

Abstract
Purpose
Peripheral blood–derived antigen-specific cytotoxic T cells (CTLs) provide a readily available source of effector cells that can be administered with minimal toxicity in an outpatient setting. In metastatic melanoma, this approach results in measurable albeit modest clinical responses in patients resistant to conventional therapy. We reasoned that concurrent cytotoxic T-cell lymphocyte antigen-4 (CTLA-4) checkpoint blockade might enhance the antitumor activity of adoptively transferred CTLs.
Patients and Methods
Autologous MART1-specific CTLs were generated by priming with peptide-pulsed dendritic cells in the presence of interleukin-21 and enriched by peptide-major histocompatibility complex multimer-guided cell sorting. This expeditiously yielded polyclonal CTL lines uniformly expressing markers associated with an enhanced survival potential. In this first-in-human strategy, 10 patients with stage IV melanoma received the MART1-specific CTLs followed by a standard course of anti–CTLA-4 (ipilimumab).
Results
The toxicity profile of the combined treatment was comparable to that of ipilimumab monotherapy. Evaluation of best responses at 12 weeks yielded two continuous complete remissions, one partial response (PR) using RECIST criteria (two PRs using immune-related response criteria), and three instances of stable disease. Infused CTLs persisted with frequencies up to 2.9% of CD8+ T cells for as long as the patients were monitored (up to 40 weeks). In patients who experienced complete remissions, PRs, or stable disease, the persisting CTLs acquired phenotypic and functional characteristics of long-lived memory cells. Moreover, these patients also developed responses to nontargeted tumor antigens (epitope spreading).
Conclusion
We demonstrate that combining antigen-specific CTLs with CTLA-4 blockade is safe and produces durable clinical responses, likely reflecting both enhanced activity of transferred cells and improved recruitment of new responses, highlighting the promise of this strategy.
INTRODUCTION
Adoptive immunotherapy involving the ex vivo expansion and reinfusion of tumor-reactive T cells is an emerging treatment modality, especially in patients for whom conventional therapy fails.1 Consequential responses have been achieved in metastatic melanoma using tumor-reactive T cells expanded from a tumor site.2 However, successful tumor-infiltrating lymphocyte therapies require sufficient accessible tumor for adequate sampling and have been confined to specialized centers by toxicities associated with high-dose preinfusion conditioning and postinfusion interleukin-2 (IL-2).3 Endogenous antigen-specific CTLs can also be obtained and expanded from peripheral blood (PB) and infused with lower-dose conditioning and a tolerable safety profile, but they have effectively reduced tumor burdens in only a limited number of patients, in part because of the short persistence of the transferred cells.4-7 In our prior studies, infused CTLs persisted beyond 42 days in 11% to 15% of patients. Median CTL persistence in vivo was fewer than 14 days, and the overall response rate (inclusive of patients achieving complete remissions [CRs] and partial responses [PRs]) was only 7%.4-7
When transferred T cells have persisted and mediated antitumor responses, the PB-derived CTLs showed characteristics associated with survival after the in vitro culture period, including CD28 expression, or exhibited or acquired these characteristics in vivo post-transfer.4,8 We hypothesized that ex vivo generation of antigen-specific CTLs with characteristics of long-lived memory may enhance cell survival after adoptive transfer, thus enhancing sustained antitumor activity.9 Therefore, we made two modifications to the methods used for T-cell generation. First, antigen-specific T cells were primed in vitro in the presence of the cytokine IL-21,10 which promotes expansion of CTLs that, in comparison with cells that have been generated in the absence of IL-21 priming, phenotypically exhibit a less terminally differentiated phenotype, with a majority of cells expressing CD28 after ex vivo culture11,12 and have been shown to exhibit enhanced persistence in murine models and humans after adoptive transfer.8,13 Second, peptide-major histocompatibility complex (pMHC) multimers and clinical-grade sorting were used to select polyclonal melanoma-specific CTLs early from in vitro–generated CD8+ T-cell lines, limiting the time required to achieve therapeutic cell numbers.14
Cytotoxic T-cell lymphocyte antigen-4 (CTLA-4) is an inducible T-cell surface protein that binds to CD80 and CD86 with a higher affinity than the costimulatory receptor CD28, intercepting the binding of the latter and providing an inhibitory signal to T cells. Thus, anti–CTLA-4 can release the brake on antigen-specific T-cell activation.15,16 Monotherapy with anti–CTLA-4 achieves disease control (CRs, PRs, and stable disease [SD] at least 12 weeks in duration) in 20% to 28% of patients with advanced melanoma and increases overall survival rates.17-20 However, the magnitude, breadth, and/or maintenance of T-cell responses triggered by anti–CTLA-4 alone is in most cases insufficient to eradicate tumors, and long-term CRs are seen in a minority of patients (range, 0% to 7%).17,21,22
We hypothesized that the adoptive transfer of melanoma-reactive CD28+ CTLs with enhanced survival properties would directly benefit from CTLA-4 blockade, promoting their antitumor reactivity in vivo. In turn, the transferred cells could facilitate the release of tumor antigens from lysed tumor cells in the proimmunogenic context fostered by anti–CTLA-4. Thus, the combination would have the potential to boost tumor-specific responses to nontargeted antigens (antigen spreading), extending the breadth of antitumor responses and reducing the outgrowth of antigen-loss tumor variants.23,24
PATIENTS AND METHODS
Clinical Protocol, Patient Characteristics, and Generation of Melanoma-Specific CTL Products
Between August 2011 and April 2013, 10 patients with progressive metastatic melanoma (Table 1) received treatment according to protocol No. 2225, approved by the Fred Hutchinson Cancer Research Center Institutional Review Board and the US Food and Drug Administration. All treated patients provided written informed consent. Eligibility required HLA-A*0201 and tumor expression of MART1.26
Table 1.
