

Abstract
A series of endosomolytic mixed micelles was synthesized from two diblock polymers, poly[ethylene glycol-b-(dimethylaminoethyl methacrylate-co-propylacrylic acid-co-butyl methacrylate)] (PEG-b-pDPB) and poly[dimethylaminoethyl methacrylate-b-(dimethylaminoethyl methacrylate-co-propylacrylic acid-co-butyl methacrylate)] (pD-b-pDPB), and used to determine the impact of both surface PEG density and PEG molecular weight on overcoming both intracellular and systemic siRNA delivery barriers. As expected, the percent PEG composition and PEG molecular weight in the corona had an inverse relationship with mixed micelle zeta potential and rate of cellular internalization. Although mixed micelles were internalized more slowly, they generally produced similar gene silencing bioactivity (~80% or greater) in MDA-MB-231 breast cancer cells as the micelles containing no PEG (100D/no PEG). The mechanistic explanation for the potent bioactivity of the promising 50 mol% PEG-b-DPB/50 mol% pD-b-pDPB (50D) mixed micelle formulation, despite its relatively low rate of cellular internalization, was further investigated as a function of PEG molecular weight (5 k, 10 k, or 20 k PEG). Results indicated that, although larger molecular weight PEG decreased cellular internalization, it improved cytoplasmic bioavailability due to increased intracellular unpackaging (quantitatively measured via FRET) and endosomal release. When delivered intravenously in vivo, 50D mixed micelles with a larger molecular weight PEG in the corona also demonstrated significantly improved blood circulation half-life (17.8 min for 20 k PEG micelles vs. 4.6 min for 5 kDa PEG micelles) and a 4-fold decrease in lung accumulation. These studies provide new mechanistic insights into the functional effects of mixed micelle-based approaches to nanocarrier surface PEGylation. Furthermore, the ideal mixed micelle formulation identified (50D/20 k PEG) demonstrated desirable intracellular and systemic pharmacokinetics and thus has strong potential for in vivo therapeutic use.
Keywords: Nanoparticle PEGylation, Pharmacokinetics, siRNA, Mixed micelles, Intracellular drug release, FRET imaging
1. Introduction
RNA interference (RNAi) has therapeutic potential to treat cancer or other pathologies whose etiology is related to aberrant gene overexpression [1–3]. However, small interfering RNA (siRNA) encounters both systemic and intracellular barriers that have continued to limit its clinical impact, especially for intravenous delivery applications. Naked molecules of siRNA are susceptible to premature renal clearance and nuclease degradation. The negative charge and hydrophilicity of siRNA also limit its permeability through cellular and endolysosomal membranes. Due to these poor pharmacokinetic properties, siRNA generally requires a carrier for effective delivery [4].
Cationic polymers and lipids are commonly used to electrostatically condense siRNA into complexes that protect it from degradation and enhance cell internalization [5–7]. However, these particles are not typically optimized to overcome both extracellular and intracellular siRNA delivery barriers. While many cationic transfection reagents produce gene silencing activity in vitro, they often have poor hemocompatibility upon intravenous injection in vivo, leading to unexpected toxicity [8]. This toxicity is the result of electrostatic complexes of cationic polymers and siRNA destabilizing in the presence of serum and/or nonspecifically interacting with blood plasma proteins and erythrocytes, causing the formation of aggregates that can occlude the pulmonary vasculature [9,10]. In addition, premature destabilization of cationic polyplexes in the circulation by anionic heparan sulfate in the glomerular basement membrane (GBM), can trigger rapid renal clearance and urinary excretion, reducing the potential for biodistribution and bioactivity within the targeted tissue [11,12]. While tight binding and colloidal stability of siRNA polyplexes may be ideal in the circulation and for initial cell internalization, overly-stable packaging may also hinder bioavailability and gene silencing activity within target cells due to inefficient siRNA release [13–17]. Thus, siRNA nanocarrier stability must be carefully tuned to optimally overcome the combined systemic and intracellular delivery barriers.
