

Abstract
Enzyme replacement therapy (ERT) has been introduced for Fabry disease and has been reported to clear some renal cell types of accumulated glycolipids and to reduce the accumulation in other cell types. We describe two patients without Fabry disease who were transplanted with kidney allografts from a male donor with Fabry disease. Biopsies were taken at transplantation and after 3 years in the first case and after 12 years in the second case. Even though these Fabry kidney allografts for many years had been exposed to normal levels of circulating α-galactosidase A (α-gal-A), the amount of accumulated lysosomal deposits in the podocytes remained unchanged. Additionally, small deposits were also found in tubular cells and glomerular endothelial cells as long as 12 years after transplantation.
Keywords: enzyme replacement therapy, Fabry disease, histological changes, kidney, transplantation
Introduction
Fabry disease is an X-linked lysosomal storage disease that may cause renal failure in addition to other organ manifestations. Accumulation of globotriaosylceramide (GL-3) and related glycolipids is demonstrated by electron microscopy in several renal cell types as osmophilic lamellar bodies in lysosomes [1,2]. It has been reported that enzyme replacement therapy (ERT) in native kidneys given for 11 months totally cleared GL-3 from some renal cell types such as endothelial cells, mesangial cells and interstitial cells [3]. Moderate clearance was found in smooth muscle cells of arterioles and small arteries. Less, but some, clearance of GL-3 was demonstrated for podocytes and distal tubular epithelial cells [3]. In another study, podocyte showed some, but not complete clearance 54 months after start of ERT [4].
We report two patients without Fabry disease who were transplanted with kidneys from a male Fabry donor. Consequently, these kidneys were exposed to normal levels of circulating α-galactosidase A (α-gal-A). In this paper, we compare the histological findings at baseline with histological findings taken 3 and 12 years after transplantation.
Case reports
In 1996, two kidney allografts were available for transplantation from a 16-year-old male donor who died from a traffic accident. Biopsies taken from both donor kidneys at transplantation showed vacuoles in the podocytes by light microscopy. As part of our standard procedure on baseline biopsies from deceased donors, electron microscopy was performed and revealed lysosomal deposits in podocytes, suggestive of Fabry disease (Figure 1). Very few deposits were also identified in glomerular endothelial cells and in some tubular cells. To confirm the diagnosis, blood sample from the donor was analysed for α-gal-A, revealing low level (0.3 nKat/l, normal: 2.3–9.9), and urine samples from the recipients revealed elevated GL-3 levels. Both tests confirmed the diagnosis of Fabry disease in the donor.
Fig. 1.

Recipient B—baseline biopsy. (a) Lipid deposits (arrows) in podocytes (toluidine blue, ×400) and (b) by electron microscopy (×5200).
Recipient A was a 48-year-old male with diabetic nephropathy; human leucocyte antigen (HLA) mismatch: 2–1–2; blood type: AB → AB; cytomegalovirus (CMV): + → +. The immunosuppression was prednisolone, cyclosporine and azathioprine. Recipient A had one early rejection and was treated with intravenous methylprednisolone; after which, s-creatinine stabilized at ∼185 μmol/l. A biopsy was taken 3 years after transplantation due to a rise in s-creatinine to ∼200 μmol/l and proteinuria (∼2 g/day). In the biopsy, abundant Fabry changes were found in podocytes, with only scattered deposits in glomerular endothelial cells and tubular cells. The amounts of lipid depositions in podocytes had apparently not changed compared to the baseline biopsy. Moreover, diabetic nephropathy and moderate interstitial fibrosis were found. The graft function gradually declined, and peritoneal dialysis was started in 2002.
Recipient B was a 25-year-old male with Wegener's granulomatosis, previously treated with prednisolone and cyclophosphamide; HLA mismatch: 2–1–1; blood type: AB → AB; CMV: + → +. There was no complication to the surgery and no rejections. S-creatinine stabilized at ∼100 μmol/l. The immunosuppression was prednisolone, cyclosporine and azathioprine. The latter was stopped in 1998 due to infertility and skin problems. A second biopsy was taken 12 years after transplantation due to increased s-creatinine (from 100 to 130 μmol/l) and proteinuria (∼3 g/day). Fabry changes were demonstrated in podocytes (Figure 2). The amounts of lysosomal deposits in podocytes were approximately similar to the baseline biopsy (Figure 1). A few deposits were found in glomerular endothelial cells, and focal accumulation of deposits was identified in scattered tubular cells (Figure 3). In addition, glomerular capillary double contour as a sign of chronic rejection, de novo immunoglobulin A (IgA) mesangial glomerulonephritis and moderate interstitial fibrosis were found.
Fig. 2.
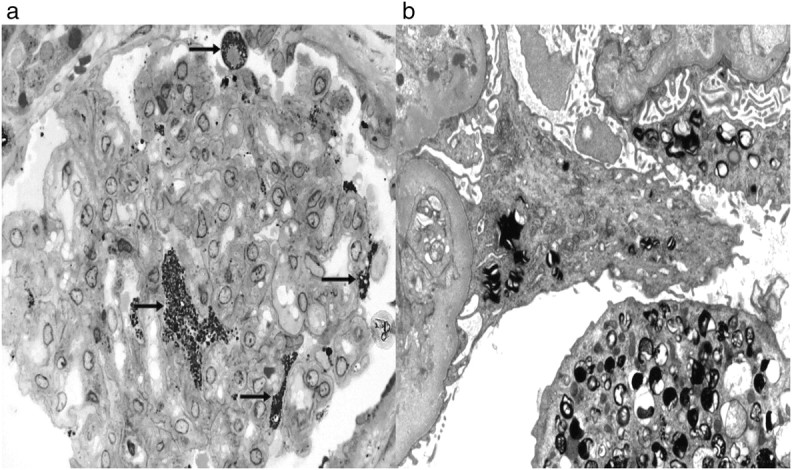
Recipient B—biopsy taken 12 years after transplantation. Lipid deposits (arrows) in podocytes (toluidine blue, ×400) and by electron microscopy (×5200).
Fig. 3.