Patient Clinical Characteristics
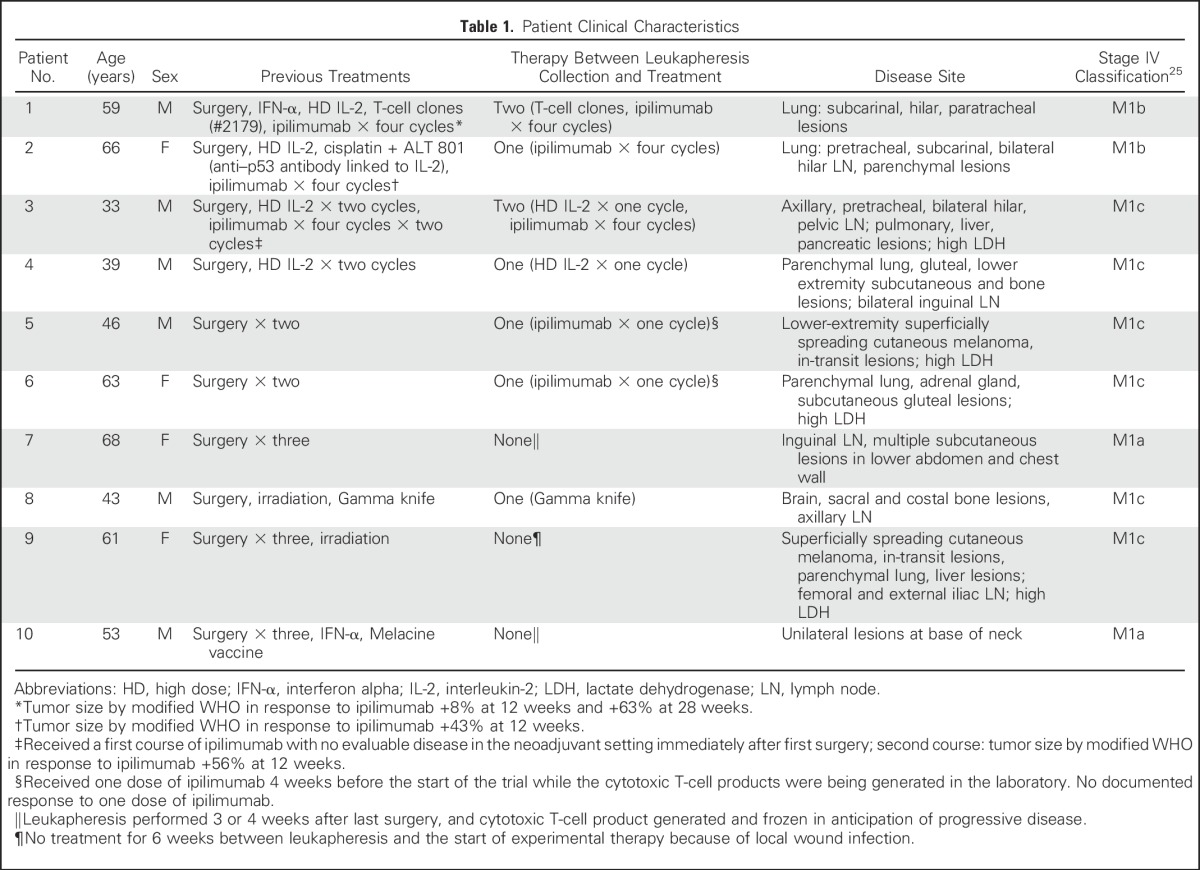
A total of 14 patients underwent leukapheresis in anticipation of entering this trial. Of the four who underwent leukapheresis and did not receive treatment as part of the trial, all started an alternate treatment while waiting for the cells to be generated: two received a BRAF inhibitor, one started a trial of IL-21 and ipilimumab, and one received high-dose IL-2. One patient remains in CR after high-dose IL-2. By the time the three remaining patients had developed progressive disease (PD), the trial was closed to accrual.
Treatment Plan
Patients received a single outpatient infusion of cyclophosphamide 300 mg/m2 48 hours before the infusion of 1010 melanoma-specific CTL/m2 (determined safe from previous studies),6 followed by low-dose subcutaneous IL-2 (250,000 IU/m2 twice daily for 14 days) to enhance the survival of transferred T cells.7 On day 1 after CTL infusion, patients received ipilimumab (anti–CTLA-4; Yervoy; Bristol-Myers Squibb, New York, NY) 3 mg/kg every 3 weeks for a total of four doses (Data Supplement). Patients were monitored for toxicities based on Common Toxicity Criteria (version 4.0).27 Staging studies were obtained 6 and 12 weeks after the infusion and then as clinically indicated.