In the current studies, formulation of mixed micelles was used as a simple yet robust approach to study the effect of varied percent composition and molecular weight of poly(ethylene glycol) (PEG) in the micelle corona on overcoming both intracellular and extracellular/circulatory delivery barriers. The mixed micelles were formulated from titration of two diblock polymers, poly[ethylene glycol-b-(dimethylaminoethyl methacrylate-co-propylacrylic acid-co-butyl methacrylate)] (PEG-b-pDPB) and poly[dimethylaminoethyl methacrylate-b-(dimethylaminoethyl methacrylate-co-propylacrylic acid-co-butyl methacrylate)] (pD-b-pDPB) in order to enable study of the structure-function relationship between the ratio of PEG and the cationic polymer poly(dimethylaminoethyl methacrylate) (pDMAEMA) in the corona of the mixed micelles. The pDPB core of these mixed micelles consisted of a pH-responsive random copolymer of butyl methacrylate (BMA), propyl acrylic acid (PAA), and DMAEMA that is known to mediate endosomal escape [18–20]. The pDPB composition has been previously utilized for successful peptide and siRNA delivery in vitro [18,19] and local siRNA delivery in vivo [21]. For siRNA delivery, pDPB has been synthesized as a diblock with pDMAEMA, which forms a cationic micelle corona that can efficiently condense anionic siRNA. However, the resulting micelleplexes have a high positive surface charge density that is incompatible with intravenous delivery and may not efficiently release its siRNA cargo intracellularly [14]. These limitations motivated our current effort to develop a series of mixed micelles that leverage the endosomolytic terpolymer core but that possess mixed coronas composed of pDMAEMA and PEG, the latter of which has well-established benefits on biocompatibility, stability, and pharmacokinetics [22–29]. This approach extends recent work that utilized mixed polymeric micelles to study the relationship between micelle structure and cytotoxicity and gene knockdown in vitro [30]. The approach described herein is unique in that it incorporates an endosomolytic micelle core, studies the effect of PEG molecular weight, applies these properties to mechanistically explore how mixed micelle structure affects intracellular unpackaging, and provides new data on the relationship between mixed micelle structure and in vivo pharmacokinetics.
Here, these new mixed micelles were characterized in vitro for stability, hemocompatibilty, cytotoxicity and intracellular delivery/bioactivity/release and in vivo for tissue biodistribution and blood circulation half-life. The effect of PEG on intracellular siRNA unpackaging was robustly explored using Förster resonance energy transfer (FRET) flow cytometry and confocal imaging, providing new insights into optimization of PEGylation to enhance intracellular release/bioavailability, in addition to its better characterized effects on systemic pharmacokinetics and enhanced permeability and retention (EPR) effect-based tumor delivery [31]. These collective studies leveraged mixed micelles to uncover new insights on the structure-function interplay of PEGylation and identified a promising new mixed micelle formulation with a desirable combination of properties tuned for overcoming both systemic and intracellular siRNA delivery barriers.
2. Materials and methods
2.1. Materials
All chemicals were purchased from Sigma–Aldrich Co. (St Louis, MO, USA) unless otherwise noted. The 10 kDa methoxy-poly(ethylene glycol) (mPEG) was purchased from CreativePEGWorks (Salem, NC, USA), and the 20 kDa mPEG was purchased from JenKem USA (Plano, TX, USA). HiPerFect transfection reagent was purchased from Qiagen (Hilden, Germany). 4-cyano-4-(ethylsulfanylthiocarbonyl) sulfanylpentanoic acid (ECT) was synthesized following previously reported literature [32].
2.2. Cell culture
MDA-MB-231 breast cancer cells and NIH-3T3 fibroblasts were purchased from ATCC (Manassas, VA, USA). MDA-MB-231 and Luc-231 (MDA-MB-231 cells that had been stably transfected with a firefly luciferase reporter gene) cells were cultured in DMEM (Gibco, Carlsbad, CA) with 10% FBS (Gibco, Carlsbad, CA) and 50 μg/mL gentamicin (Gibco, Carlsbad, CA). NIH 3T3 and Luc-3T3 (NIH 3T3 cells that had been stably transfected with a firefly luciferase reporter gene) cells were cultured in DMEM + 10% FBS and 1% P/S.
2.3. Polymer synthesis and characterization
All polymers were synthesized by reversible addition fragmentation chain transfer (RAFT) polymerization.
