creatinine: 7.3 mg/dl (0.6–1.3) | None |
| 7 | 36/F | Active lupus | Lupus nephritis. Stage IV + V–global (acute/chronic) | SLE diagnosed with polyarthritis, joint pain, and generalized edema | Proteinuria: 6718 mg/24 h; lymphocytes: 4600/mm3 (<1500); complement: C3, 47 (90–170); C4, 5 (12–36); ANA: 1/80 (>1/80); α-DNA: 25 UI/ml (>20); α-SSA: − | Micophenolate mofetil, Prednisone |
| 8 | 52/F | Active lupus | Lupus nephritis. Stage IV–global (acute) | SLE-diagnosed nephrotic syndrome | Proteinuria: 1451 mg/24 h; lymphocytes: 2300/ mm3 (<1500); complement: C3, 72 (90–170); C4, 24 (12–36); ANA: +; α−DNA: 431 UI/ml (>20), 29 UI/ml (<7); α-SSA/Ro, 60 kDa: 29 UI/ml (<7); creatinine: 1.9 mg/dl (0.6–1.3) | Myfortic, Prednisone |
| 9 | 65/M | Partial remission | Lupus nephritis. Stage V (chronic) | Nephrotic syndrome | Proteinuria: 3222 mg/24 h; lymphocytes: 4500/mm3 (<1500) | None |
| 10 | 23/F | Moderately active | Lupus nephritis. Stage IV–global (chronic) | Chronic kidney disease, ANA+, anti-DNA+ | Proteinuria: 2500 mg/24 h; creatinine: 1.7 mg/dl (0.61.3) | NSAIDs |
| 11 | 67/F | Partial remission | Lupus nephritis. Stage III + V (chronic) | Nephrotic syndrome with generalized edema | Proteinuria: 10,007 mg/24 h; lymphocytes: 2300/mm3 (<1500); complement: C3, 118 (90–170); C4, 46 (12–36); ANA: 1/80 (>1/80) | Furosemide, Micophenolate mofetil, Prednisone |
ANA, antinuclear antibodies; anti-La (anti-SSB), anti-Sjögren's syndrome antigen B antibodies; anti-Ro/α-SSA, anti-Sjögren's syndrome antigen A antibodies; anti-Sm, anti-Smith antibody; NSAIDs, non-steroidal anti-inflammatory drugs; α−, anti.
Immunohistochemistry and immunofluorescence antibodies and reagents
Antibodies to the following antigens were used for immunohistochemistry and immunofluorescence stainings: NKp46 (AF2225), Rae-1γ (AF1136), and MICA (BAF1300; all from R&D Systems, Minneapolis, MN, USA); ULBP1 (NBP1-80856; Novus Biologicals, Littleton, CO, USA); and Synaptopodin (163-002; Synaptic Systems, Goettingen, Germany). Rat anti-mouse Mult-1 was a kind gift from Dr. Stipan Joncic (University of Rijeka, Croatia) [19], aged NZBxNZW(F1) OCT-embedded kidney tissue sections were a kind gift from Dr. Shozo Izui (University of Geneva, Switzerland), and 3-mo-old female BALB/c kidney tissue sections were a kind gift from Dr. Manuela Zonca (CNB).
Immunohistochemistry and confocal microscopy
Spleens and kidneys were removed and snap frozen in tissue-freezing medium (Jung). Sections were acetone fixed and after blocking endogenous peroxidase, incubated with primary antibody, followed by rabbit EnVision+ System-HRP reagent (Dako, Glostrup, Denmark) or rat or goat Histofine Simple Stain kits (Nichirei Biosciences, Tokyo, Japan). Sections were stained with AEC+ Substrate-Chromogen (Dako) and hematoxylin counterstained. HRP-conjugated polymer-stained sections and control isotype-incubated slides were used as negative controls.
To ascertain if NKG2D ligands were also present in the kidneys of diseased SLE patients, we performed specific immunohistochemical staining for the presence of the NKG2D ligands MICA and ULBP1 in formalin-fixed paraffin sections of 11 patients with a diagnosis of lupus nephritis, Classes II–V, with active and/or chronic lesions. As healthy controls, formalin-fixed paraffin sections of healthy parenchyma of radical nephrectomies were used.
Paraffin-embedded sections or renal biopsies from patients with lupus nephritis and human kidney controls were deparaffinized and rehydrated and washed in TBS 1×, and heat-induced antigen retrieval was performed in a water steamer for 30 min. Sections were washed, endogenous peroxidase was blocked, and slides were incubated overnight with primary antibody, followed by rabbit EnVision+ System-HRP reagent or the Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA, USA). Sections were stained with AEC+ and hematoxylin counterstained. HRP-conjugated, polymer-stained sections and control isotype-incubated slides were used as negative controls. Immunohistochemical staining for MICA and ULBP1 was analyzed with the use of an Olympus BX-45 microscope, and the intensity of staining was graded, ranging from 0 through 3+ (0, no staining; 1+, mild staining; 2+, moderate staining; 3+, strong staining).
Confocal analysis was performed on a Leica SP5 confocal microscope. Whole-tissue section pictures were analyzed via immunofluorescence by use of a Leica DMI6000 B inverted microscope and the Leica Application Suite microscope software to create a full, processed image. All samples include appropriate antibody-staining controls.
Quantification of Rae-1 staining intensity in glomeruli of glomerular infiltrates
Chromogen deposition was measured by quantitative immunohistochemistry by use of an established method [20]. In brief, images of glomeruli (×100 magnification) were acquired in a Leica microscope (vertical Leitz DM RB) with an adapted Olympus (DP70) camera; image files were saved in a tagged-image file format. The amount of chromogen/pixel was determined by selecting glomeruli (25 glomeruli/group) in a 200 × 200 pixel region and subtracting the mathematical energy (EM) of the control slide (not exposed to primary antibody) from that of a homologous glomerulus on the experimental slide (exposed to Rae-1 antibody). Chromogen quantity (EM) is expressed as energy units/pixels. To quantify the percentage of glomeruli with NKp46+ infiltration, 25 random field sections were analyzed for 5 mice/group. In each section, the number of glomeruli that showed at least 1 positive-stained cell inside of the glomerulus was counted and divided by the total number of glomeruli counted/field.
Cell isolation
Single-cell suspensions were prepared from mouse spleen and BM (tibiae and femur). To obtain kidney lymphocytes, decapsulated kidneys were minced, digested with 10 μg/ml collagenase D (Roche Diagnostics, Indianapolis, IN, USA), passed through a cell strainer, washed, overlaid on Ficoll (GE Healthcare, Pittsburgh, PA, USA), and centrifuged. Kidney lymphocytes were isolated from the interface, washed, and counted, and the absolute number of NK cells was calculated by multiplying the total number of kidney lymphocytes by the percentage of positive NK cells (live CD45+CD3−NKp46+ cells), as determined by flow cytometry.
Flow cytometric analysis
Antibodies to the following antigens were used for staining: B220 (RA3-6B2; Beckman Coulter, Brea, CA, USA); CD4 (GK1.5), CD8 (53-6.7), CD3 (145-2C11), NKG2D (A10), Eomes (Dan11mag), and T-bet (4B10; all from eBioscience, San Diego, CA, USA); Ly-6G/Ly-6C (RB6-8C5) and NKp46 (29A1.4; both from BioLegend, San Diego, Ca, USA); CD2 (12-15; Southern Biotechnology, Birmingham, AL, USA); CD11a (2D7), CD11b (3A33), CD27 (LG.3A10), CD43 (S7), CD49b (DX5), CD51 (RMV-7), CD107a (1D4B), CD122 (5H4), CD45 (I3/2.3), IFN-γ (XMG1.2), TER119 (TER119), TNF-α (MP6-XT22), and pSTAT5 (pY694; all from BD Biosciences, San Jose, CA, USA); and CXCR3 (220803) and CXCR4 (247506; both from R&D Systems). Dead cells were distinguished by use of the Live/Dead Fixable Dead Cell Stain kit (Invitrogen, Carlsbad, CA, USA).
NKP cells were gated on the CD122+LIN− (lineage) gate, which included the following antibodies: B220, CD3, CD11b, Ly-6G/Ly-6C, and TER119.
Cells were stained for Eomes and T-bet following the forkhead box p3 staining kit (eBioscience). For intracellular IFN-γ and TNF-α staining, cells were cultured in medium containing PMA (25 ng/ml; Sigma, St. Louis, MO, USA) and ionomycin (1 μg/ml; Sigma) or with murine rIL-15 (20 mM) and rIL-12 (10 ng/ml; both from PeproTech, Rocky Hill, NJ, USA), and BFA (5 μg/ml; BioLegend) was added for the last 4 h at 37°C, permeabilized with 0.5% saponin, and stained for IFN-γ and TNF-α (30 min, room temperature). For pSTAT5 staining, cells were incubated (15 min) in medium alone or with 20 nM murine rIL-15 (PeproTech); cells were then fixed, permeabilized with BD Phosphflow Perm Buffer III (BD Biosciences), and stained for pSTAT5, according to the manufacturer’s instructions. Stained cells were analyzed in a Gallios (Beckman Coulter) flow cytometer and data analyzed with FlowJo software, v. 8.0 (TreeStar, Ashland, OR, USA).