Assessment of Clinical Responses
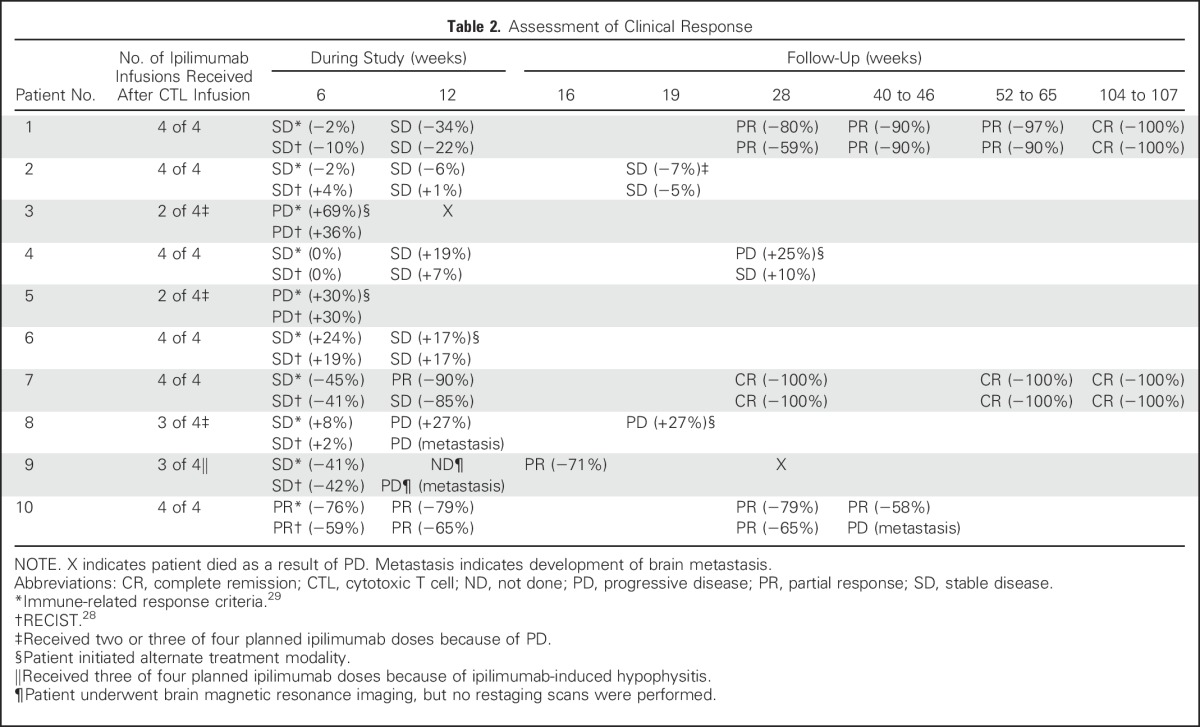
Both immune-related response criteria (irRC) and RECIST criteria were used to assess clinical responses.28,29 Discrepancies (Table 2) were the result of differences in the specific measures of tumor size and classification of new lesions,29 with the latter being incorporated into the global tumor burden in irRC but classified as PD in RECIST criteria. A detailed description of the materials and methods used is provided in the Data Supplement.
Table 2.
Assessment of Clinical Response
RESULTS
Polyclonal IL-21–Primed MART1-Specific CTLs Demonstrate Ex Vivo Antitumor Activity and Express Phenotypic Characteristics Associated With In Vivo Survival
All polyclonal CTL products demonstrated intracellular production of interferon gamma (IFN-γ) in response to antigen-presenting targets (HLA-A*0201-positive, transporter for antigen presentation–deficient, B lymphoblastoid cell lines [ie, T2 B-LCLs]), pulsed with the HLA-A*0201–restricted MART126-35 peptide (Data Supplement), as well as specific lysis (median, 54%; range, 27% to 66%) of the HLA A*0201-positive, MART1-positive MEL-526 cell line30 (Data Supplement). Immediately before infusion, multimer-binding CTLs expressed CD45RO (median, 98.9%), consistent with an antigen-experienced phenotype (Data Supplement). In accordance with our previous studies,11,12 a subset of the expanded CTLs retained expression of CD27, CD28, and CD127 (with medians of 63.5%, 72.4%, and 36.2%, respectively), consistent with IL-21 exposure during priming.8 Little or no expression of the lymph node homing markers CD62L and CCR7 or the activation/exhaustion markers CD57 and PD1 was detected.
Concurrent Transfer of MART1-Specific CTLs Does Not Alter Safety and Toxicity Profiles of Anti–CTLA-4
Ten patients with metastatic melanoma whose disease progressed after previous systemic or surgical therapy, including ipilimumab monotherapy in three patients (Table 1), received combined CTLs plus anti–CTLA-4 treatment (Data Supplement). Seven patients experienced transient (< 24 hours) sterile fevers (≥ 38.3°C) with or without chills, associated with a CTL infusion–induced cytokine-release syndrome (Data Supplement). Lymphopenia lasting 10 days or fewer8 and self-limiting moderately erythematous maculopapular skin rashes were observed in nine of 10 patients. Ipilimumab therapy was associated with self-limited nausea/diarrhea (grade ≤ 1) in eight of 10 patients, and transient (< 14 days) moderately elevated (grade ≤ 2) liver enzymes were detected in five of 10 patients. Patient 9 developed pituitary insufficiency secondary to ipilimumab-induced hypophysitis,31 and patient 5 experienced a drug fever attributed to the combined effect of ipilimumab plus vemurafenib introduced for PD.32,33 Both patients received systemic steroids as treatment for immune-related adverse events.31 Overall, the toxicities could be attributed to either CTL infusion or ipilimumab alone, but no unexpected toxicities were associated with the combination.
Adoptive Transfer of MART1-Specific CTLs With Concurrent Anti–CTLA-4 Can Produce Sustained Clinical Responses
Two of 10 patients achieved sustained CRs as defined by irRC and RECIST criteria26,34 at 12 and 104 weeks after CTL infusion, respectively (Table 2; Figs 1A [green lines,] and 1B). Patient 1 had experienced PD after salvage ipilimumab monotherapy initiated 7 months earlier (Chapuis et al, manuscript submitted for publication). The time to response after ipilimumab alone was outside the expected range, and the appearance of new lesions during the interim precluded any beneficial effect of ipilimumab monotherapy.29 Neither patient received additional antitumor therapy, and both were alive and disease free 220 and 169 weeks (as of November 1, 2015), respectively, after the start of treatment.
Fig 1.