2.3.1. Synthesis of 5 k, 10 k and 20 k Y-shaped PEG macro chain transfer agent (macroCTA)
Each PEG macroCTA was synthesized by adding dicyclohexylcarbodimide (DCC, 1 mmol) to a stirring solution of mPEG (250 μmol), ethyl cyanovaleric trithiocarbonate (ECT, 500 μmol) and 4-Dimethylaminopyridine (DMAP, 50 μmol) in anhydrous dichloromethane. The reaction mixture was stirred at room temperature for 48 h under a nitrogen atmosphere. The precipitated cyclohexyl urea was removed by filtration, and the dichloromethane layer was concentrated by rotary evaporation and precipitated into chilled diethyl ether twice. The precipitated polymer was washed three times with diethyl ether and dried under vacuum overnight. 1H nuclear magnetic resonance (NMR) spectra showed that 88% of the 20 k Y-shaped PEG was conjugated with ECT, 90% of the 10 k PEG was conjugated with ECT, and 84% of the 5 k PEG was conjugated with ECT.
2.3.2. Synthesis of diblock PEG-b-(DMAEMA-co-PAA-co-BMA)
The diblock polymer 5 k PEG-b-(DMAEMA-co-PAA-co-BMA) (PEG-b-pDPB) was synthesized via RAFT polymerization. The monomers were added to the 5 k mPEG macroCTA at stoichiometric quantities of 50% BMA, 25% DMAEMA, and 25% PAA at a monomer to CTA molar ratio of 400. The initiator azobisisobutyronitrile (AIBN) was added at a CTA to initiator molar ratio of 20 in a mixture of 70% dioxane and 30% DMF as the solvent. The polymerization was stirred for 24 h at 70 C under a nitrogen atmosphere. The resulting polymer was precipitated in 50% ether and 50% pentane three times, followed by drying under vacuum overnight. The same conditions were used to synthesize and precipitate the 10 k PEG-b-pDPB.
The 20 k PEG-b-pDPB was synthesized by RAFT by adding monomers in a stoichiometric ratio of 50% BMA, 25% DMAEMA, and 25% PAA to the 20 k PEG macroCTA at a monomer to CTA ratio of 800. The initiator AIBN was added at a CTA to initiator ratio of 8.33. The polymerization was stirred over 24 h in dioxane and DMF under a nitrogen atmosphere at 70 °C. The resulting polymer was precipitated in the same way as the 5 k PEG-b-pDPB and 10 k PEG-b-pDPB.
2.3.3. Synthesis of pDMAEMA macroCTA
The pDMAEMA macroCTA was synthesized via RAFT polymerization as previously described [19,32]. Briefly, DMAEMA monomer was added to the ECT at a monomer to CTA ratio of 100, and the initiator AIBN was added at a CTA to initiator ratio of 10. The reaction was stirred over 6 h at 70 °C in DMF under a nitrogen atmosphere. The resulting polymer was precipitated in pentane three times and dried under vacuum overnight.
2.3.4. Synthesis of diblock DMAEMA-b-(DMAEMA-co-PAA-co-BMA)
The diblock polymer poly[DMAEMA-b-(DMAEMA-co-PAA-co-BMA)] (pD-b-pDPB) was synthesized via a RAFT polymerization as previously described [19,32]. Briefly, monomers were added to pDMAEMA macroCTA at stoichiometric quantities of 50% BMA, 25% DMAEMA and 25% PAA at a monomer to CTA ratio of 250. The initiator AIBN was added at a CTA to initiator ratio of 5. Degassed solution was stirred at 70 °C for 24 h under a nitrogen atmosphere. The resulting polymer was precipitated in 50% ether and 50% pentane three times and dried overnight under vacuum. It was then dissolved in a minimal volume of 100% ethanol, further diluted into dH2O, and purified through a PD10 desalting column (GE Healthcare). The eluant was lyophilized.
2.3.5. Polymer characterization
To determine molecular composition of the polymers, 1H NMR was performed in CDCl3 using a Bruker 400 MHz spectrometer. To determine absolute molecular weight and polydispersity, gel permeation chromatography (GPC) was performed on the polymers in filtered N,N-dimethyl formamide (DMF) containing 0.1 M lithium bromide (LiBr) on an Agilent Infinity GPC equipped with refractive index (RI) and a Wyatt miniDAWN TREOS light scattering detector. The dn/dc values used to calculate absolute molecular weight were determined on the same instrument by direct, off-line injections of polymer solutions into the RI detector.
