

Introduction
Eating disorders with severe electrolyte abnormalities are serious diseases often encountered in the clinical practice [1]. Their association with chronic kidney disease has been defined as a prescription for disaster, thus underlining the mutual interactions between salt-losing interstitial nephropathies, most commonly encountered in this setting, and the combination of malnutrition and diuretic abuse [2].
There are several life-threatening complications of electrolyte imbalance, including cardiac arrhythmia and involvement of the peripheral and central nervous systems [1–5]. The presence of renal failure and of contracted vascular volume may enhance the risks and render the correction of the complex electrolyte alterations particularly difficult.
Furthermore, the differential diagnosis of the complications may be difficult and the clinical nephrologist should be aware of the main features of the life-threatening neurological diseases, as exemplified by the present case [6–9].
Case presentation
A 40-year-old woman came to the emergency room, accompanied by her mother, complaining of the progressive development of severe asthaenia, diffuse muscular pain and malaise. ‘I feel like I’m going to die’, she said repeatedly, ‘even if I do not know why’.
At admission, the clinical evaluation revealed a collaborative, suffering, lean and hypotensive woman (blood pressure 90/60 mmHg, last reported body weight 55 kg, height 175 cm), with normal heart rate (76 bpm, rhythmic), who was severely asthaenic. She looked dehydrated and her skin was diffusely hyperpigmented with hypertrichosis.
She denied any major problem in her past clinical history. The recent clinical history was also uneventful, except for an episode of gastroenteritis, about 10 days previously, after which she slowly improved for a few days, followed by the progressive development of the presenting complaints. Renal ultrasounds, performed in the emergency room, revealed kidneys of normal size, without signs of obstruction. Chest X-ray was normal. The patient was oligoanuric; she reported decreasing urinary output in the last few days and catheterization yielded <50 mL of urine. The EKG is shown in Figure 1.
Fig. 1.
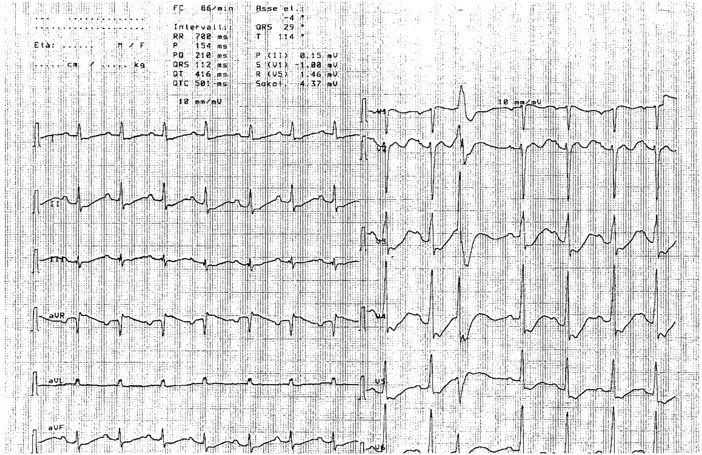
EKG showing diffuse alterations of the repolarization phase; the Q–U prolongation is extreme and the distinction between T wave and U wave is lost, one ventricular extrasystole only is observed, in line with a chronic adaptation.
No previous blood test was available and the patient recalled having performed the last ones a few years before, when, on the occassion of the death of her father, she had undergone a general evaluation for having lost ‘considerable weight’.
The first blood tests performed in the emergency room revealed: serum creatinine, 5.55 mg/dL, active inflammatory signs [C-reactive protein, 41.2 mg/dL (normal < 0.8 mg/dL); fibrinogen, 938 mg/dL], massive leukocytosis (WBC, 67 000), suggestive of both severe haemoconcentration and infection, with severe sodium and potassium imbalance (Na, 107 mmol/L; K, 1.61 mmol/L), moderate, compensated metabolic acidosis (pH, 7.34; HCO3, 19.4 mmol/L; base excess, −4 mmol/L) and moderate hyperglycaemia (160 mg/dL). Haemoglobin level (15 g/dL) was normal. Elevated levels of creatinine–phosphokinase (26 186 UI/L) indicated concomitant rhabdomyolysis, probably as a result of infection and severe electrolyte imbalance, potentially contributing to the acute renal failure.