Tumor regressions after melanoma-reactive polyclonal cytotoxic T cells (CTLs) combined with anti–cytotoxic T-cell lymphocyte antigen-4. (A) Spider plot of all treated patients showing changes from baseline in the tumor burden (y-axis), measured as the products of the perpendicular diameters of all target lesions, assessed weeks after the CTL infusion (x-axis). The dashed line above the solid line indicates 25% progression (modified WHO progressive disease [PD]), and the dashed line below the solid line indicates 50% reduction (modified WHO partial response [PR]). Red lines indicate patients with PD, purple lines indicate patients with stable disease, blue lines indicate patients with PRs, and green lines indicate patients with complete remissions. Red squares indicate the occurrence of new lesions, asterisks indicate the start of an alternate treatment, pound signs indicate disease progression sufficient to transition to comfort care, and blue horizontal arrows indicate continued monitoring. (B) Serial images of computed tomography scans performed before infusion (left) and 64 and 55 weeks after treatment (right) for patients 1 (top) and 7 (bottom), respectively. Arrows indicate the location of the largest index lesions for each patient.
Two patients experienced PRs as best responses by irRC (Fig 1A [blue lines]). After demonstrating a 41% reduction in tumor burden at 6 weeks, patient 9 was diagnosed with new subcentimetric brain metastasis at 7 weeks, along with ipilimumab-induced hypophysitis treated with systemic steroids. Despite the new brain lesions, the patient exhibited a reduction in overall tumor burden of 71% at 16 weeks, associated first with flattening then central blanching of numerous cutaneous metastases (Data Supplement). However, as a possible consequence of systemic steroids, or because of the characteristics of the brain parenchyma in which CTLs may be excluded in the absence of inflammation,35 the brain lesions progressed. The patient elected to receive comfort care and died 2 months later. Patient 10 experienced a PR at 6 weeks (by irRC), which was maintained at 28 weeks (−79%), but developed new brain metastases at 46 weeks. Patients 2, 4, and 6 exhibited SD at 12 weeks (Fig 1A [purple lines]); patients 2 and 6 elected to undergo alternate treatments because of persistent disease. Patients 3, 5, and 8 experienced PD at 6, 6, and 12 weeks, respectively (Fig 1A [red lines]).
Overall, by irRC, seven of 10 patients achieved best responses of CR, PR, or SD. Of three patients who experienced PD after four doses of ipilimumab monotherapy (patients 1, 2, and 3), one achieved a CR, one had SD, and one experienced PD. With a median follow-up of 187 weeks (range, 140 to 220 weeks), five of 10 patients were alive, and two of 10 remained in sustained CRs (Data Supplement).
Polyclonal MART1-Specific IL-21–Primed CTLs Persist In Vivo When Transferred With Concurrent Anti–CTLA-4
A majority of patients had nearly undetectable pre-existing PB mononuclear cell (PBMC) frequencies of endogenous MART1-specific multimer-binding CTLs (median, ≤ 0.05%; range, ≤ 0.05 to 0.21%). Persistence of the infused CTLs was documented for 10 of 10 patients at 6 weeks and for seven of seven evaluable patients at 12 weeks, with median frequencies of 1.6% (range, 0.3% to 2.9%) and 1.1% (range, 0.3% to 2.2%), respectively (Fig 2). The transferred CTLs could be detected for as long as the patients donated PBMCs for analysis, regardless of their tumor response at 12 weeks, with the exceptions of patients 9 and 5, who each received the equivalent of prednisone 1 mg/kg. Both experienced a gradual decline in the frequency of transferred cells to undetectable levels at 6 and 17 weeks, respectively.
Fig 2.

Kinetics of in vivo persistence of melanoma-reactive polyclonal cytotoxic T cells (CTLs). (A) Percent multimer-positive CD8+ T cells (y-axis) in peripheral-blood mononuclear cells (solid circles) collected 7 days (± 2 days) before and at defined time points after infusions is shown for patients who achieved complete remissions, partial responses, or stable disease after treatment. Green arrows indicate CTL infusions, black vertical arrows indicate anti–cytotoxic T-cell lymphocyte antigen-4 infusions, asterisks indicate the start of an alternate treatment, pound signs indicate comfort care, orange arrows indicate concurrent corticosteroid therapy, and blue horizontal arrows indicate ongoing monitoring. (B) The same analysis performed for patients who experienced disease progression after treatment. IL-2, interleukin-2.
Transferred CTLs Detected in PB Exhibit or Acquire Phenotypic and Functional Characteristics of Long-Lived Memory T Cells in Patients Achieving CRs, PRs, or SD
Regardless of the levels detected on multimer-positive CTLs at the time of infusion (Data Supplement), CTL tracking in patients who achieved CRs, PRs, or SD documented a significant increase in the frequency of multimer-positive cells expressing CD28 (P < .05), CD27 (P < .05), CD127 (P < .005), CD62L (P < .05), and CCR7 (P < .005) at 12 weeks (Fig 3A [left column]). By contrast, although the statistical power to detect differences was limited because of the small sample size (n = 3), the expression of CD28 was lost (P < .05), the expression of CD27 and CD127 decreased, and CD62L and CCR7 were never acquired on the transferred cells in patients with PD (Fig 3A [right column]). The expression of CD57 and programmed death-1 (PD1), associated with an exhausted or activated or exhausted phenotype, respectively, was increased on transferred cells found in PBMCs of patients with PD, but not in patients who achieved CRs, PRs, or SD (Fig 3B). We were unable to obtain tumor tissue after treatment to assess the expression of these markers on cells that had reached the tumor microenvironment.
Fig 3.