2.4. Micelle formation and characterization
Micelles were formed by solvent exchange. Lyophilized diblock polymers of the desired characteristics and stoichiometry were weighed out and co-dissolved in a minimal volume of pure ethanol. PBS was then slowly added to the dissolved polymers using a syringe pump at a rate of 8 mL/h. The final micelle solution was 2 mg/mL and contained 1% ethanol. Micelles were filtered through a 0.45 μm syringe filter and dynamic light scattering (DLS) measurements were performed on a Malvern Zetasizer Nano-ZS (Malvern Instruments Ltd, Worcestershire, U.K). Zeta (ζ) potential was also determined on the same instrument. Transmission electron microscopy (TEM) images were acquired using a FEI Tecnai Osiris TEM (Hillsboro, Oregon, USA). TEM samples were prepared by adding a drop of 0.5 mg/mL micelle solution on a copper grid and blotting the grid, then staining the grid briefly with 3% uranyl acetate and blotting the grid dry. The grid was then allowed to dry under vacuum overnight before imaging.
2.5. Cellular internalization by flow cytometry
Cellular internalization of the micelles by MDA-MB-231 mammary epithelial cells was determined by flow cytometry as previously described [33]. Briefly, MDA-MB-231 cells were seeded onto 24-well plates in media supplemented with 1% FBS and were allowed to adhere overnight. Micelles were loaded with AlexaFluor488-labeled dsDNA at an N+/P− ratio of 6 for 1 h. Cells were then treated with micelleplexes at a dsDNA concentration of 100 nM for 6, 12, and 24 h. Immediately after treatment, cells were washed with PBS and trypsinized. Following trypsinization, cells were centrifuged, and cell pellets were re-suspended in 0.05% trypan blue solution to quench extracellular fluorescence. Cell fluorescence was measured via flow cytometry (FACSCalibur, BD Biosciences, Franklin Lakes, NJ) and analyzed using FlowJo software.
2.6. siRNA gene knockdown
Luc-231 cells were seeded at a density of 6000 cells/well in a black, clear-bottom 96-well plate in media supplemented with 1% FBS and allowed to adhere overnight. Micelles were loaded with anti-luciferase siRNA (Ambion model #AM4629) at an N+/P− ratio of 6, and cells were treated with micelleplexes for 6, 12, and 24 h. At the end of the designated treatment times, cells were washed with PBS, fresh media containing 1% FBS was added, and the cells were incubated for an additional 48 h. Following incubation, media was replaced with luciferin-containing media, and bioluminescence was measured using an IVIS Imaging System (Xenogen Corporation, Alameda, CA, USA). Bioluminescence data was normalized to total cellular protein as determined using a Bradford Assay (Bio-Rad Laboratories Inc., Hercules, CA, USA).
2.7. Cytotoxicity assessment
Cellular viability was measured in Luc-3T3s after treatment with micelleplexes. Luc-3T3 cells were seeded on a black, clear bottom 96-well plate and allowed to adhere overnight. Micelles were loaded with scrambled, negative control siRNA at a N+/P− ratio of 6. Cells were treated for 6, 12, and 24 h at a concentration of 100 nM siRNA. Immediately following treatments, media was removed, and cells were washed once with PBS. Fresh media containing luciferin was added to the cells, and the bioluminescence was measured on the IVIS Imaging System (Xenogen Corporation, Alameda, CA, USA). Luciferase was used to calculate cell number relative to untreated cells.
2.8. Mechanistic studies of intracellular bioavailability
2.8.1. Measurement of micelleplex stability in the presence of heparin
Heparin sodium salt was used as a model for competing polyanions that are present in vivo and within cells. Micelles were complexed with double stranded siRNA at an N+/P− ratio of 6 for an hour. The micelleplexes were then added to a black, clear-bottom 96 well plate at a final concentration of 100 nM with 250 U/L heparin sodium salt in PBS in a total volume of 100 μL. Dilute Quant-iT RiboGreen RNA Reagent (Life Technologies, Grand Island, NY, USA) was added per manufacturer’s instructions to the mixture for a total final volume of 200 μL. RiboGreen cannot access siRNA when it is stably packaged within micelleplexes and does not fluoresce unless it interacts with the free siRNA. Increase in Ribogreen fluorescence (excitation of 488 nm, emission of 520 nm) was measured over 2 h on a plate reader (Tecan Group Ltd, Mannedorf, Switzerland), and percent siRNA released from the micelles was determined by calculating percent fluorescence normalized to fluorescence of an equivalent concentration of free siRNA.