Total organ ELISA
Mice were killed and the kidneys weighed and finely minced. The preparations were plated in 2 ml RPMI, supplemented with 10% FBS, 4 mM L-glutamine, 10 mM HEPES, 1% 100× nonessential amino acids, 1 mM sodium pyruvate, 100 U/ml penicillin/100 μg/ml streptomycin, and 50 nM 2-mercaptoethanol for 24 h at 37°C, 5% CO2. Supernatants were collected, spun down 3 times to remove cellular debris, and analyzed by ELISA, at least in triplicates, for the presence of IL-15/IL-15R (mouse IL-15/IL-15R complex ELISA Ready-SET-Go!; eBioscience), CX3CL1 (mouse CX3CL1/Fractalkine DuoSet; R&D Systems), IL-12 [BD OptEIA Mouse IL-12 (p70) ELISA set] and IFN-γ (BD OptEIA Mouse IFN-γ ELISA set; both BD Biosciences), and TNF-α (MiniELISA development kit; PeproTech). The final concentration of cytokine/chemokine was then normalized to the total organ weight.
RESULTS
The human NKG2D ligand MICA is expressed in kidneys of patients with lupus nephritis
NKG2D ligand involvement in SLE pathogenesis, particularly in the glomerulonephritic process, has not been studied. Several genetic studies have shown an association between MICA gene polymorphisms and susceptibility to SLE, suggesting that this human NKG2D ligand could contribute to the pathogenesis of SLE [21, 22]. To ascertain if NKG2D ligands were present in the kidneys of diseased SLE patients, we performed specific immunohistochemistry staining for the presence of the NKG2D ligands MICA and ULBP1 in formalin-fixed paraffin sections of 11 patients with a diagnosis of lupus nephritis, Classes II–V, with active and/or chronic lesions (Table 1). Formalin-fixed paraffin sections of healthy renal parenchyma from 6 nonlupus patients that had undergone radical nephrectomy were used as controls. The analysis of kidney biopsies from patients with lupus nephritis revealed focal-positive cytoplasmic staining for MICA (of variable intensity) in nonatrophic tubular epithelial cells of 8 out of 11 patients, whereas slight MICA staining was observed in only 1 control sample (P < 0.05 Mann-Whitney test on MICA score staining, lupus nephritis case vs. control; Fig. 1, Table 2, and Supplemental Figs. 1 and 2). In cases of mild positivity (1+ and 2+), staining was found predominantly restricted to the basal region of tubular epithelial cells. One-half of these 8 patients also showed focal and segmental glomerular immunoreactivity in subepithelial regions of glomerular capillaries. Patient 2 had focal lupus nephritis with active lesions (Class III-A), Patient 3 was affected by global diffuse lupus nephritis (Class IV-G) and showed active lesions, Patient 9 suffered membranous lupus nephritis (Class V), and Patient 11 showed global diffuse lupus nephritis coexisting with membranous lupus nephritis (Class III + V).
Figure 1. Quantification of NKG2D ligand expression in kidneys of patients with lupus nephritis. (A) Immunohistochemical analysis of MICA and ULBP1 expression in kidney biopsies from patients with lupus nephritis and healthy kidneys used as controls. Representative images are shown (×200). Neg Ct, Negative controls. (B) MICA intensity scores were plotted in control and lupus nephritis cases. *P < 0.05 Mann-Whitney test (control vs. lupus nephritis cases).

TABLE 2.
Quantitative analysis of the intensity for MICA by use of Grades 0–3+
| Patient | Type of nephropathy | ULBP1 | MICA |
|---|---|---|---|
| 1 | Stage III (acute/chronic) | – | 1+ |
| 2 | Stage III (acute) | – | 2+ |
| 3 | Stage IV–global (acute) | – | 3+ |
| 4 | Stage IV–global (chronic) | – | 3+ |
| 5 | Stage IV–segmental (acute) | – | 1+ |
| 6 | Stage IV–global (acute) | – | 0 (+/−) |
| 7 | Stage IV + V (acute/chronic) | – | 0 |
| 8 | Stage IV–global (acute) | – | 1+ |
| 9 | Stage V (chronic) | – | 2+ |
| 10 | Stage IV–global (chronic) | – | 0 (+/−) |
| 11 | Stage II + V (chronic) | – | 1+ |
0, No staining; 1+, mild staining; 2+, moderate staining; 3+, strong staining.
Immunoreactivity for ULBP1 was only found in the cell membrane of scattered erythrocytes of all patients and was considered as a nonspecific finding. No interstitial, tubular, glomerular, or vascular staining was found (Fig. 1A and Supplemental Fig. 1).
Rae-1 expression in glomeruli of murine SLE models
To assess whether the MRL lupus-prone genetic background also causes NKG2D ligand up-regulation and thus, participates in glomerulonephritis onset, we tested NKG2D ligand expression in the MRL mouse kidney. C57BL/6 mice were used as a genotype-negative control strain, in which no ligand expression was anticipated.
We assessed antibodies to various NKG2D ligands for validity in immunohistochemistry, including H60, Rae-1, and Mult-1, but only the Rae-1γ antibody stained appropriately. Rae-1γ stained clearly in glomeruli of prediseased and diseased MRL/MpJ and MRL/lpr mice but not in equivalent C57BL/6 controls (Fig. 2A). Rae-1γ staining was significantly more intense in glomeruli of diseased than of prediseased MRL/lpr and MRL/MpJ mice (Fig. 2B).
Figure 2. Analysis of Rae-1γ and Mult-1 NKG2D ligands in murine SLE models. (A) Immunohistochemical analysis of Rae-1γ expression in kidney-tissue cryosections from MRL/MpJ (prediseased and diseased), MRL/lpr (prediseased and diseased), C57BL/6, BALB/c, and NZBxNZW(F1) (diseased) mice. Specific staining (red chromogen stains) was observed in the glomeruli of all MRL genotype and diseased NZBxNZW(F1) mice, whereas staining was absent in C57BL/6 and BALB/c controls. Representative images of kidneys from 5 mice/group. (B) Quantification of Rae-1γ staining in MRL mouse glomeruli. Data were analyzed with a 2-tailed, unpaired Student’s t-test (mean ± sd; n = 5/group; **P < 0.0001; *P < 0.0025). (C) Cryosections of diseased MRL/lpr kidney (10 μm) were costained with rabbit anti-mouse synaptopodin (green) and goat anti-mouse Rae-1γ antibody (red), followed by appropriate Alexa-conjugated secondary antibodies and DAPI (blue). All synaptopodin-positive cells corresponded to Rae-1γ-positive cells, indicating glomerulus-specific expression of this NKG2D ligand. (D) Confocal analysis of synaptopodin and Mult-1 expression in C57BL/6 and diseased MRL/lpr kidney sections. Kidney cryosections were stained with rabbit anti-mouse synaptopodin (red) and rat anti-mouse Mult-1 (green), followed by secondary antibodies and DAPI (blue). Mult-1 expression was absent in the C57BL/6 strain.

To determine whether Rae-1γ is expressed in glomeruli of other SLE-like, disease-prone mice and not in healthy strains, we analyzed Rae-1γ in kidney sections of diseased NZBxNZW(F1) (another SLE-like murine model) and healthy, 3-mo-old BALB/c mice. NZBxNZW(F1) mice showed Rae-1γ expression in glomeruli, whereas no Rae-1γ was observed in the glomeruli of BALB/c mice (Fig. 2A).
Rae-1γ and Mult-1 expression is limited and specific to glomeruli of the MRL genotype
To confirm specific Rae-1γ expression in glomeruli and to show that all glomeruli expressed this NKG2D ligand, we stained whole kidney sections for Rae-1γ and synaptopodin, a differentiated glomerular cell marker [23, 24]. Rae-1γ expression was specific to synaptopodin-expressing cells (Fig. 2C), and all glomeruli were Rae-1γ positive.
We evaluated the expression of other NKG2D ligands on the MRL background. With the use of an anti-Mult-1 antibody of known efficiency in immunofluorescence analysis, we tested kidney sections from MRL and C57BL/6 mice [19]. Confocal analysis of cryosections showed strong glomerulus-specific Mult-1 expression in MRL but not in C57BL/6 mice (Fig. 2D). No anti-H60 antibodies tested showed positive staining (not shown).
NKp46 infiltration in MRL and C57BL/6 mouse glomeruli
To ascertain whether NK cell infiltration correlates with glomerular infiltrates, we used immunohistochemical techniques to analyze the percentage of glomeruli that showed NK cell (NKp46+) infiltration. Glomeruli of diseased MRL/lpr and MRL/MpJ mice showed significantly more NKp46+ cell infiltrates than prediseased MRL/MpJ and MRL/lpr mice (Fig. 3). These data suggest that NKG2D ligand expression and NK cell infiltration could play a role in lupus nephritis.
Figure 3. Quantification of NKp46 infiltrates in glomeruli. (A) Representative images of immunohistochemical staining of NK (NKp46). Positive staining inside glomeruli (arrows). (B) Quantification of the percentage of glomeruli with cell infiltrates/field (×100). Data were analyzed with a 2-tailed, unpaired Student’s t-test (mean ± sd; n = 5/group; 25 kidney sections/mouse; **P < 0.0001).

NK cell characterization in the MRL mouse strain
As NK cell participation in SLE is poorly understood, we decided to characterize NK cell populations at the site of precursor generation (BM), in the periphery (spleen), and in a target organ of the disease (kidney) in MRL murine models.
As there is only limited information on NK cells in a SLE environment, we characterized the NK cell population in BM and spleen from prediseased and diseased MRL/MpJ and MRL/lpr mice. We analyzed the percentage and total number of NK cells (gated on the NKp46+CD3− cell population by use of the gating strategy shown in Fig. 4A in prediseased MRL/lpr (9-wk-old), diseased MRL/lpr (3- to 4-mo-old), prediseased MRL/MpJ (3- to 4-mo-old), and diseased MRL/MpJ (1-yr-old) mice.