Phenotypic and functional characteristics of transferred melanoma-reactive cytotoxic T cells (CTLs). (A) Expression of CD27, CD28, CD127, CD62L, and CCR7 (y-axis) on gated multimer-positive cells for CD8+ CTL products immediately before infusion and after 3, 6, 9, and 23 weeks in vivo for patients who achieved complete remissions (CRs), partial responses (PRs), or stable disease (SD). (B) The same analysis in panel A for expression of CD57 and programmed death-1 (PD1; y-axis) performed for patients who experienced progressive disease (PD). (C) Open symbols indicate patients who achieved CRs, PRs, or SD (left column); solid symbols indicate patients who experienced PD (right column). Left plots: percentage of cells within the infusion products producing interferon gamma (IFN-γ) in response to MART1 peptide and in peripheral-blood mononuclear cells (PBMCs) before and 6 and 12 weeks after the CTL infusion; right plots: respective percentages of tumor necrosis factor alpha (TNF-α) and interleukin-2 (IL-2) cells among IFN-γ–positive cells. (D) Mean intranuclear Ki67 expression of endogenous CD8+ multimer-negative cells (black columns) and preinfusion CTL products and multimer-positive CD8+ T cells (gray columns) at indicated time points for all patients combined. Two-tailed paired t tests were used for statistical analysis. NS, not significant. (*) P < .05. (†) P < .005. (‡) P < .001.
The functional profile of transferred CTLs was determined by gating for IFN-γ–positive cells (Fig 3C). Before cyclophosphamide infusions, less than 0.2% of IFN-γ–producing cells could be detected in PBMCs, including in patients with low but detectable levels of MART1-specific cells, attesting to the lack of pre-existing, functional PB CTLs. Consistent with the maintenance of CD28 expression on the generated cells, preinfusion CTLs secreted IL-2 in addition to IFN-γ and tumor necrosis factor alpha (TNF-α) on exposure to cognate antigen.36,37 In patients with CRs, PRs, or SD, transferred CTLs continued to produce all three cytokines 6 and 12 weeks post-transfer (Fig 3C [left column]) and for as long as transferred CTLs could be detected by multimer staining (Data Supplement). However, no IFN-γ–secreting cells could be detected post-transfer in patients who had PD (Fig 3C [right column]).
We then investigated whether the infused CTLs maintained the potential to divide, as reflected by the expression of Ki-67 in vivo.38 Before infusions, multimer-negative CD8+ T cells contained a mean of 1.1% Ki-67–positive cells, and polyclonal multimer-positive CTL products contained a mean of 81.2% Ki-67–positive cells 14 days after in vitro stimulation (Fig 3D [left columns]). Between 0 and 11 weeks after transfer, Ki-67 expression in transferred multimer-positive CTLs progressively decreased, and expression in host multimer-negative CD8+ T cells transiently increased (Fig 3D [middle columns]). By 12 weeks after CTL infusion, after the completion of exogenous subcutaneous IL-2 and anti–CTLA-4, a small fraction of the transferred CTLs were Ki-67 positive (mean, 3.9%; Fig 3D [right columns]). Expression of this proliferation marker in the multimer-positive cells was significantly higher compared with that in host multimer-negative CD8 T cells, which had returned to pretreatment levels (mean, 1.14%; P < .001). When directly compared, cell products administered to patients with PD did not seem less functional than products infused into patients with CRs, PRs, or SD (Data Supplement). Although no correlation with overall survival could be detected, the quantity of TNF-α secreted by CTL products in response to cognate antigen was associated with a decreased likelihood of progression-free survival, and CD57 expression on infused cells after 3 weeks in vivo was associated with an increased likelihood of progression-free survival (Data Supplement).
Evidence of Epitope Spreading in Patients Who Achieved CRs, PRs, or SD
We sought to identify and assess reactivity pre- and post-therapy to melanoma antigen epitopes that could bind to all expressed MHC antigens and could be recognized by host CD4+ and CD8+ T cells. ELISpot analyses were used to assess reactivity of patient-derived T cells toward overlapping peptides spanning the melanoma-associated proteins MART1, NY-ESO1, gp100, tyrosinase, and MAGE A3 (Appendix Fig A1, online only). All patients who achieved CRs, PRs, or SD demonstrated a significantly increased reactivity to the melanoma-associated proteins at one or more time point after CTL infusion (Appendix Fig A1A). In contrast, patients who experienced PD (Appendix Fig A1B) either developed no new reactivity (patient 3) or did not have a significant increase in reactivity (patients 5 and 8). Detectable reactivity to melanoma-associated antigens was also not observed in three other patients with metastatic melanoma who declined therapy (Data Supplement) or in five patients who received CTL clones alone (Data Supplement).
DISCUSSION
We report, to our knowledge, the first prospective use in an ambulatory setting of antigen-specific PB-derived CTLs in combination with CTLA-4 blockade and demonstrate induction of durable antitumor responses in patients with metastatic melanoma.39 Although the number of patients who participated in this initial study was limited, the results are encouraging compared with those of each individual modality.4,17,21 Seven of 10 patients achieved a CR, PR, or SD as best response by irRC. Two patients remained in sustained CRs without additional therapy more than 3 and 4 years later, respectively. These results were achieved with a toxicity profile comparable to that of ipilimumab monotherapy.31
Compared with our previous trials, infused CTLs in this study consisted of polyclonal, pMHC multimer-sorted T-cell lines that had undergone shorter ex vivo manipulation (≤ 6 weeks) and fewer cell divisions.4,6,7,14 The transferred CTLs persisted in vivo (seven of seven evaluable patients at 12 weeks), which is in sharp contrast with our previous results (two of 11 CTL clones beyond 2 weeks).4 CTLs were exposed to IL-21 during primary stimulation, which has been shown to promote retention of CD27, CD28, and CD127 on naïve T cells.8,10-13 Although this cannot be precisely determined retrospectively, it is likely that the infused cells in our trial originated mostly from the autologous IL-21–responsive naïve T-cell pool, because few if any MART1-specific multimer-positive cells were detected preinfusion.