2.8.2. FRET flow cytometry measurement of intracellular siRNA release
Intracellular siRNA unpackaging was measured by modifying a previously reported protocol [34]. MDA-MB-231 cells were seeded at a density of 60,000 cells/mL onto 24-well plates in media supplemented with 1% FBS and were allowed to adhere overnight. Mixed micelles were complexed with an equimolar ratio of FRET pair labeled dsDNA (AlexaFluor 488-labeled dsDNA and AlexaFluor 546-labeled dsDNA) at an N+/P− ratio of 6. Cells were treated at a concentration of 100 nM for 4 h and all treatments were removed. Cell fluorescence was measured via flow cytometry (FACSCalibur, BD Biosciences, Franklin Lakes, NJ) 0, 2, 8 and 24 h after removal of treatments. Cells were excited with a 488 ± 10 nm laser, and emission was measured with 530 ± 3- nm (FL1) and 585 ± 42 nm (FL2) filters. Analysis and compensation on the fluorescent channels was performed using FlowJo software. Compensation was performed using single-fluorophore control for every micelleplex.
2.8.3. Confocal FRET microscopy imaging of intracellular siRNA release
MDA-MB-231 cells were seeded onto 8-well chamber slips at a density of 60,000 cells/mL in media supplemented with 1% FBS and were allowed to adhere overnight. Mixed micelles were complexed with an equimolar ratio of FRET pair labeled dsDNA (AlexaFluor 488-labeled dsDNA and AlexaFluor 546-labeled dsDNA) at an N+/P− ratio of 6. Cells were treated at a concentration of 100 nM for 4 h and all treatments were removed. Fluorescent images were then taken using confocal microscopy (Zeiss LSM 710Meta, Oberkochen, Germany) equipped with a Carl Zeiss plan apochromat 20× objective with N.A. = 0.8. Images were acquired immediately after removal of treatments and two washes with PBS −/−. Cells were excited with an argon 488 nm laser (2.0%), and emission was collected between 493 nm–543 nm for the green (donor) channel and 575 nm–680 nm for the red (acceptor) channel. Emission and gain settings for both channels were chosen such that: (i) no donor fluorescence was found in the acceptor channel, (ii) no acceptor fluorescence was found in the donor channel, and (iii) no cellular autofluorescence occurred in the unlabeled micelle control group. FRET efficiency (I546/I488) was calculated and analyzed using MATLAB. Briefly, the donor channel’s nonzero pixels were used as a mask to avoid divide by zero errors from extracellular pixels. I546/I488 was then calculated. FRET acceptor bleaching and fluorometry were performed to ensure confocal images reflect FRET phenomena (Supplementary Materials).
2.8.4. Confocal microscopy imaging of endosomal escape
Confocal microscopy was performed as previously described [35]. MDA-MB-231 cells were seeded onto 8-well chamber slips at a density of 60,000 cells/mL in media supplemented with 1% FBS and were allowed to adhere overnight. Micelles were complexed with AlexaFluor488-labeled dsDNA at an N+/P− ratio of 6 for 1 h. Cells were then treated with micelleplexes at a dsDNA concentration of 100 nM for 24 h. Immediately after treatment, media was replaced with Lysotracker (Invitrogen Life Sciences, Grand Island, NY, USA) containing media (75 nM), and cells were incubated for 1 h. Following incubation, cells were imaged with confocal microscopy (Zeiss LSM 710Meta, Oberkochen, Germany). Images were analyzed using JACOP, an extension of ImageJ, as previously described [36].
2.9. Micelle interactions with whole blood components
The whole blood inertness of the micelles was determined by performing a whole blood aggregation assay as previously described [35]. Micelles were complexed with Cy5-labeled double stranded DNA (dsDNA) (Sigma–Aldrich Co., St. Louis, MO, USA) at an N+/P− ratio of 6. The micelleplexes were then added to dilute human whole blood at a final siRNA concentration of 200 nM in a 96-well plate. As a control, the micelles were also added to PBS at the same concentration. The formulations were placed on a shaker for 5 min before incubation at 37 °C for 1 h. The plates were then centrifuged at 500 × g for 5 min. Then, 100 μL of supernatant from each well was transferred to a black, clear bottom 96-well plate, and Cy5 fluorescence was measured on a plate reader (Tecan Group Ltd, Mannedorf, Switzerland).