Figure 4. Phenotypic characterization of freshly isolated NK cells from mouse spleen and BM. (A) For all FACS analysis presented herein, cells were gated by doublet discrimination, followed by Live/Dead discrimination, CD45 positivity, and finally, by debris discrimination. Cells were then gated on the CD3−NKp46+ subset for NK cell-subset analysis. FSC (A) and (W), Forward-scatter area and width, respectively; SSC, side-scatter. (B and C) NK cells were gated as CD45+NKp46+CD3−, and percentages and absolute numbers of NK cells were calculated in fresh spleen (B) and BM (C). (D and E) Gates for each surface marker were set by use of unstained or nonspecific mAb isotype controls; a representative staining of each marker is shown. The median fluorescence intensity for each marker was analyzed in spleen (D) and BM (E). Data were analyzed with a 2-tailed, unpaired Student’s t-test (mean ± sd; n = 4–8 mice/group in 3–6 independent experiments; *P < 0.05).

We found a significant reduction in the percentage of spleen NK cells in diseased compared with prediseased mice in both groups (Fig. 4B). Diseased MRL/MpJ mice showed significantly lower total spleen NK cell numbers compared with prediseased MRL/MpJ mice (Fig. 4B). There were no differences in total numbers or percentage of NK cells in BM (Fig. 4C).
Surface-marker expression in NK cells
To determine whether the SLE-like environment affects NK cell maturation, we analyzed expression of developmental markers in fresh NK cells from BM and spleen. We focused on markers that characterize phenotypically mature murine NK cells, including CD122 (IL-2R and IL-15R β-chain), NKG2D, CD49b (α2 integrin), CD11b (Mac-1), CD2 (LFA-2), CD11a (LFA-1), and CD43 (leukosialin) [25, 26].
CD2, CD11b, CD43, CD49b, and CD122 were down-regulated in spleen NK cells in diseased compared with prediseased NK cells in both groups (Fig. 4D). In BM, only CD43 down-regulation was observed in diseased compared with prediseased MRL/MpJ mice (Fig. 4E). NKG2D and CD11a levels were unchanged in BM and spleen NK cells in both mouse groups (Fig. 4D and E).
iNK cells accumulate in the spleen of diseased, SLE-like mice
Based on the observed decrease in mature NK cell markers on mature peripheral NK cells, we postulated a defect in NK cell differentiation in the periphery. NKP cells are thought to arise in the BM; they can differentiate fully and mature in the BM or travel to other organs and complete differentiation in situ. NKP cells have been reported in peripheral organs of mice, including spleen [27]. NK cell differentiation is a complex process that begins in the BM with the generation of NKP cells, which are CD122+LIN−CD49b−NKp46−. These cells follow a developmental pattern that gives rise to iNK cells when they begin to express some but not all NK-specific developmental markers: CD122+LIN−CD49b−NKp46− (Stage 1) → CD122+LIN−NKp46+CD49b− (Stage 2) → CD122+LIN−NKp46+CD49b+ (Stage 3) [26–29]. After Stage 3, NK cells begin to express the NK cell marker CD11b and become CD122+LIN−NKp46+CD49b+CD11b+ (Stage 4), considered mature NK cells. Analysis of total NKP and iNK cell numbers in prediseased and diseased MRL/lpr and MRL/MpJ mice showed an increase in Stage 1 and Stage 2 cell numbers in the spleen but not in the BM of diseased MRL/lpr and MRL/MpJ mice compared with prediseased counterparts (Fig. 5A and B). Stage 3 cells were increased only in the spleens of diseased MRL/MpJ but not MRL/lpr mice compared with prediseased counterparts (Fig. 5A and B).
Figure 5. Characterization of iNK and CD11bCD27 NK cell subsets from mouse spleen and BM. (A) Representative flow cytometric density plots show the proportion of CD122+LIN− cells. Plots show proportions of Stage 1 (CD122+LIN−NKp46−CD49b−), Stage 2 (CD122+LIN−NKp46+CD49b−), and Stage 3 (CD122+LIN−NKp46+CD49b+) iNK cell groups in spleen and BM. (B) Absolute numbers were calculated for Stages 1–3 iNK cell groups. (C) Representative flow cytometric density plots show the proportion of CD11bCD27 NK cell subsets in spleen and BM from all 4 mouse groups. (D) Percentages of CD11bHighCD27Low, CD11bHighCD27High, and CD11bLowCD27High cells were calculated for splenocytes and BM cells. Gates for each population were set by use of unstained or nonspecific mAb isotype controls. Data were analyzed with a 2-tailed, unpaired Student’s t-test (mean ± sd; n = 4–8 mice/group in 3–6 independent experiments; **P < 0.01; *P < 0.05).

CD11bHighCD27Low NK populations are greatly reduced in diseased, SLE-like mice
NK cell maturation can be subdivided further into another 4-stage process, dependent on CD27 and CD11b expression [28]. NK cell effector functions correlate with the progressive acquisition of the following markers in mature NK cells: CD11bLowCD27Low → CD11bLowCD27High → CD11bHighCD27High → CD11bHighCD27Low.
Freshly isolated BM and spleen cells were gated in NKp46+ CD3− and analyzed for CD11b and CD27 expression. We found a significant reduction in CD11bHighCD27Low NK cells and an increase in CD11bLowCD27High NK cells in spleens from diseased MRL/lpr and MRL/MpJ compared with their prediseased counterparts (Fig. 5C and D). There were no significant differences in CD11bCD27 subsets in BM NK cells.
Phenotypic characterization of infiltrating kidney NK cells
Given our finding of major NK cell defects in diseased, SLE-like mouse spleens, we studied NK cells in the kidneys of these mice. Percentages and absolute numbers of CD45+NKp46+CD3− cells were calculated in the kidneys of all mouse groups. The absolute number of infiltrating CD45+NKp46+CD3− cells was higher in diseased than in prediseased MRL/lpr mice but not in diseased MRL/MpJ mice compared with prediseased counterparts (Fig. 6A and B). The percentage of CD45+NKp46+CD3− cells was significantly lower in diseased MRL/MpJ and MRL/lpr mice compared with their prediseased counterparts (Fig. 6A and B). The decrease in the percentage of CD45+NKp46+CD3− cells was concomitant with an increase in other lymphoid subsets, such as CD45+CD3+CD4+ cells (not shown).
Figure 6. Phenotypic characterization of freshly isolated Ficoll-isolated kidney lymphocytes. (A) Representative flow cytometric density plots show the proportion of NK cells in the 4 mouse groups. (B) Cumulative data representative of the mean percentage and total number of CD45+NKp46+CD3− cells in all 4 groups. (C) NK cells were gated as CD45+NKp46+CD3− and stained for various NK cell markers. Gates were set by use of unstained or nonspecific mAb isotype controls; a representative staining of each marker is shown. The median fluorescence intensity for each marker was analyzed. (D) NK cells were analyzed for CD11bCD27 subsets, and the percentages of CD11bHighCD27Low, CD11bHighCD27High, and CD11bLowCD27High cells were calculated. Gates for each population were set by use of unstained or nonspecific mAb isotype controls. Data were analyzed with a 2-tailed, unpaired Student’s t-test (mean ± sd; n = 4–6 mice/group in 3–4 independent experiments; **P < 0.01; *P < 0.05).

In contrast to spleen and BM, CD45+NKp46+CD3−-gated cells in diseased MRL/MpJ and MRL/lpr mouse kidneys showed significantly higher CD11b levels than their prediseased counterparts (Fig. 6C). CD43 was also increased in diseased compared with prediseased MRL/lpr mice (Fig. 6C). There were no differences in NKG2D, CD122, CD2, or CD49b expression in any of the mouse groups (Fig. 6C).
The CD11b expression increase in diseased MRL/lpr and MRL/MpJ mice correlated with significantly larger percentages of the more mature CD11bHighCD27Low cells than the other CD11bCD27 subsets (Fig. 6D). We also observed a tendency toward a larger percentage of CD11bLowCD27High NK cells in prediseased compared with diseased mice (Fig. 6D). To our knowledge, this is the first characterization of CD11bCD27 NK subsets in kidney. This analysis shows that regardless of disease state, kidney NK cells are comprised mainly of more mature CD11bHighCD27Low subsets.