In the face of the complex metabolic disorder, a hierarchy of interventions was defined, starting from potassium supplementation with very slow sodium infusion, aimed at raising the sodium level by no more than 10–12 mEq/day. As it will be further discussed, the working diagnostic hypotheses are of an acute renal failure superimposed on a chronic kidney disease in the context of diuretic abuse. In spite of the need for rapid correction of the potassium and sodium levels, each of the interventions has, however, important limits and drawbacks and the anuric status limits the use of the most widely employed formulae guiding assessment and therapy in severe sodium imbalance (Table 1)
Table 1.
Major problems and risks and hierarchy of interventions need in the patient
| Major problem and major risks | Hierarchy main features of interventions | Management and limits |
|---|---|---|
| Hyponatraemia | First, at the same time of potassium repletion. | The most widely used formulae employed in urinary assessment (Na, osmolarity or both), which are not applicable to anuric patients and are often of limited value in patients with chronic renal damage, as they presuppose that the kidneys have maintained their concentration ability, frequently lost in interstitial nephropathies, such as those frequently encountered in eating disorders and diuretic abuse. |
| Relative increase in water: cerebral oedema | Very slow rate indicated (8–20 mEq/day), slower in nonsymptomatic patients or in patients with chronic disorders. | |
| The effect of volume repletion on electrolyte levels in a presumably chronic hypovolaemic state is difficult to predict. | ||
| Rapid correction: osmotic demyelination | The laboratory errors for sodium reach 5% at low sodium levels, thus rendering management guided by serial sodium levels quite difficult. | |
| Hypokalaemia | First, together with very slow sodium correction. | No indications for the velocity of correction reported. |
| Unpredictable effect of volume repletion on potassium levels. | ||
| Cardiac arrhythmia; peripheral paralysis | Monitoring via EKG changes. There is a practical limit to the use of high potassium concentration when the correction starts on a peripheral vein for phlebitis and thrombosis. A central venous catheter is of great use, but often not immediately available in the emergency room. | |
| Acidosis | Last, after potassium correction. | The presence of acidosis points to the presence of chronic kidney disease (in the absence of recent diarrhoea), as alkalosis is more commonly associated with diuretic use and volume contraction; acidosis should be corrected slowly and only after potassium correction with normalization of the EKG alterations. |
| Not life-threatening; impairment of kidney function | While there are signs of rhabdomyolysis and alkalinization of the urine is effective for improving excretion of toxic metabolites, the risks of hypokalaemia clearly overcome those of not correcting a potential factor for acute kidney failure. | |
| Volume depletion | Slow correction, with intravenous isotonic saline; the low sodium level suggests that water is still in excess as compared with sodium. | The patient is anuric; diuretics are contraindicated in the case of volume and potassium depletions, even if they are occasionally used in hyponatraemic patients, on the account of the relative excess of water with respect to sodium. |
| Acute cardiovascular collapse, in particular in combination with severe hypokalaemia | The entity of volume depletion is difficult to assess, as only clinical parameters are available, in the wait for central vein catheterization, which may help in guiding infusion rate, based upon central venous pressure. | |
| The urine output is the clue for tailoring interventions, for the risk of a relatively rapid shift between and hypovolaemic the hypervolaemic states. | ||
| Renal failure | Postpone dialysis, when needed, at least for partial sodium correction. | Dialysis may trigger a too rapid correction of electrolytes, with an increased risk for central pontine demyelinization; dialysis with hypotonic and isotonic solutions has been performed occasionally, but the management is still based on empiric and scattered experiences. |
| No immediate threats, as the patient is hypovolaemic. | Careful volume management | |
| Tetraparesis | Immediate need for intubation. | The need for intubation and sedation impairs basing or tailoring the clinical management upon symptoms of hyponatraemia (such as nausea, asthaenia and confusion). |
| Respiratory failure |
About 12 h after admission, in the absence of cognitive deficits, she suddenly developed flaccid tetraplegia and restrictive respiratory failure. A prompt transfer to the intensive care unit (ICU) was required because of respiratory failure. She was sedated and mechanical ventilation was started.