Patients who achieved CRs, PRs, or SD shared a number of biologic correlates. Compared with the infused product, persisting CTLs expressed or acquired phenotypic and functional characteristics associated with long-lived memory T cells (including markers associated with survival [CD28, CD27, and CD127]40,41; lymph-node homing [CD62L and CCR7]; and production of IFN-γ, TNF-α, and IL-2),41,42 suggesting the preferential survival or expansion of this subset. These favorable characteristics may also have been facilitated by CTLA-4 blockade such that infused CD28+ CTLs experienced unopposed stimulation or enhanced signaling through binding of the costimulatory ligands CD80 and/or CD86. Although these ligands are present at low levels on more than 80% of melanoma cells,43 higher expression can be induced by inflammatory cytokines, such as IFN-γ, which would have been provided by the responding transferred CTLs.44 Consequently, transferred CD28+ cells may have gained proliferative or survival advantages related to B-cell lymphoma extra large (Bcl-XL) expression and autocrine production of IL-2.40 In contrast, no IFN-γ–secreting cells could be detected posttransfer in patients with PD.
Epitope spreading was observed in all patients with CRs, PRs, and SD, likely a consequence of transferred CTL–induced tumor lysis and heightened T-cell activation fostered by anti–CTLA-4.24 Released tumor antigens presented by local antigen-presenting cells may have promoted activation of new responses to nontargeted melanoma-associated proteins.23,24,45 Whether such T-cell responses, toward wild-type or nonevaluated tumor-specific mutations,46 induced the decrease in tumor size observed in some patients cannot be ascertained. However, the combination may represent a strategy to specifically increase the number and strength of T cells targeting multiple antigens of the patient’s own tumor, which may be particularly relevant when targeting nononcogenic antigens, such as MART1.47
Although the intrapatient pre- and postinfusion immunobiologic results suggest the combination of anti–CTLA-4 plus adoptive T-cell therapy mediates the observed effects, tumor regressions observed in this single-arm phase I study cannot be definitely attributed to either component because of the lack of matched comparison groups. Further study is warranted to determine the specific contributions of anti–CTLA-4, persistence of memory T cells (potentially facilitated by anti–CTLA-4), and epitope spreading. Long-term CRs and PRs were not observed in all patients, suggesting that mechanisms limiting antitumor efficacy are still operative. Indeed, transferred CTLs isolated from patients with PD expressed the exhaustion or activation markers CD57 and PD1,36,37 suggesting interventions that block the PD1–PD1 ligand axis may further enhance therapeutic activity.48 The finding that CTLs from the PB of patients with SD, PRs, or CRs did not express PD1 is distinct from reports that PD1 expression in the tumor environment reflects the presence of activated functional CTLs.49 This could, however, reflect the fact that tumor-reactive CTLs in PB represent a renewable source of memory cells that can migrate and function in the tumor environment. Overall, the continued development of improved cell-culture methods and immune-modulatory antibodies holds great promise for designing immunotherapeutic combinations that have increased efficacy and reduced toxicity.
Additional material
Supplementary data supplied by authors.
Appendix
Fig A1.

Reactivity to nontargeted epitopes. Interferon gamma (IFN-γ) spots per 105 peripheral-blood mononuclear cells (PBMCs; y-axis) at indicated time points (x-axis) before and after the infusion of polyclonal cytotoxic T cells (CTLs) generated in the presence of interleukin-21 for patients who (A) achieved complete remissions, partial responses, or stable disease or (B) experienced disease progression after receiving treatment. Pound signs indicate patients who had received prior ipilimumab, green arrows indicate CTL infusions, and horizontal lines indicate ipilimumab administration with the total number of doses received indicated immediately above. The scales of the y-axis for graphs for patients 1 (maximum, 700 spots/105 PBMCs) and 4 (maximum, 3,000 spots/105 PBMCs) are different from all others (maximum, 400 spots/105 PBMCs). Two-tailed paired t tests were used for statistical analysis. NS, not significant. (*) P < .05. (†) P < .005.
Footnotes
Supported by the Cancer Research Institute; by a National Institutes of Health Career Development in Pediatric and Medical Oncology Award K12, and an American Association for Cancer Research (AACR) –American Society of Clinical Oncology Conquer Cancer Foundation Young Investigator Translational Cancer Research Award (A.G.C.); and by a Burroughs Wellcome Fund Translational Scientist Award, Cancer Prevention Research Institute of Texas (CPRIT R1301), and NRF of Korea (NRF-2007-00107 and NRF-2013M3A9D3045719) (C.Y., who is co-leader of the Stand Up To Cancer–AACR/Cancer Research Institute Immunology Dream Team).
Authors’ disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
Clinical trial information: NCT 00871481.