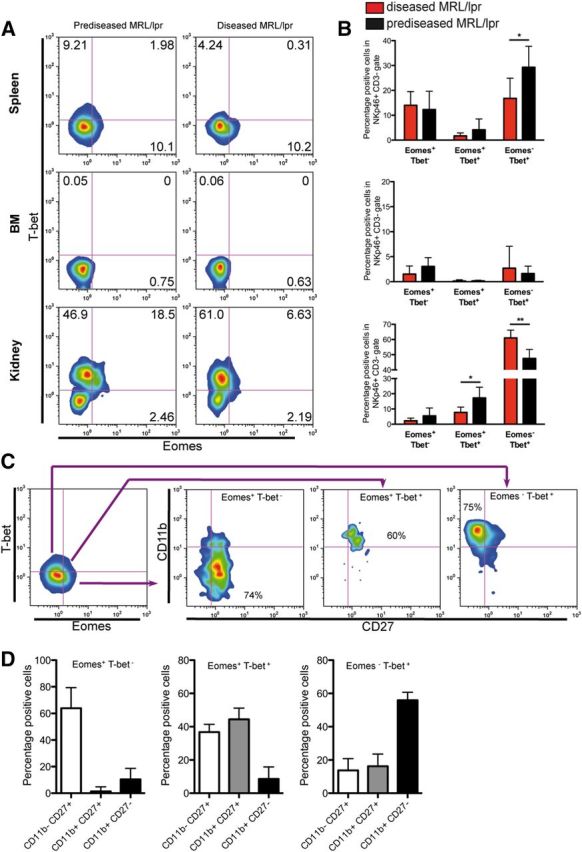
We next analyzed the expression of the transcription factors T-bet and Eomes in NK cells obtained from spleen, BM, and kidney of MRL/lpr mice (Fig. 7A and B). These transcription factors have been linked with NK cell development and maturation [30, 31]. Two main subsets of NK cells regarding the expression of these transcription factors were observed in spleen: Eomes+ T-bet− NK cells, in which the percentage did not vary with disease stage, and Eomes− T-bet+ NK cells, in which the percentage decreased in diseased MRL/lpr mice. Expression of both transcription factors in NK cells was low in the BM. In kidneys, most NK cells were T-bet+ in accordance with their more mature phenotype. We detected an increased population of Eomes+ T-bet+ NK cells in prediseased mice in this organ, whereas Eomes− T-bet+ NK cells were increased in diseased animals. We also examined the expression of CD11b and CD27 in the different NK cell subsets expressing Eomes and T-bet (Fig. 7C and D). We found in spleen (not shown) and in kidneys that expression of CD11b and CD27 closely matched that of T-bet and Eomes, respectively. Most Eomes+ T-bet− NK cells were also CD27highCD11blow, most Eomes+ T-bet+ NK cells were CD27highCD11blow/high, and most Eomes− T-bet+ cells were CD27lowCD11bhigh. Taken together, the analysis of T-bet and Eomes expression confirmed a decrease maturation of NK cells in the spleen concomitantly with a more mature phenotype in the kidneys of diseased animals. In addition, CXCR4 expression was decreased in the diseased kidney, confirming the more mature phenotype of these cells (Supplemental Fig. 3) [32].
Figure 7. Expression of T-bet and Eomes in NK cells from MRL/lpr mice. Lymphocytes purified from spleen, BM and kidney were analyzed for the expression of T-bet and Eomes by flow cytometry. (A) Representative density plot of NKp46+CD3− cells stained for T-bet and Eomes. (B) NK cells were analyzed for Eomes and T-bet subsets, and the percentage of Eomes+T-bet−, Eomes+T-bet+, and Eomes−T-bet+ cells was calculated. (C) Representative staining for CD27 and CD11b in T-bet/Eomes NK cell subsets in spleen from MRL/lpr mice. (D) The percentage of CD11bCD27 subsets in Eomes+T-bet−, Eomes+T-bet+, and Eomes−T-bet+ NK cell subsets in kidneys was calculated. Similar results were obtained in spleen. Data were analyzed by use of a Mann-Whitney test (diseased vs. prediseased); *P < 0.05; **P < 0.01. Data are presented as mean ± sd of 3 independent experiments (n = 7 mice/group).
NK cells in diseased kidneys produce more IFN-γ
To determine whether kidney-infiltrating NK cells behave differently in an autoimmune and a nonautoimmune environment, we tested their ability to produce IFN-γ after PMA/ionomycin stimulation. Kidney NK cells from diseased mice had a larger percentage of PMA/ionomycin-responding cells that secreted more IFN-γ than their prediseased counterparts. The difference in the percentage of IFN-γ producers was more pronounced in CD11bHighCD27Low but not in the CD27High cells (Fig. 8A and B). When kidney NK cells from MRL/lpr mice were treated with IL-12 and IL-15, which induces the expression of IFN-γ more specifically than the PMA/ionomycin treatment, we observed similar levels of IFN-γ production in diseased and prediseased mice (Fig. 8C and D). No significant production of TNF-α was detected in NK cells (data not shown).
Figure 8. IFN-γ secretion and pSTAT5 of Ficoll-isolated kidney lymphocytes. (A) Ficoll-purified kidney lymphocytes were incubated with PMA, ionomycin, and BFA and stained with anti-NKp46, -CD45, -CD3, -CD11b, -CD27, and -IFN-γ mAb (4 h). Representative flow cytometric density plots show the percentage of gated CD45+NKp46+CD3− and CD45+NKp46+CD3−CD11b+CD27− cells positive for IFN-γ. (B) Percentages of CD11bCD27 subsets positive for IFN-γ, and the median fluorescence intensity (MFI) of IFN-γ in NK cells were calculated. (C) As in A, lymphocytes from MRL/lpr mice were stimulated with IL-12 and IL-15 for 6 h, and BFA added in the last 4 h of incubation. Cells were stained as in A. Representative flow cytometry density plots are shown. (D) Percentage of IFN-γ−positive cells in total NKp46+ CD3− and in CD11bCD27 NK subsets and median fluorescence intensity of IFN-γ+ NK cells was calculated. (E) Fresh single-cell suspensions of spleen, BM, and Ficoll-isolated kidney lymphocytes were activated with murine rIL-15 (15 min), fixed, permeabilized, and stained with anti-NKp46, -CD45, -CD3, and -pSTAT5 mAb. Representative flow cytometric histograms show pSTAT5 expression on gated CD45+ NK cells for diseased MRL/MpJ (blue line), prediseased MRL/MpJ (green), diseased MRL/lpr (red), and prediseased MRL/lpr (black) mice. Isotype control staining (gray) is shown in each panel. (F) NK cells were gated for CD45+NKp46+CD3− and the median fluorescence intensity calculated for total pSTAT5 expression in BM, spleen, and kidney. Gates for each population were set by use of unstained or nonspecific mAb isotype controls. Data were analyzed with a 2-tailed, unpaired Student’s t-test (mean ± sd; n = 3–6 mice/group in 3–4 independent experiments; *P < 0.05).

Organ-specific pSTAT5 induction in NK cells
As NK cells suffer from impaired maturation in diseased mouse spleen but not in the BM or kidney, we postulated an alteration in the pSTAT5 pathway in diseased NK cells. STAT5 is a member of the STAT family of proteins, which signal via the JAK/STAT pathway. STAT5 is central to modulation of the biologic response to cytokines, such as IL-15, and pSTAT levels were correlated recently to SLE activity [33, 34]. To determine whether NK cells from these organs have a differential capacity to phosphorylate STAT5, we used IL-15 to stimulate fresh kidney lymphocytes, as well as spleen and BM cells from prediseased and diseased MRL/lpr and MRL/MpJ mice. FACS analysis of the BM and spleen showed no differences between groups in pSTAT5 expression levels after IL-15 stimulation (Fig. 8E and F). In contrast, after IL-15 stimulation, kidney NK cells from diseased MRL/MpJ and MRL/lpr mice showed significantly higher pSTAT5 levels than their prediseased counterparts (Fig. 8E and F). Furthermore, kidney NK cells phosphorylated more STAT5 than their spleen and BM counterparts (Fig. 8E and F).
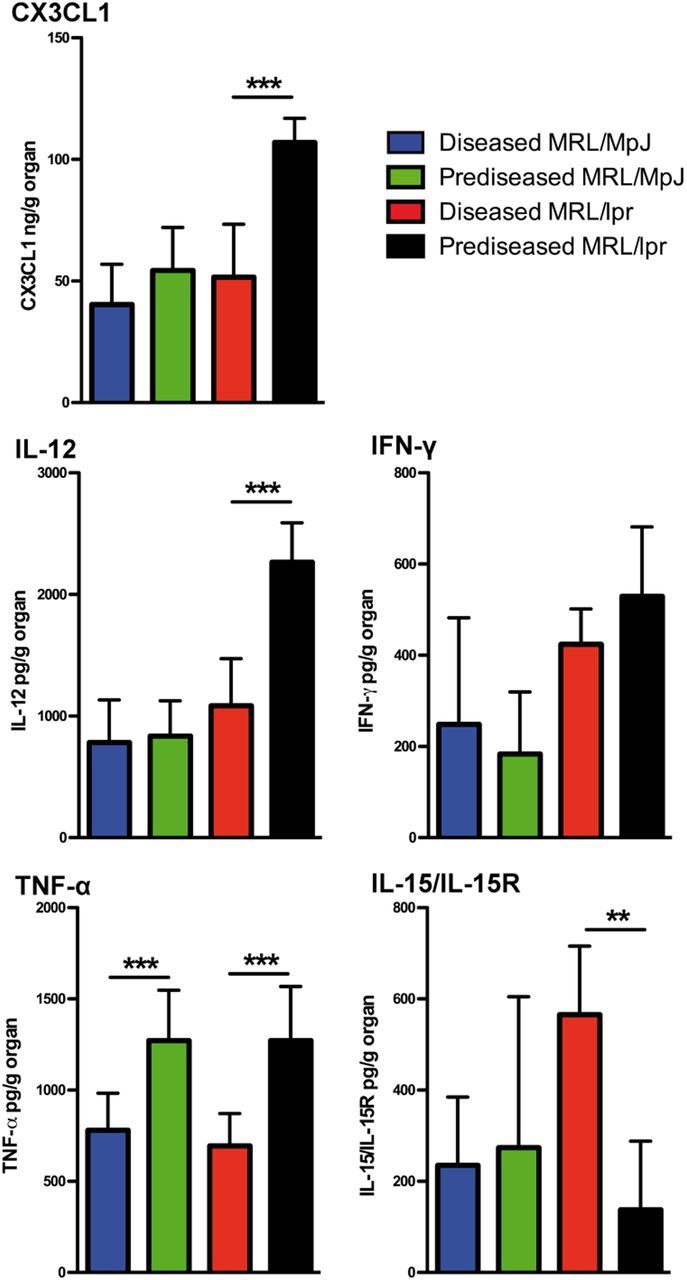
To understand some of the factors underlying the activation of NK cells in kidneys, we examined by ELISA the production of cytokines and chemokines important in the inflammatory process (Fig. 9). We observed an increased production of the chemoattractant chemokine CX3CL1 and the proinflammatory cytokine IL-12 in prediseased MRL/lpr mice when compared with diseased counterparts. We did not observe differences in IFN-γ production between diseased and prediseased animals, although MRL/lpr mice appear to produce larger amounts than MRL/MpJ mice. Both prediseased MRL/MpJ and MRL/lpr kidneys produced greater amounts of TNF-α than their diseased counterpart. Interestingly, diseased MRL/lpr mouse kidneys produced large amounts of the IL-15/IL-15R complex essential for NK cell effector function. These data suggest that NK cells could be actively recruited to the kidney and play a role in the maintenance of the inflammatory process in lupus nephritis.