At transfer to the ICU and immediately after intubation (pO2, 193 mmHg; pCO2, 35.4 mmHg), the major electrolyte levels were the following: Na, 114 mmol/L; K, 0.8 mmol/L; HCO3, 15 mmol/L; lactate, 0.8 mmol/L; chloride, 83 mmol/L; ionized calcium, 1.16 mmol/L; pH, 7.255; base excess, −12 mmol/L; anion gap, 17 mmol/L. Calculated osmolarity was 278 mOsm/kg (according to the simplified formula: Osmolality = Sodium × 2 + Glucose/18 + BUN/2.8), the effect of the low sodium being partly counterbalanced by the high BUN (BUN, 116 mg/dL).
On account of the recent gastrointestinal viral disease, the flaccid paralysis with intact sensorium and the rapid course, the neurologist suspected Guillain–Barré syndrome (GBS) and performed a lumbar puncture, leading to a picture of albuminocytological dissociation (WBC, 7 mm3; proteins, 63 mg/dL; glucose, 90 mg/dL). In parallel with the slow correction of the electrolyte imbalance (Figure 2), leading also to slow volume repletion, high-dose immunoglobulins were started (0.4 mg/kg/day for 5 days), with prompt clinical improvement and discharge from the ICU 7 days later, to be transferred to the internal medicine ward.
Fig. 2.

The main biochemical parameters during hospitalization in the emergency room.
Diuresis gradually restarted in the following 12 h, reaching 800 mL by the second day. In keeping with the hypothesis of chronic kidney disease with impaired concentration ability, urinalysis showed low urine specific gravity (1010) and relatively high spot urinary sodium (39 mEq/L, with serum sodium, 121 mEq/L).
At discharge from the internal medicine ward 2 weeks later, serum creatinine was 1.38 mg/dL, BUN 16 mg/dL, creatinine clearance (24-h urine collection) ranged from 50 to 60 mL/min, proteinuria ranged from 0.2 to 0.3 g/day, Na 141 mmol/L, K 4.5 mmol/L, HCO3 24 mEq/L and PCR 0.04; urinalysis was normal. Renal ultrasounds revealed a decreased corticomedullary differentiation and slightly decreased thickness of the renal cortex, in keeping with the presence of chronic parenchymal disease; the presence of markedly hyperechogenic papillae is suggestive of nephrocalcinosis.
Discussion
The case discussed here regards a woman presenting with severe and combined electrolyte disorder, referred to the emergency room after an intercurrent acute disease, who, a few hours after hospitalization, suddenly developed a severe neurologic disorder, with the need for intubation.
The interest in the case resides in the fact that it gathers three life-threatening conditions, hyponatraemia, hypokalaemia and tetraparesis, with complex clinical management, and two major diagnostic clues.
The first diagnostic clue regards the origin of the complex electrolyte disorder, in combination with renal failure; the quota of chronic renal failure is critical for the further management of the patient and for relying on the common formulae for electrolyte replacement (Table 1).
As it was recently underlined in a comprehensive review, there are two major diagnostic and therapeutic approaches to the hyponatraemic patient: a traditional one, based upon a combination of clinical and laboratory parameters often centred on the use of diagnostic algorithms, and a physiology-based approach, centred on the mechanisms that might have led to the development of hyponatraemia [10]. The authors found, in an extensive systematic literature search, 10 different formulae, eight out of 10 including extracellular fluid volume and eight out of 10 employing urinary sodium excretion or urinary osmolarity or both [10]. As, however, our patient was anuric and, as it will be discussed, chronic kidney disease was likely to be present, the urine-based formulae are of limited or no use. Furthermore, the assessment of extracellular fluid volume is mainly clinical (estimated upon blood pressure, reported to be low in the past, and weight loss, not measured in the last period, a relatively common attitude in eating disorders) with the support of urinary osmolarity or urinary sodium (Table 1). No reliable formula is available for calculating the water deficit in severely hyponatraemic patients, in contrast to hypernatraemic patients [10–12]. The most recent review by Hoorn and Zieste, indeed, specifically warns against relying ‘too heavily’ on the assessment of the extracellular fluid volume [12].