AUTHOR CONTRIBUTIONS
Conception and design: Aude G. Chapuis, Philip D. Greenberg, Cassian Yee
Financial support: Philip D. Greenberg
Provision of study materials or patients: Kim A. Margolin, Jedd D. Wolchok
Collection and assembly of data: Aude G. Chapuis, Ilana M. Roberts, John A. Thompson, Kim A. Margolin, Shailender Bhatia, Sylvia M. Lee, Heather L. Sloan, Ivy P. Lai, Erik A. Farrar, Felecia Wagener, Kendall C. Shibuya, Jianhong Cao, Cassian Yee
Data analysis and interpretation: Aude G. Chapuis, Ilana M. Roberts, Kim A. Margolin, Erik A. Farrar, Jedd D. Wolchok, Philip D. Greenberg, Cassian Yee
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
T-Cell Therapy Using Interleukin-21–Primed Cytotoxic T-Cell Lymphocytes Combined With Cytotoxic T-Cell Lymphocyte Antigen-4 Blockade Results in Long-Term Cell Persistence and Durable Tumor Regression
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Aude G. Chapuis
Research Funding: Juno Therapeutics
Patents, Royalties, Other Intellectual Property: Patent (not related to this research)
Ilana M. Roberts
No relationship to disclose
John A. Thompson
Consulting or Advisory Role: Seattle Genetics, Genentech, Eisai, Celldex
Research Funding: Bristol-Myers Squibb (Inst), Merck (Inst), Agensys (Inst), Seattle Genetics (Inst), Pfizer (Inst)
Kim A. Margolin
Honoraria: Genentech
Consulting or Advisory Role: ImaginAb, Amgen, Bristol-Myers Squibb
Travel, Accommodations, Expenses: Bristol-Myers Squibb
Shailender Bhatia
Research Funding: Bristol-Myers Squibb (Inst), EMD Serono (Inst), Merck (Inst), NantKwest (Inst), Immune Design (Inst), OncoSec (Inst), Amgen (Inst)
Sylvia M. Lee
Research Funding: Juno Therapeutics, MedImmune
Heather L. Sloan
Research Funding: Multiple industry studies support salary
Ivy P. Lai
No relationship to disclose
Erik A. Farrar
No relationship to disclose
Felecia Wagener
No relationship to disclose
Kendall C. Shibuya
No relationship to disclose
Jianhong Cao
No relationship to disclose
Jedd D. Wolchok
Stock or Other Ownership: Potenza Therapeutics, Tizona Therapeutics
Consulting or Advisory Role: Bristol-Myers Squibb, Merck, MedImmune, Genentech, Polaris, BeiGene, Adaptive Biotechnologies, Amgen, Polynoma, ZIOPHARM Oncology
Research Funding: Bristol-Myers Squibb (Inst), Merck (Inst), Genentech (Inst)
Patents, Royalties, Other Intellectual Property: Patent on oncolytic Newcastle disease virus; patent on xenogeneic DNA vaccines
Travel, Accommodations, Expenses: Bristol-Myers Squibb, Genentech
Philip D. Greenberg
Employment: Juno Therapeutics, Inovio Pharmaceuticals, Innate Pharma
Stock or Other Ownership: Juno Therapeutics, Inovio Pharmaceuticals, Innate Pharma
Consulting or Advisory Role: Juno Therapeutics, Inovio Pharmaceuticals, Innate Pharma
Research Funding: Juno Therapeutics
Patents, Royalties, Other Intellectual Property: Juno Therapeutics
Travel, Accommodations, Expenses: Juno Therapeutics, Inovio Pharmaceuticals, Innate Pharma
Cassian Yee
Honoraria: Bristol-Myers Squibb
Consulting or Advisory Role: Oxis Biotech, Adaptive Biotechnologies, Immatics US
Patents, Royalties, Other Intellectual Property: Patent held by MD Anderson Cancer Center and licensed to Immatics US; patent held by MD Anderson Cancer Center and licensed to Immatics US (Inst)
REFERENCES
- 1.Stromnes IM, Schmitt TM, Chapuis AG, et al. Re-adapting T cells for cancer therapy: From mouse models to clinical trials. Immunol Rev. 2014;257:145–164. doi: 10.1111/imr.12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang JC. The adoptive transfer of cultured T cells for patients with metastatic melanoma. Clin Dermatol. 2013;31:209–219. doi: 10.1016/j.clindermatol.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 4.Chapuis AG, Thompson JA, Margolin KA, et al. Transferred melanoma-specific CD8+ T cells persist, mediate tumor regression, and acquire central memory phenotype. Proc Natl Acad Sci USA. 2012;109:4592–4597. doi: 10.1073/pnas.1113748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunder NN, Wallen H, Cao J, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallen H, Thompson JA, Reilly JZ, et al. Fludarabine modulates immune response and extends in vivo survival of adoptively transferred CD8 T cells in patients with metastatic melanoma. PLoS One. 2009;4:e4749. doi: 10.1371/journal.pone.0004749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yee C, Thompson JA, Byrd D, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: In vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. 2002;99:16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapuis AG, Ragnarsson GB, Nguyen HN, et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med. 2013;5:174ra27. doi: 10.1126/scitranslmed.3004916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klebanoff CA, Gattinoni L, Restifo NP. Sorting through subsets: Which T-cell populations mediate highly effective adoptive immunotherapy? J Immunother. 2012;35:651–660. doi: 10.1097/CJI.0b013e31827806e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hinrichs CS, Spolski R, Paulos CM, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008;111:5326–5333. doi: 10.1182/blood-2007-09-113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y, Bleakley M, Yee C. IL-21 influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. J Immunol. 2005;175:2261–2269. doi: 10.4049/jimmunol.175.4.2261. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Yee C. IL-21 mediated Foxp3 suppression leads to enhanced generation of antigen-specific CD8+ cytotoxic T lymphocytes. Blood. 2008;111:229–235. doi: 10.1182/blood-2007-05-089375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui W, Liu Y, Weinstein JS, et al. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity. 2011;35:792–805. doi: 10.1016/j.immuni.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pollack SM, Jones RL, Farrar EA, et al. Tetramer guided, cell sorter assisted production of clinical grade autologous NY-ESO-1 specific CD8(+) T cells. J Immunother Cancer. 2014;2:36. doi: 10.1186/s40425-014-0036-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4:336–347. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- 16.Sharma P, Wagner K, Wolchok JD, et al. Novel cancer immunotherapy agents with survival benefit: Recent successes and next steps. Nat Rev Cancer. 2011;11:805–812. doi: 10.1038/nrc3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ribas A, Kefford R, Marshall MA, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31:616–622. doi: 10.1200/JCO.2012.44.6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 20.Thompson JA, Hamid O, Minor D, et al. Ipilimumab in treatment-naive and previously treated patients with metastatic melanoma: Retrospective analysis of efficacy and safety data from a phase II trial. J Immunother. 2012;35:73–77. doi: 10.1097/CJI.0b013e31823735d6. [DOI] [PubMed] [Google Scholar]