Figure 9. Cytokine and chemokine detected in kidneys of SLE-prone mice. Kidneys were isolated, minced, and cultured in complete media overnight. Culture supernatants were spun down to remove cell debris, and amounts of IL-15/IL-15R complex, CX3CL1, IL-12, IFN-γ, and TNF-α were measured by ELISA. The final concentrations were normalized to the total organ weight. Data were analyzed with a 2-tailed, unpaired Student’s t-test (mean ± sd; n = 3/group; **P < 0.01; ***P < 0.001).
DISCUSSION
Here, we report that NKG2D ligand expression was found in SLE patients. We analyzed the expression of the human NKG2D ligands MICA and ULBP1 in kidney biopsies of patients with active and chronic lupus nephritis and found mild (1+) to strong MICA (3+) cytoplasmic staining in epithelial cells of nonatrophic tubules in 8 patients. No relationship was found among the positivity for MICA and the stage of lupus nephritis (focal, diffuse, or membranous), either among the intensity of the staining or the presence of active and/or chronic lesions.
The pattern of expression of MICA in patients with lupus nephritis resembles what has been reported previously in kidney sections from patients suffering from Wegener's granulomatosis [35], which is an inflammatory disease of unknown origin, whose most life-threatening symptom is necrotizing glomerulonephritis; patients in an active disease state show increased numbers of circulating MICA+ cells in the blood and positive MICA expression in glomerular and peritubular epithelium in the kidneys [35]. Thus, the expression of MICA seems to be up-regulated specifically during the active stages of inflammatory-autoimmune kidney manifestations. These observations, combined with reports that link MICA polymorphism to SLE pathogenesis via modulation of CD4+ cells, NK cells, and/or monocytes, provide an interesting link to possible effects of MICA expression in kidneys of patients with active SLE [18, 22, 36].
A small, immunosuppressive NKG2D+CD4+ T cell subset correlates inversely with disease activity in juvenile-onset lupus [17]. These cells are proposed to have regulatory properties, and their reduction in patients with active disease correlates with disease severity. The same study also found soluble MICA in SLE patient serum but not in that of controls; the authors nonetheless did not identify the MICA-expressing tissue, as a result of lack of tissue biopsies. It is tempting to postulate that the increased NKG2D ligand expression that we found in diseased kidneys could cause the release of a soluble form of these ligands. Soluble NKG2D ligands are reported to impair NKG2D expression and might explain the reduction in this NKG2D+CD4+ cell subset during the active disease state [37].
To assess whether the expression of NKG2D ligands also occurs in SLE-prone mice, we investigated their expression in MRL mouse strains. We observed specific Rae-1 and Mult-1 expression in the glomeruli of MRL/MpJ and MRL/lpr mice and Rae-1 expression in the glomeruli of diseased NZBxNZW(F1) mice. Expression of these ligands in kidney is specific to the glomeruli, which allows us to postulate that this SLE-prone background is the cause of NKG2D ligand expression, not present in C57BL/6 or BALB/c control strains. In addition, we found that heightened Rae-1 expression correlates with active disease. The glomeruli of mice in an active disease state might thus be subject to a broader stress-induced environment, which could be responsible for increased expression of this NKG2D ligand.
In accordance with the reported correlation of site-specific expression of NKG2D ligands with specific NK cell accumulation at the same site [38, 39], we observed that the expression NKG2D ligands in SLE-prone mouse kidneys correlated with an increase in NK cell infiltration as the disease progressed. Thus, we decided to investigate NK cell differentiation and activity in these murine models. MRL/lpr mice are commonly used to study SLE, given the rapid onset of SLE-like symptoms; nonetheless, this accelerated phenotype is mainly a result of a recessive, autosomal mutation that alters transcription of the FasR [40]. This model mimics many features found in SLE patients, and defective Fas signaling correlates with the development of autoimmune lymphoproliferative syndrome, which shares many symptoms with SLE. Notwithstanding, the extreme autoimmune manifestations in these mice, such as massive accumulation of double-negative and B220+ T cells, are neither typical of nor common in SLE patients [41]. The lpr mutation itself is not able to cause glomerular nephritis, and Fas-sufficient MRL/MpJ mice have background genes that promote development of SLE-like symptoms in a more “natural,” albeit slower, manner than MRL/lpr mice [40, 42]. Only the MRL/lpr mouse strain has been used in the limited studies of NK cells in SLE, and no information is available regarding the role of NK cells in diseased MRL/MpJ mice.
The only information on peripheral NK cells in human SLE patients indicates reduced expression and number of CD122+ NK cells and a reduction in total NK cell number and in their cytotoxicity [9, 10]. Proliferation, cytotoxicity, and differentiation defects are reported for NK cells cultured in vitro from SLE patient hematopoietic BM cells [9], but fresh BM samples have not been tested and to our knowledge, the NK cell phenotype has not been characterized in a SLE/SLE-like environment. Here, we confirm previous data indicating a reduction in the percentage of NK cells in diseased MRL/lpr mice [10] and show a similar reduction in diseased, 1-yr-old MRL/MpJ mice, indicating their usefulness for the study of NK cells in SLE. Comparison of percentages and absolute numbers of NKp46+CD3− cells in BM from prediseased and diseased MRL/lpr and MRL/MpJ mice suggests that the reduction of these cells in the periphery in diseased mice is not a result of defective BM production of NK cells.
Several reports show a nonmature, functional phenotype in peripheral NK cells from SLE patients and from diseased MRL/lpr mice, characterized by reduced CD122 expression and cytotoxicity [9, 12, 13]. In line with this, we observed that surface expression of typical markers of NK cell was down-regulated significantly in diseased spleen NK cells and to a lesser extent, in BM. The slight differences in the mature profile identified here in fresh BM samples resemble data of a study that showed reduced cytotoxicity/proliferation of human hematopoietic stem cells differentiated in vitro to NK cells [9].
We next analyzed NKP cell numbers and developmental markers in BM and in spleen. The 4-stage developmental process is far better understood in mice than in humans (the NK1.1 marker, a defining marker for NK cell development, was not analyzed, as it is not expressed in the MRL strain). As NKP cells have been found in various organs other than BM, they are thought to arise in the BM and travel to other organs where they develop fully. Thus, we hypothesized that in diseased mice, NK cells develop differently in spleen than in the BM. Our data showed no differences in total NKP or iNK cell numbers in the BM. In diseased mouse spleen, we observed larger numbers of iNK and NKP cells in Stages 1–, which led us to postulate that NK cells do not develop correctly in the periphery in diseased mice. NK cells in diseased patients are thought to suffer impaired maturation and differentiation, and NK cell activation is influenced by microenvironmental factors, such as the cytokine milieu or BM stromal cells [13, 43, 44]. These results correlated with our analysis of CD11bCD27 NK cell subsets in spleens from SLE-like mice, showing an accumulation of CD11bLowCD27High and a decrease in the percentage of CD11bHighCD27Low NK cells in diseased mouse spleen and demonstrating further that NK cells do not develop a completely mature phenotype. This was also confirmed by the reduction in T-bet+ NK cells in the spleen of diseased MRL/lpr mice.
The phenotypic and functional aspects of NK cells in SLE target organs have not been studied extensively. To date, only 3 studies have been published, 2 of which reported a more cytotoxic phenotype for liver and lung NK cells from diseased MRL/lpr mice than from prediseased MRL/MpJ or MRL/lpr mice [45, 46]. In a 3rd study, PMA/ionomycin-stimulated NK cells from diseased MRL/lpr mice produced more cytotoxic granules than prediseased MRL/lpr or MRL/MpJ mice [10]. These data suggest phenotype reversal of NK cells in target organs compared with NK cells in the periphery. Cytometric staining of freshly isolated NK cells from diseased kidneys showed an increase in the percentage of mature CD11bHighCD27Low cells. This was confirmed when the expression of the transcription factors Eomes and T-bet was analyzed. Intriguingly, in our hands, the expression pattern of Eomes and T-bet matched that of CD27 and CD11b, respectively. Eomes expression in NK cells has been associated with a more mature NK cell phenotype [30, 47], although recently, both transcription factors have been shown to instruct different NK cell lineages [31], suggesting that their expression is likely to depend on the microenvironment in which the NK cell matures. In CD4+ T cells, T-bet expression induced chromatin modification at the Eomes locus, probably leading to its repression [48]. It has also been suggested that early T-bet expression could repress Eomes expression in NK cells (giving rise to Eomes- T-bet+ NK cells), but once Eomes is expressed, T-bet is probably unable to repress its expression [31] (giving rise to Eomes+ NK cells, which can become T-bet+). In line with this, it could be speculated that the Eomes+ T-bet+ NK cell population, mostly detected in prediseased kidneys, could represent a peripheral NK cell population recruited to the organ, whereas the Eomes− T-bet+ NK cells could represent a locally differentiated NK cell population as a result of the proinflammatory environment of the MRL/lpr mouse kidneys. As the disease progresses, the decrease in Eomes+ T-bet+ NK cells in favor of Eomes- T-bet+ NK cells could reflect an accentuated change in the kidney microenvironment, driving T-bet overexpression and NK cell differentiation toward a more mature phenotype. Further work is required to identify the microenvironmental factors that can drive T-bet and Eomes expression in nephritis and clarify these findings.