In keeping with a physiopathology-based, diagnosis-oriented approach, therefore, we focused on the first diagnostic clues: what was the most likely diagnosis and whether there was an underlying chronic kidney disorder. Several recent articles and reviews point to the importance of the rapidity of development of the electrolyte disorder (where acute is usually defined as evolving within 48 h). In our patient, however, no previous test was available.
The hypothesis of an acute problem superimposed on chronic kidney disease was, therefore, based upon the clinical history of malaise and progressive clinical impairment in the last days, the recent gastroenteritis and the signs of rhabdomyolysis. Furthermore, while the haemoconcentration might have partially masked anaemia, the presence of normal serum ionized calcium and haemoglobin suggest the presence of a milder (if any) kidney disease.
Conversely, there were two major clues for a chronic electrolyte disorder: the profound electrolyte imbalance was remarkably well tolerated, both from the cardiac point of view (Figure 1), as, in spite of the presence of polarization abnormalities, a potassium level of around 1 mEq/L led to one ventricular extrasystole only, and from the neurological point of view, as the patient was slowed, but entirely conscious, with a sodium level of <110 mEq/L. Secondly, serum creatinine needs several days to reach 5.5 mg/dL, even if the presence of an underlying condition may affect the kinetic pattern.
Therefore, the most likely diagnosis was an acute effect of a gastrointestinal disease, with acute sodium and potassium losses, in the context of chronic sodium and potassium depletion and chronic kidney disease.
The most likely cause of the chronic depletion of both sodium and potassium encountered in a very thin, relatively young, otherwise healthy woman, is an eating disorder, combined with chronic diuretic abuse. The chronic adjustment to a less severe electrolyte imbalance is a feature of anorexia and related eating disorders [1–3]. In this setting, it is not surprising that the patient initially denied diuretic use and a history of anorexia–bulimia, as denial is part of the usual, at least initial, behavioural pattern of patients with severe eating disorders.
Several kidney diseases have been described in patients with eating disorders; these include electrolyte disturbances; chronic tubulointerstitial nephritis, in particular in the two forms of hypokalaemic nephropathy and nephrocalcinosis; renal stone disease; unexplained chronic renal failure, eventually linked also to the exposure to toxic substances, has also been described, such as ‘Chinese herbs nephropathy’ or interstitial nephropathy related to Senna abuse [4,5].
Thus, we initially targeted the metabolic correction to a slow rise in serum sodium, in keeping with the hypothesis of a chronic oligosymptomatic picture, starting with slow infusion of normal saline solution with 40 mEq/L of potassium, the maximum usually allowed in a peripheral vein, and scheduling electrolyte and acid base controls every 4 h; the empiric indication of 100 mL/h of normal saline per day in the first 2 days was initially halved to 50 mL/h (Table 1; [11]).
The sudden development of neurologic problems, however, prompted a rapid re-evaluation of the electrolyte correction, with a second major diagnostic clue. In fact, the correction of severe hyponatraemia calls for a differential diagnosis with central pontine myelinolysis, a rare disease, potentially causing tetraplegia. This is characterized by central pontine, and occasionally also extrapontine, demyelinization, usually occurring in the context of rapid correction of hyponatraemia. The disease has, however, been described also in cases in which sodium correction was apparently in keeping with the guidelines, suggesting the increase in serum sodium by no more than 8–10 mmol/day [8,9].
Several predisposing factors are described, including malnutrition, chronic alcohol abuse and pregnancy. This diagnosis covers several aspects, present in our case, including quadriplegia, onset after correction of electrolyte imbalance and malnutrition [8,9].