- 21.Prieto PA, Yang JC, Sherry RM, et al. CTLA-4 blockade with ipilimumab: Long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res. 2012;18:2039–2047. doi: 10.1158/1078-0432.CCR-11-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolchok JD, Weber JS, Maio M, et al. Four-year survival rates for patients with metastatic melanoma who received ipilimumab in phase II clinical trials. Ann Oncol. 2013;24:2174–2180. doi: 10.1093/annonc/mdt161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ribas A, Timmerman JM, Butterfield LH, et al. Determinant spreading and tumor responses after peptide-based cancer immunotherapy. Trends Immunol. 2003;24:58–61. doi: 10.1016/s1471-4906(02)00029-7. [DOI] [PubMed] [Google Scholar]
- 24.Kvistborg P, Philips D, Kelderman S, et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med. 2014;6:254ra128. doi: 10.1126/scitranslmed.3008918. [DOI] [PubMed] [Google Scholar]
- 25.Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawakami Y, Eliyahu S, Sakaguchi K, et al. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. J Exp Med. 1994;180:347–352. doi: 10.1084/jem.180.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.National Cancer Institute. National Institutes of Health . Common Terminology Criteria for Adverse Events (CTCAE), Version 4.0. http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf. [Google Scholar]
- 28.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 29.Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin Cancer Res. 2009;15:7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 30.Liao X, Li Y, Bonini C, et al. Transfection of RNA encoding tumor antigens following maturation of dendritic cells leads to prolonged presentation of antigen and the generation of high-affinity tumor-reactive cytotoxic T lymphocytes. Mol Ther. 2004;9:757–764. doi: 10.1016/j.ymthe.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 31.Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30:2691–2697. doi: 10.1200/JCO.2012.41.6750. [DOI] [PubMed] [Google Scholar]
- 32.Ribas A, Hodi FS, Callahan M, et al. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365–1366. doi: 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- 33.Harding JJ, Pulitzer M, Chapman PB. Vemurafenib sensitivity skin reaction after ipilimumab. N Engl J Med. 2012;366:866–868. doi: 10.1056/NEJMc1114329. [DOI] [PubMed] [Google Scholar]
- 34.Ho WY, Nguyen HN, Wolfl M, et al. In vitro methods for generating CD8+ T-cell clones for immunotherapy from the naïve repertoire. J Immunol Methods. 2006;310:40–52. doi: 10.1016/j.jim.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 35.Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. 2012;12:623–635. doi: 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- 36.Ragheb JA, Deen M, Schwartz RH. CD28-Mediated regulation of mRNA stability requires sequences within the coding region of the IL-2 mRNA. J Immunol. 1999;163:120–129. [PubMed] [Google Scholar]
- 37.Topp MS, Riddell SR, Akatsuka Y, et al. Restoration of CD28 expression in CD28- CD8+ memory effector T cells reconstitutes antigen-induced IL-2 production. J Exp Med. 2003;198:947–955. doi: 10.1084/jem.20021288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 39.Butler MO, Friedlander P, Milstein MI, et al. Establishment of antitumor memory in humans using in vitro-educated CD8+ T cells. Sci Transl Med. 2011;3:80ra34. doi: 10.1126/scitranslmed.3002207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McAdam AJ, Schweitzer AN, Sharpe AH. The role of B7 co-stimulation in activation and differentiation of CD4+ and CD8+ T cells. Immunol Rev. 1998;165:231–247. doi: 10.1111/j.1600-065x.1998.tb01242.x. [DOI] [PubMed] [Google Scholar]
- 41.Kimura MY, Pobezinsky LA, Guinter TI, et al. IL-7 signaling must be intermittent, not continuous, during CD8⁺ T cell homeostasis to promote cell survival instead of cell death. Nat Immunol. 2013;14:143–151. doi: 10.1038/ni.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: Implications for vaccine development. Nat Rev Immunol. 2002;2:251–262. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 43.Bernsen MR, Håkansson L, Gustafsson B, et al. On the biological relevance of MHC class II and B7 expression by tumour cells in melanoma metastases. Br J Cancer. 2003;88:424–431. doi: 10.1038/sj.bjc.6600703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hersey P, Si Z, Smith MJ, et al. Expression of the co-stimulatory molecule B7 on melanoma cells. Int J Cancer. 1994;58:527–532. doi: 10.1002/ijc.2910580413. [DOI] [PubMed] [Google Scholar]
- 45.Robert L, Tsoi J, Wang X, et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin Cancer Res. 2014;20:2424–2432. doi: 10.1158/1078-0432.CCR-13-2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 47.Chandran SS, Paria BC, Srivastava AK, et al. Persistence of CTL clones targeting melanocyte differentiation antigens was insufficient to mediate significant melanoma regression in humans. Clin Cancer Res. 2015;21:534–543. doi: 10.1158/1078-0432.CCR-14-2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gros A, Robbins PF, Yao X, et al. PD-1 identifies the patient-specific CD8⁺ tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124:2246–2259. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.