Overall, our data support the existence of a more mature NK cell phenotype in diseased kidneys than in the periphery. Modifications in NK cell-activating and -inhibitory receptor levels are associated with other autoimmune diseases, such as psoriasis and type I diabetes, and are thought potentially to alter the NK cell activation threshold [49–52]. The organ- and disease-specific alterations in NK cell subsets and receptor expression that we describe during active SLE might reflect profound changes in NK cell activation and thus, their functional properties and possibly contribute to the autoimmune environment.
Increased CD11b expression in diseased mouse kidneys correlated with a larger percentage of kidney-infiltrating NK cells, which respond to PMA/ionomycin stimulation and thus, produce more IFN-γ. There were significant differences in the percentage of CD11bHighCD27Low cells that produced IFN-γ between diseased and prediseased mice, suggesting that these more mature, infiltrating NK cells are responsible for the increased IFN-γ production. The fact that no differences were detected between diseased and prediseased MRL/lpr mice in IFN-γ production by NK cells stimulated with IL-12 and IL-15 (a more specific IFN-γ-inducing stimulus) indicates that the potential for IFN-γ production of NK cells in prediseased mice is intact. On the other hand, the increased IFN-γ production induced by PMA/ionomycin treatment in NK cells from diseased kidneys also suggests that the activation threshold of these cells is lowered as the disease advances, resulting in a potential increase for overall cytokine production by these cells. The role of IFN-γ in the nephritic lupus process is well documented [53–55]. As such, these NK cells, which are activated and infiltrate the kidney during active stages of the disease, could be among the main IFN-γ producers and thus, responsible for maintaining and promoting the high degree of inflammation in nephritic lupus kidney.
Various still-unknown, kidney-specific factors could underlie this mature NK cell accumulation, and an active autoimmune environment might be able to alter specific pathways, fundamental for NK cell development, such as the STAT5 signaling pathway [56]. STAT5 has a crucial function in NK cell signaling, given its role in NK cell development and survival, and in transducing and activating myriad biologic processes after NK cell stimulation by cytokines, such as IL-2, IL-15, and IL-21 [34, 56]. We found in NK cells stimulated with IL-15 increased pSTAT5 in kidneys but not in BM or spleen of diseased MRL/MpJ and MRL/lpr mice compared with prediseased counterparts. Additionally, kidney NK cells showed higher levels of pSTAT5 than spleen or BM NK cells. These data suggest a difference between the mechanisms involved in NK cell regulation in affected and unaffected tissues/organs during active and inactive SLE, especially in the microenvironment in which the NK cells are found. Studies to date have focused mainly on analyzing peripheral blood NK cells in SLE patients, but other autoimmune diseases have highlighted the importance of studying tissue-infiltrating NK cells; in rheumatoid arthritis, synovium-infiltrating NK cells produce more IFN-γ than blood NK cells from the same patients [57, 58]. These more mature, IFN-γ-producing NK cells in diseased kidneys might have a key role in direct or indirect promotion (via recruitment/activation of other immune cells) of the glomerulonephritic process. The spleen, kidney, or BM factors that could impair or foster NK cell maturation/activation in a SLE-like environment nonetheless remain to be identified.
We also observed increased production of the chemoattractant chemokine CX3CL1 and the proinflammatory cytokine IL-12 in the kidney of prediseased MRL/lpr mice, both of which have been linked with the onset of kidney injury [59–61]. Although both of these inflammatory mediators have been shown to drive IFN-γ secretion [60, 62], we only detected increased IFN-γ production in MRL/lpr mice when compared with MRL/MpJ but not between the prediseased and diseased groups. A more localized role for IFN-γ in the tissue could explain this discrepancy. We also detected increased TNF-α production in prediseased MRL mice. Intrarenal TNF-α expression correlated with disease activity in SLE-prone models [63, 64], although high TNF-α doses can exert a protective effect on renal injury [64], and anti-TNF-α therapy can induce lupus nephritis-like syndrome in autoimmunity patients [65]. This dual role of TNF-α in lupus nephritis is not fully understood [66], although p38 MAPK activation status is likely involved in the outcome for renal injury dictated by TNF-α [67]. The increased TNF-α production detected in kidneys of prediseased mice suggests that TNF-α is likely to promote kidney damage in MRL strains. Finally, we detected increased production of the IL-15/IL-15R complex, essential for NK cell activity [43], in the kidneys of diseased MRL/lpr mice. Increased levels of IL-15 have been described in the serum of SLE patients [68], although the action of this cytokine on NK cells has been shown to depend mostly on its transpresentation [43]. Importantly, elevated expression of transmembrane IL-15 on immune cells has been associated with the development of murine lupus [69], suggesting that the presence of IL-15/IL-15R complexes in the diseased kidneys is likely to maintain NK cell activity.
Taken together, these data indicate that circulating NK cells and resident NK cells are recruited and activated very early into the prediseased kidneys (based on the production of CX3CL1 and IL-12, low expression of chemokine receptors CXR3 and CXCR4, and increased CD11b and T-bet expression), where the microenvironment allows for their maturation and maintenance in an active state, where they can exacerbate lupus nephritis by producing IFN-γ. Indeed, long-term depletion of NK cells (by anti-NK1.1 administration) has been shown to delay the onset of glomerular injury in NZBxNZW(F1) mice [70].
We propose that NK cells participate in glomerulonephritis, based on analysis of their infiltration into kidneys. Notwithstanding, we found that diseased MRL/lpr mice were the only group that showed an increase in the total number of kidney-infiltrating NK cells, which might thus have a qualitative rather than a quantitative role (as suggested by increased IFN-γ production and pSTAT5 in NK cells isolated from diseased kidneys) during glomerulonephritis, as there is no notable difference in total NK cell numbers between diseased MRL/MpJ and control mice. Nonetheless, this contrasts with a previous study, in which larger numbers of NK cells were found in kidneys of prediseased rather than of diseased MRL/lpr mice [10]. Regardless of whether total NK cell numbers increase in diseased or prediseased kidneys, our data show a correlation between infiltration of mouse glomeruli and an active disease state.
In summary, our results provide evidence for microenvironmental, organ-specific development and regulation of NK cells, which could clarify previous reports showing impaired terminal differentiation in peripheral NK cells from human SLE patients. Our data emphasize a larger role for NK cells in SLE target organs than previously proposed. The results show that NK cells infiltrate the glomeruli of glomerulonephritic SLE-like mice, which is concomitant with an increase in localized glomerular expression of NKG2D ligands. In conjunction with our finding of MICA expression in human SLE patient kidneys, these observations could implicate NKG2D ligands as possible therapeutic or diagnostic markers of SLE. Further work is needed to determine the potential of NK cells and the NKG2D receptor and its ligands in the development of lupus-based glomerulonephritis to identify potential therapeutic targets.
AUTHORSHIP
R.S. performed most of the experiments. R.S., J.M.R., and D.F.B. designed the experiments and wrote the manuscript. J.M.R., S.P.-Y., and V.M. performed some experiments. P.C.-O. and R.B. provided and analyzed patients’ samples. D.F.B. directed the work.
ACKNOWLEDGMENTS
This work was supported, in part, by grants from the Spanish Ministry of Economy and Competitiveness (SAF-2008-00471 and SAF-2011-23639; to D.F.B.), the Madrid regional government (CCG08-CSIC/SAL-3451; to D.F.B.), and the Spanish Ministry for Health, Social Services and Equality, Carlos III Health Institute Cooperative Research Thematic Network program: Research Network in Inflammation and Rheumatic Diseases (RIER; RD08/0075/0015; to D.F.B.) and Spanish National Biobank Network (RD09/0076/00101 to the FJD). R.S. was supported by a FPI predoctoral fellowship from the Spanish Ministry of Economy and Competitiveness. J.M.R. is supported by a postdoctoral JAE-Doc contract from the Consejo Superior de Investigaciones Científicas, cofunded by the European Social Fund. V.M. is supported by a predoctoral fellowship from Fundación La Caixa-CNB program. The authors thank Drs. Stipan Joncic for the anti-Mult-1 antibody, Shozo Izui for supplying OCT-embedded kidneys from NZBxNZW(F1) mice, and Manuela Zonca for OCT-embedded kidneys from BALB/c mice. The authors also thank Sylvia Gutiérrez Erlandsson and the CNB Confocal Microscopy Service and Catherine Mark for editorial assistance.
Glossary
- BFA
brefeldin A, BM = bone marrow
- CNB
Centro Nacional de Biotecnología
- Eomes
eomesodermin
- FJD
Fundación Jiménez Díaz
- IIS
Instituto de Investigación Sanitaria
- iNK
immature NK
- MICA/B
MHC class I polypeptide-related sequence AB
- MRL/lpr
MRL/MpJlpr
- MRL/MpJ
Murphy Roths Large mice
- Mult-1
murine UL16-binding protein-like transcript 1
- NKP
NK precursor
- pSTAT
phosphorylated STAT
- Rae-1
retinoic acid early inducible 1
- SLE
systemic lupus erythematosus
- ULBP1
UL16-binding protein-like transcript 1
Footnotes
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
DISCLOSURES
The authors claim no financial conflict of interest.