However, the development of myelinolysis is usually less sudden (and the same is a distinguishing feature from the different forms of hypokalaemic paralysis, which, in contrast with our case, only rarely affect respiratory muscles); furthermore, as a rule, myelinolysis occurs after an initial improvement of general status, following the correction of hyponatraemia. In our patient, the onset of the neurological disorder was almost immediate, thus pointing to the search for an alternative diagnosis.
Most importantly, the initial symptoms of myelinolysis are usually mutism and dysarthria and, in keeping with a central origin, an increase of deep tendon reflexes; on the contrary, our patient was fully conscious, albeit severely asthaenic, and displayed severe hyporeflexia, indicating a peripheral nervous problem or muscular problem.
Our patient suffered from GBS, a relatively rare neurological disease, occasionally linked with severe electrolyte imbalance; the main point of interest for the nephrologist is indeed in the differential diagnosis with central pontine myelinolysis.
GBS is a rare disease (reported incidence, 1–3/100 000 individuals per year) and an important cause of acute neuromuscular paralysis. While often self-limited, GBS is a severe disease, whose mortality ranges from 3 to over 10%, with a risk of up to 30% of long-term sequelae [6,7].
The recent infection was a major diagnostic clue for the diagnosis. The onset of GBS, in fact, often follows an acute infection, but is reported also after other immunological challenges, including vaccines; Campylobacter jejuni is the infection most commonly reported. The main diagnostic hallmarks are rapidly progressive symmetrical weakness in the arms and legs with or without sensory disturbances, hyporeflexia or areflexia and the combination of increased protein concentration and a normal cell count in the cerebrospinal fluid, called also albuminocytological dissociation [6,7]. The disease may progress in a few days, but, especially after gastrointestinal illness, it may present a fulminant onset, after a few days of vague complaints, as occurred in our patient.
As a confirmation to the diagnosis, the diagnostic magnetic resonance of myelinolysis was normal and no positivity for neuronal antibodies was found (GM1, GQ1b, GD1, sulfatides, asialo GM1).
While various therapeutic approaches have been reported for GBS, intravenous immunoglobulin and plasma exchange are considered as the most effective treatments; in the absence of clear indications towards either therapy, immunoglobulins are usually preferred due to their easier management [6,7].
Interestingly, there are a few reports of the use of high-dose immunoglobulins in myelinolysis, with promising results indirectly suggesting a dysimmune pathogenesis in this acute disease as well [13,14]. Conversely, GBS has been occasionally reported also in the context of profound hyponatraemia [15], thus pointing to a possible link between these two diagnoses [14].
Conclusion
In conclusion, the case here described reports an uncommon association between the complex metabolic alterations of chronic diuretic abuse and eating disorder with GBS, only occasionally described in a similar context, thus underlining the importance of early integrated multidisciplinary evaluation of patients with acute renal failure and complex electrolyte imbalances or neurological disorders [14].
Teaching points
Eating disorders, in combination with chronic diuretic abuse, are a common background for acute, severe and potentially deadly electrolyte imbalances, as they determine chronic depletion in particular of the sodium and potassium body pools.
Several chronic kidney diseases have been described in patients with eating disorders; the most common ones are chronic tubulointerstitial nephropathies, including hypokalaemic nephropathy and nephrocalcinosis. Tubulointerstitial nephropathies may in turn enhance electrolyte losses, thus increasing the risks of profound depletion. In the presence of anuria or in the case of chronic kidney disease, the management should mainly rely on clinical grounds and on a strict metabolic surveillance, as the most commonly employed formulae require urinary assessment of osmolarity and urinary electrolytes. Slow sodium correction (<10 mEq/day, slower in the presence of a chronic disorder) is the major therapeutic clue, to avoid severe neurological side effects.
The onset of tetraplegia, in the context of severe electrolyte imbalance, in particular hyponatraemia, calls for differential diagnosis between two main diseases, a peripheral one, GBS, and a central one, central pontine myelinolysis. The latter is linked with rapid correction of sodium levels (more than 10 mEq/day) and bears a poor prognosis.