Recipient B—biopsy at 12 years after transplantation. (a) Tubular cell with lipid deposition (arrows) (toluidine blue, ×400). (b) Myelin-like substructure of lipid deposits in tubular cell (electron microscopy, ×7000).
Discussion
In this report, we present two kidneys with Fabry disease from a male donor that were transplanted to recipients without Fabry disease. Biopsies were taken at baseline and after 3 and 12 years. To our knowledge, only one case of deceased donation of Fabry kidneys has been previously reported. The donor was a 54-year-old woman with Fabry disease, and biopsies were taken from the recipients 6 and 9 months after transplantation [5]. In kidneys from patients with Fabry disease, accumulation of GL-3 and related glycolipids can be found in most renal cell types [1–4]. Podocytes reveal deposits at early stages of the disease and are usually heavily affected [1,2]. ERT in patients with Fabry disease has been reported to reverse microscopic changes in some renal cell types and to decrease lipid deposition in other renal cell types, including podocytes [3,4]. In our cases, the donor kidneys at baseline had extensive GL-3 accumulation in podocytes, but surprisingly, few deposits in other cell types (glomerular endothelial cells and tubular cells). Podocytes are probably the renal cell type with the slowest turnover rate [5]. Therefore, GL-3 clearing in these cells is expected to be slow. In our cases, no clear cut reduction of GL-3 accumulation in podocytes was found after 3 years and even as long as 12 years after transplantation. Deposits in tubular cells were evident as long as 12 years after transplantation (Figure 3). The presence of deposits in this cell type, which has much higher cell turnover, may indicate ongoing deposition of GL-3.
The Fabry kidney allografts in this report have been exposed to normal circulating concentrations of α-gal-A. In ERT, on the other hand, the enzyme is usually given every 2 weeks, which probably exposes Fabry cells to way higher concentrations of enzyme, especially during the first few days after infusion. Even though a comparison between these two ‘options for treatment’ is impossible, it is of interest to note that most renal cells, in our patients, showed no obvious sign of reduction of GL-3 accumulation after as long as 12 years, suggesting that the normal systemic enzyme environment that this kidney has been exposed to might have prevented GL-3 accumulation. The rate of renal cell turnover may also be of importance in the clearing of deposits from Fabry cells and is probably one of the reasons why GL-3 clearing differs between renal cell types [3].
Previous studies have shown that ERT may increase glomerular filtration rate (GFR) in Fabry patients if treatment starts at GFR >55 ml/min [6]. We have no accurate information on the donor's GFR. However, the donor had normal s-creatinine and no proteinuria. Recipient A lost his graft due to other reasons than Fabry nephropathy. In Recipient B, the Fabry allograft was functioning adequately after 12 years. The reduction in GFR and the proteinuria that was observed in Recipient B after 12 years was probably not caused by Fabry changes.
To conclude, exposing well-functioning homozygous Fabry kidneys to physiological concentrations of circulating enzyme for up to 12 years in non-Fabry recipients did not clear accumulated glycolipids from the podocytes, glomerular endothelial cells or tubular cells. On the other hand, Fabry disease did not progress in these kidneys. Therefore, a well-functioning Fabry kidney from a deceased donor might be accepted as donor to non-related recipients.
Acknowledgments
We thank Dr Jan-Eric Månsson at Sahlgrenske Hospital, Sweden, who performed the biochemical analysis.
Conflict of interest statement. None declared.
References
- 1.Sessa A, Meroni M, Battini G, et al. Renal pathological changes in Fabry disease. J Inherit Metab Dis. 2001;24:66–70. doi: 10.1023/a:1012423924648. [DOI] [PubMed] [Google Scholar]
- 2.Gubler M-C, Lenoir G, Grünfeld J-P, Ulmann A, Droz D, Habib R. Early renal changes in hemizygous and heterozygous patients with Fabry's disease. Kidney Int. 1978;13:223–235. doi: 10.1038/ki.1978.32. [DOI] [PubMed] [Google Scholar]
- 3.Thurberg BL, Rennke H, Colvin RB, et al. Globotriaosylceramide, accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. 2002;62:1933–1946. doi: 10.1046/j.1523-1755.2002.00675.x. [DOI] [PubMed] [Google Scholar]
- 4.Germain DP, Waldek S, Banikazemi M, et al. Sustained, long-term renal stabilization after 54 months of agalsidase β therapy in patients with Fabry disease. 2007;18:1547–1557. doi: 10.1681/ASN.2006080816. [DOI] [PubMed] [Google Scholar]
- 5.Marshall CB, Shankland S. Cell cycle regulatory proteins in podocyte health and disease. Nephron Exp Nephrol. 2007;106:e51–e59. doi: 10.1159/000101793. [DOI] [PubMed] [Google Scholar]
- 6.Basic-Jukic N, Coric M, Kes P, Bubic-Filipi LJ, Pasinin J, Kokos I. Anderson–Fabry disease in kidneys from diseased donor. Am Transplant. 2007;7:2829–2833. doi: 10.1111/j.1600-6143.2007.02003.x. [DOI] [PubMed] [Google Scholar]