REFERENCES
- 1.Flodström-Tullberg M., Bryceson Y. T., Shi F. D., Höglund P., Ljunggren H. G. (2009) Natural killer cells in human autoimmunity. Curr. Opin. Immunol. 21, 634–640. [DOI] [PubMed] [Google Scholar]
- 2.Gillgrass A., Ashkar A. (2011) Stimulating natural killer cells to protect against cancer: recent developments. Expert Rev. Clin. Immunol. 7, 367–382. [DOI] [PubMed] [Google Scholar]
- 3.Lanier L. L. (2005) NK cell recognition. Annu. Rev. Immunol. 23, 225–274. [DOI] [PubMed] [Google Scholar]
- 4.Pisetsky D. S., Rönnblom L. (2009) Systemic lupus erythematosus: a matter of life and death. Arthritis Rheum. 60, 1567–1570. [DOI] [PubMed] [Google Scholar]
- 5.Mills J. A. (1994) Systemic lupus erythematosus. N. Engl. J. Med. 330, 1871–1879. [DOI] [PubMed] [Google Scholar]
- 6.Sekine H., Watanabe H., Gilkeson G. S. (2004) Enrichment of anti-glomerular antigen antibody-producing cells in the kidneys of MRL/MpJ-Fas(lpr) mice. J. Immunol. 172, 3913–3921. [DOI] [PubMed] [Google Scholar]
- 7.Balow J. E. (1991) Kidney disease in systemic lupus erythematosus. Rheumatol. Int. 11, 113–115. [DOI] [PubMed] [Google Scholar]
- 8.Park M. H., D’Agati V., Appel G. B., Pirani C. L. (1986) Tubulointerstitial disease in lupus nephritis: relationship to immune deposits, interstitial inflammation, glomerular changes, renal function, and prognosis. Nephron 44, 309–319. [DOI] [PubMed] [Google Scholar]
- 9.Park Y. W., Kee S. J., Cho Y. N., Lee E. H., Lee H. Y., Kim E. M., Shin M. H., Park J. J., Kim T. J., Lee S. S., Yoo D. H., Kang H. S. (2009) Impaired differentiation and cytotoxicity of natural killer cells in systemic lupus erythematosus. Arthritis Rheum. 60, 1753–1763. [DOI] [PubMed] [Google Scholar]
- 10.Huang Z., Fu B., Zheng S. G., Li X., Sun R., Tian Z., Wei H. (2011) Involvement of CD226+ NK cells in immunopathogenesis of systemic lupus erythematosus. J. Immunol. 186, 3421–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erkeller-Yuksel F. M., Lydyard P. M., Isenberg D. A. (1997) Lack of NK cells in lupus patients with renal involvement. Lupus 6, 708–712. [DOI] [PubMed] [Google Scholar]
- 12.Pan L. Z., Dauphinée M. J., Ansar Ahmed S., Talal N. (1986) Altered natural killer and natural cytotoxic cellular activities in lpr mice. Scand. J. Immunol. 23, 415–423. [DOI] [PubMed] [Google Scholar]
- 13.Hervier B., Beziat V., Haroche J., Mathian A., Lebon P., Ghillani-Dalbin P., Musset L., Debré P., Amoura Z., Vieillard V. (2011) Phenotype and function of natural killer cells in systemic lupus erythematosus: excess interferon-γ production in patients with active disease. Arthritis Rheum. 63, 1698–1706. [DOI] [PubMed] [Google Scholar]
- 14.Bottino C., Castriconi R., Moretta L., Moretta A. (2005) Cellular ligands of activating NK receptors. Trends Immunol. 26, 221–226. [DOI] [PubMed] [Google Scholar]
- 15.Lanier L. L. (2008) Up on the tightrope: natural killer cell activation and inhibition. Nat. Immunol. 9, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eagle R. A., Trowsdale J. (2007) Promiscuity and the single receptor: NKG2D. Nat. Rev. Immunol. 7, 737–744. [DOI] [PubMed] [Google Scholar]
- 17.Dai Z., Turtle C. J., Booth G. C., Riddell S. R., Gooley T. A., Stevens A. M., Spies T., Groh V. (2009) Normally occurring NKG2D+CD4+ T cells are immunosuppressive and inversely correlated with disease activity in juvenile-onset lupus. J. Exp. Med. 206, 793–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang D., Wang H., Ni B., He Y., Li J., Tang Y., Fu X., Wang Q., Xu G., Li K., Yang Z., Wu Y. (2009) Mutual activation of CD4+ T cells and monocytes mediated by NKG2D-MIC interaction requires IFN-gamma production in systemic lupus erythematosus. Mol. Immunol. 46, 1432–1442. [DOI] [PubMed] [Google Scholar]
- 19.Krmpotic A., Hasan M., Loewendorf A., Saulig T., Halenius A., Lenac T., Polic B., Bubic I., Kriegeskorte A., Pernjak-Pugel E., Messerle M., Hengel H., Busch D. H., Koszinowski U. H., Jonjic S. (2005) NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J. Exp. Med. 201, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matkowskyj K. A., Schonfeld D., Benya R. V. (2000) Quantitative immunohistochemistry by measuring cumulative signal strength using commercially available software Photoshop and Matlab. J. Histochem. Cytochem. 48, 303–312. [DOI] [PubMed] [Google Scholar]
- 21.Xu W. D., Zhang Y. J., Li R., Pan H. F., Ye D. Q. (2012) New evidence for a role of MICA in the pathogenesis of systemic lupus erythematosus: comment on the article by Yoshida et al. Arthritis Rheum. 64, 1294–1295. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida K., Komai K., Shiozawa K., Mashida A., Horiuchi T., Tanaka Y., Nose M., Hashiramoto A., Shiozawa S. (2011) Role of the MICA polymorphism in systemic lupus erythematosus. Arthritis Rheum. 63, 3058–3066. [DOI] [PubMed] [Google Scholar]
- 23.Ueda H., Miyazaki Y., Matsusaka T., Utsunomiya Y., Kawamura T., Hosoya T., Ichikawa I. (2008) Bmp in podocytes is essential for normal glomerular capillary formation. J. Am. Soc. Nephrol. 19, 685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barutta F., Corbelli A., Mastrocola R., Gambino R., Di Marzo V., Pinach S., Rastaldi M. P., Perin P. C., Gruden G. (2010) Cannabinoid receptor 1 blockade ameliorates albuminuria in experimental diabetic nephropathy. Diabetes 59, 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vosshenrich C. A., Samson-Villéger S. I., Di Santo J. P. (2005) Distinguishing features of developing natural killer cells. Curr. Opin. Immunol. 17, 151–158. [DOI] [PubMed] [Google Scholar]
- 26.Di Santo J. P. (2006) Natural killer cell developmental pathways: a question of balance. Annu. Rev. Immunol. 24, 257–286. [DOI] [PubMed] [Google Scholar]
- 27.Huntington N. D., Vosshenrich C. A., Di Santo J. P. (2007) Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat. Rev. Immunol. 7, 703–714. [DOI] [PubMed] [Google Scholar]
- 28.Chiossone L., Chaix J., Fuseri N., Roth C., Vivier E., Walzer T. (2009) Maturation of mouse NK cells is a 4-stage developmental program. Blood 113, 5488–5496. [DOI] [PubMed] [Google Scholar]
- 29.Narni-Mancinelli E., Chaix J., Fenis A., Kerdiles Y. M., Yessaad N., Reynders A., Gregoire C., Luche H., Ugolini S., Tomasello E., Walzer T., Vivier E. (2011) Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc. Natl. Acad. Sci. USA 108, 18324–18329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordon S. M., Chaix J., Rupp L. J., Wu J., Madera S., Sun J. C., Lindsten T., Reiner S. L. (2012) The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity 36, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daussy C., Faure F., Mayol K., Viel S., Gasteiger G., Charrier E., Bienvenu J., Henry T., Debien E., Hasan U. A., Marvel J., Yoh K., Takahashi S., Prinz I., de Bernard S., Buffat L., Walzer T. (2014) T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J. Exp. Med. 211, 563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernardini G., Benigni G., Antonangeli F., Ponzetta A., Santoni A. (2014) Multiple levels of chemokine receptor regulation in the control of mouse natural killer cell development. Front. Immunol. 5, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang X., Guo Y., Bao C., Shen N. (2011) Multidimensional single cell based STAT phosphorylation profiling identifies a novel biosignature for evaluation of systemic lupus erythematosus activity. PLoS ONE 6, e21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lieberman L. A., Tsokos G. C. (2010) The IL-2 defect in systemic lupus erythematosus disease has an expansive effect on host immunity. J. Biomed. Biotechnol. 2010, 740619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holmén C., Elsheikh E., Stenvinkel P., Qureshi A. R., Pettersson E., Jalkanen S., Sumitran-Holgersson S. (2005) Circulating inflammatory endothelial cells contribute to endothelial progenitor cell dysfunction in patients with vasculitis and kidney involvement. J. Am. Soc. Nephrol. 16, 3110–3120. [DOI] [PubMed] [Google Scholar]
- 36.Schrambach S., Ardizzone M., Leymarie V., Sibilia J., Bahram S. (2007) In vivo expression pattern of MICA and MICB and its relevance to auto-immunity and cancer. PLoS ONE 2, e518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Groh V., Wu J., Yee C., Spies T. (2002) Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 419, 734–738. [DOI] [PubMed] [Google Scholar]