GBS has been occasionally associated with severe sodium depletion and more commonly with immunological challenges (vaccines) or infectious diseases. The therapy is based upon plasma exchanges or high doses of immunoglobulins. The prognosis is overall favourable in most of the cases.
Acknowledgments
The authors thank E.M. for the trust and the friendship.
Conflict of interest statement. None declared.
References
- 1.Williams PM, Goodie J, Motsinger C D. Treating eating disorders in primary care. Am Fam Physician. 2008;77:187–195. [PubMed] [Google Scholar]
- 2.Luthra M, Davids MR, Shafiee MA, Halperin ML. Anorexia nervosa and chronic renal insufficiency: a prescription for disaster. QJM. 2004;9:167–178. doi: 10.1093/qjmed/hch031. [DOI] [PubMed] [Google Scholar]
- 3.Takakura S, Nozaki T, Nomura Y, et al. Factors related to renal dysfunction in patients with anorexia nervosa. Eat Weight Disord. 2006;11:73–77. doi: 10.1007/BF03327754. [DOI] [PubMed] [Google Scholar]
- 4.Vanhaelen M, Vanhaelen-Fastre R, But P, Vanherweghem JL. Identification of aristolochic acid in Chinese herbs. Lancet. 1994;343:174. doi: 10.1016/s0140-6736(94)90964-4. [DOI] [PubMed] [Google Scholar]
- 5.Abdel-Rahman EM, Moorthy AV. End-stage renal disease (ESRD) in patients with eating disorders. Clin Nephrol. 1997;47:106–111. [PubMed] [Google Scholar]
- 6.Hahn A F. Guillain–Barré syndrome. Lancet. 1998;352:635–641. doi: 10.1016/S0140-6736(97)12308-X. [DOI] [PubMed] [Google Scholar]
- 7.van Doorn P, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain–Barré syndrome. Lancet Neurol. 2008;7:939–950. doi: 10.1016/S1474-4422(08)70215-1. [DOI] [PubMed] [Google Scholar]
- 8.Laureno R, Illowsky Karp L. Myelinolysis after correction of hyponatremia. Ann Intern Med. 1997;126:57–62. doi: 10.7326/0003-4819-126-1-199701010-00008. [DOI] [PubMed] [Google Scholar]
- 9.Kleinschmidt-DeMasters BK, Rojiani AM, Filley CM. Central and extrapontine myelinolysis: then and now. J Neuropathol Exp Neurol. 2006;65:1–11. doi: 10.1097/01.jnen.0000196131.72302.68. [DOI] [PubMed] [Google Scholar]
- 10.Hoorn EJ, Halperin ML, Zietse R. Diagnostic approach to a patient with hyponatremia: traditional versus physiology-based options. Q J Med. 2005;98:529–540. doi: 10.1093/qjmed/hci081. [DOI] [PubMed] [Google Scholar]
- 11.Yeates KE, Singer M, Morton RA. Salt and water, a simple approach to hyponatremia. CMAJ. 2004;170 [PMC free article] [PubMed] [Google Scholar]
- 12.Hoorn EJ, Zietse R. Hyponatremia revisited: translating physiology to practice. Nephron Physiol. 2008;108:46–59. doi: 10.1159/000119709. [DOI] [PubMed] [Google Scholar]
- 13.Deleu D, Salim K, Mesraoua B, El Siddig A, Al Hail H, Hanssens Y. “Man-in-the-barrel” syndrome as delayed manifestation of extrapontine and central pontine myelinolysis: beneficial effect of intravenous immunoglobulin. J Neurol Sci. 2005;237:103–106. doi: 10.1016/j.jns.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 14.Finsterer J, Engelmayer E, Trnka E, Stiskal M. Immunoglobulins are effective in pontine myelinolysis. Clin Neuropharmacol. 2000;23:110–113. doi: 10.1097/00002826-200003000-00009. [DOI] [PubMed] [Google Scholar]
- 15.Colls BM. Guillain–Barre syndrome and hyponatraemia. Intern Med J. 2003;33:5–9. doi: 10.1046/j.1445-5994.2002.00322.x. [DOI] [PubMed] [Google Scholar]