- 38.Diefenbach A., Jensen E. R., Jamieson A. M., Raulet D. H. (2001) Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 413, 165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Markiewicz M. A., Wise E. L., Buchwald Z. S., Pinto A. K., Zafirova B., Polic B., Shaw A. S. (2012) RAE1ε ligand expressed on pancreatic islets recruits NKG2D receptor-expressing cytotoxic T cells independent of T cell receptor recognition. Immunity 36, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jabs D. A., Prendergast R. A. (1988) Murine models of Sjögren’s syndrome. Immunohistologic analysis of different strains. Invest. Ophthalmol. Vis. Sci. 29, 1437–1443. [PubMed] [Google Scholar]
- 41.Teachey D. T., Seif A. E., Grupp S. A. (2010) Advances in the management and understanding of autoimmune lymphoproliferative syndrome (ALPS). Br. J. Haematol. 148, 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vidal S., Kono D. H., Theofilopoulos A. N. (1998) Loci predisposing to autoimmunity in MRL-Fas lpr and C57BL/6-Faslpr mice. J. Clin. Invest. 101, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lucas M., Schachterle W., Oberle K., Aichele P., Diefenbach A. (2007) Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 26, 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roth C., Rothlin C., Riou S., Raulet D. H., Lemke G. (2007) Stromal-cell regulation of natural killer cell differentiation. J. Mol. Med. (Berl) 85, 1047–1056. [DOI] [PubMed] [Google Scholar]
- 45.Magilavy D. B., Steinberg A. D., Latta S. L. (1987) High hepatic natural killer cell activity in murine lupus. J. Exp. Med. 166, 271–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nilsson N., Carlsten H. (1996) Enhanced natural but diminished antibody-mediated cytotoxicity in the lungs of MRLlpr/lpr mice. Clin. Exp. Immunol. 105, 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tayade C., Fang Y., Black G. P., Paffaro V. A. Jr., Erlebacher A., Croy B. A. (2005) Differential transcription of Eomes and T-bet during maturation of mouse uterine natural killer cells. J. Leukoc. Biol. 78, 1347–1355. [DOI] [PubMed] [Google Scholar]
- 48.Zhu J., Jankovic D., Oler A. J., Wei G., Sharma S., Hu G., Guo L., Yagi R., Yamane H., Punkosdy G., Feigenbaum L., Zhao K., Paul W. E. (2012) The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity 37, 660–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin M. P., Nelson G., Lee J. H., Pellett F., Gao X., Wade J., Wilson M. J., Trowsdale J., Gladman D., Carrington M. (2002) Cutting edge: susceptibility to psoriatic arthritis: influence of activating killer Ig-like receptor genes in the absence of specific HLA-C alleles. J. Immunol. 169, 2818–2822. [DOI] [PubMed] [Google Scholar]
- 50.Van der Slik A. R., Koeleman B. P., Verduijn W., Bruining G. J., Roep B. O., Giphart M. J. (2003) KIR in type 1 diabetes: disparate distribution of activating and inhibitory natural killer cell receptors in patients versus HLA-matched control subjects. Diabetes 52, 2639–2642. [DOI] [PubMed] [Google Scholar]
- 51.French A. R., Yokoyama W. M. (2004) Natural killer cells and autoimmunity. Arthritis Res. Ther. 6, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bilbao J. R., Martín-Pagola A., Pérez De Nanclares G., Calvo B., Vitoria J. C., Vázquez F., Castaño L. (2003) HLA-DRB1 and MICA in autoimmunity: common associated alleles in autoimmune disorders. Ann. N. Y. Acad. Sci. 1005, 314–318. [DOI] [PubMed] [Google Scholar]
- 53.Seery J. P., Carroll J. M., Cattell V., Watt F. M. (1997) Antinuclear autoantibodies and lupus nephritis in transgenic mice expressing interferon gamma in the epidermis. J. Exp. Med. 186, 1451–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee S. K., Silva D. G., Martin J. L., Pratama A., Hu X., Chang P. P., Walters G., Vinuesa C. G. (2012) Interferon-γ excess leads to pathogenic accumulation of follicular helper T cells and germinal centers. Immunity 37, 880–892. [DOI] [PubMed] [Google Scholar]
- 55.Pollard K. M., Cauvi D. M., Toomey C. B., Morris K. V., Kono D. H. (2013) Interferon-γ and systemic autoimmunity. Discov. Med. 16, 123–131. [PMC free article] [PubMed] [Google Scholar]
- 56.Eckelhart E., Warsch W., Zebedin E., Simma O., Stoiber D., Kolbe T., Rülicke T., Mueller M., Casanova E., Sexl V. (2011) A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood 117, 1565–1573. [DOI] [PubMed] [Google Scholar]
- 57.Aramaki T., Ida H., Izumi Y., Fujikawa K., Huang M., Arima K., Tamai M., Kamachi M., Nakamura H., Kawakami A., Origuchi T., Matsuoka N., Eguchi K. (2009) A significantly impaired natural killer cell activity due to a low activity on a per-cell basis in rheumatoid arthritis. Mod. Rheumatol. 19, 245–252. [DOI] [PubMed] [Google Scholar]
- 58.De Matos C. T., Berg L., Michaëlsson J., Felländer-Tsai L., Kärre K., Söderström K. (2007) Activating and inhibitory receptors on synovial fluid natural killer cells of arthritis patients: role of CD94/NKG2A in control of cytokine secretion. Immunology 122, 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Inoue A., Hasegawa H., Kohno M., Ito M. R., Terada M., Imai T., Yoshie O., Nose M., Fujita S. (2005) Antagonist of fractalkine (CX3CL1) delays the initiation and ameliorates the progression of lupus nephritis in MRL/lpr mice. Arthritis Rheum. 52, 1522–1533. [DOI] [PubMed] [Google Scholar]
- 60.Schwarting A., Tesch G., Kinoshita K., Maron R., Weiner H. L., Kelley V. R. (1999) IL-12 drives IFN-gamma-dependent autoimmune kidney disease in MRL-Fas(lpr) mice. J. Immunol. 163, 6884–6891. [PubMed] [Google Scholar]
- 61.Kikawada E., Lenda D. M., Kelley V. R. (2003) IL-12 deficiency in MRL-Fas(lpr) mice delays nephritis and intrarenal IFN-gamma expression, and diminishes systemic pathology. J. Immunol. 170, 3915–3925. [DOI] [PubMed] [Google Scholar]
- 62.Yoneda O., Imai T., Nishimura M., Miyaji M., Mimori T., Okazaki T., Domae N., Fujimoto H., Minami Y., Kono T., Bloom E. T., Umehara H. (2003) Membrane-bound form of fractalkine induces IFN-gamma production by NK cells. Eur. J. Immunol. 33, 53–58. [DOI] [PubMed] [Google Scholar]
- 63.Boswell J. M., Yui M. A., Burt D. W., Kelley V. E. (1988) Increased tumor necrosis factor and IL-1 beta gene expression in the kidneys of mice with lupus nephritis. J. Immunol. 141, 3050–3054. [PubMed] [Google Scholar]
- 64.Brennan D. C., Yui M. A., Wuthrich R. P., Kelley V. E. (1989) Tumor necrosis factor and IL-1 in New Zealand Black/White mice. Enhanced gene expression and acceleration of renal injury. J. Immunol. 143, 3470–3475. [PubMed] [Google Scholar]
- 65.Williams V. L., Cohen P. R. (2011) TNF alpha antagonist-induced lupus-like syndrome: report and review of the literature with implications for treatment with alternative TNF alpha antagonists. Int. J. Dermatol. 50, 619–625. [DOI] [PubMed] [Google Scholar]
- 66.Yung S., Cheung K. F., Zhang Q., Chan T. M. (2013) Mediators of inflammation and their effect on resident renal cells: implications in lupus nephritis. Clin. Dev. Immunol. 2013, 317682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iwata Y., Wada T., Furuichi K., Sakai N., Matsushima K., Yokoyama H., Kobayashi K. (2003) p38 Mitogen-activated protein kinase contributes to autoimmune renal injury in MRL-Fas lpr mice. J. Am. Soc. Nephrol. 14, 57–67. [DOI] [PubMed] [Google Scholar]
- 68.Baranda L., de la Fuente H., Layseca-Espinosa E., Portales-Pérez D., Niño-Moreno P., Valencia-Pacheco G., Abud-Mendoza C., Alcocer-Varela J., González-Amaro R. (2005) IL-15 and IL-15R in leucocytes from patients with systemic lupus erythematosus. Rheumatology (Oxford) 44, 1507–1513. [DOI] [PubMed] [Google Scholar]
- 69.Bo H., Wei X. Q., Dong H., Zhang Y., Lv P., Liu W., Koutoulaki A., Gao X. M. (2009) Elevated expression of transmembrane IL-15 in immune cells correlates with the development of murine lupus: a potential target for immunotherapy against SLE. Scand. J. Immunol. 69, 119–129. [DOI] [PubMed] [Google Scholar]
- 70.Postól E., Meyer A., Cardillo F., de Alencar R., Pessina D., Nihei J., Mariano M., Mengel J. (2008) Long-term administration of IgG2a anti-NK1.1 monoclonal antibody ameliorates lupus-like disease in NZB/W mice in spite of an early worsening induced by an IgG2a-dependent BAFF/BLyS production. Immunology 125, 184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]