2.10. Micelle protection of siRNA from RNase degradation
The hyperchromic effect is the increase in absorbance at 260 nm that occurs when nucleic acids degrade. This phenomenon was used to measure the ability of the mixed micelles to protect siRNA against nuclease degradation as previously described [37,38]. Micelleplexes at a N+/P− ratio of 6 were diluted in 100 μL of dH2O to a final siRNA concentration of 500 nM and placed in a small volume quartz cuvette. Then, 300 nU of Riboshredder RNAse Blend (Epicentre, Madison, WI, USA) was added, and absorbance at 260 nm was kinetically monitored for 20 min on a Cary 100 UV-vis spectrophotometer (Agilent Technologies, Santa Clara, CA, USA).
2.11. In vivo blood circulation half-life determination
Micelles were complexed to IRDye800-labeled dsDNA for an hour at a N+/P− ratio of 6. Five BALB/c mice (6–8 weeks old) (Charles Rivers Laboratories, Wilmington, MA, USA) were injected in the tail vein with 2 mg/kg (normalized to dsDNA dose) micelleplexes. Blood was retro-orbitally collected at 5 and 10 min post-injection (not to exceed two collections per animal). At 20 min post-injection, animals were sacrificed, and blood was immediately collected from the renal vein. Collected blood was centrifuged at 500 × g for 5 min, and 5 μL of plasma supernatant was collected and diluted into 95 μL dH2O. Fluorescence was measured on a plate reader (Tecan Group Ltd., Mannedorf, Switzerland) at an excitation wavelength of 790 ± 5 nm and an emission wavelength of 810 ± 2.5 nm. A standard curve was produced by measuring the fluorescence of the micelleplexes in dH2O over the range of 200%–6.25% of initial injected dose. This standard was utilized to calculate the percent of injected dose in each blood sample, and these calculated values were used to determine blood circulation half-life.
2.12. In vivo biodistribution
Animals were injected with dsDNA micelleplexes as stated for the determination of blood circulation half-life. At 20 min post-injection, animals were sacrificed and organs of interest were excised. Lungs, heart, spleen, liver, and kidneys were fluorescently imaged on the IVIS Imaging System (Xenogen Corporation, Alameda, CA, USA). The different organs were separated into regions of interest (ROI), and the total fluorescence in each organ was quantified.
2.13. Statistical analysis
All data are reported as mean ± standard error of the mean (SEM). ANOVA was used for statistical analysis, and Tukey’s post-hoc test was used to determine pairwise significance based on an alpha value of 0.05.
2.14. Ethics statement
All animals in the study were treated humanely under conditions outlined by National Health Institute (NIH). Experiments performed on the animals were approved by Vanderbilt University’s Institutional Animal Care and Use Committee (IACUC). Human blood was collected from anonymous donors following a protocol approved by the Vanderbilt Institutional Review Board (IRB).
3. Results and discussion
3.1. Polymer synthesis and characterization
Four different diblock polymers were synthesized by RAFT polymerization. All four polymers contained an ampholytic/hydrophobic, pH-responsive block consisting of random copolymer of approximately 50% BMA, 25% PAA, and 25% DMAEMA. The other block of each polymer was a hydrophilic, corona-forming segment consisting of either PEG (5, 10, or 20 kg/mol) or pDMAEMA (Table 1). The hydrophobic block was approximately 20 kDa for all polymers except for the polymer containing the 20 kDa PEG, which was made with a slightly bigger hydrophobic block to compensate for the large hydrophilic block.
Table 1.
Molecular weight and composition of polymers used to formulate mixed micelles.
| Polymer name | Mn (kg/mol) | PDI | Mn pDMAEMA (kg/mol) | Mn PEG (kg/mol) | % BMA (core) | % DMAEMA (core) | % PAA (core) |
|---|---|---|---|---|---|---|---|
| pD-b-pDPB | 32.4 | 1.1 | 12.2 | N/A | 52 | 21 | 27 |
| 5 k PEG-b-pDPB | 26.6 | 1.3 | N/A | 5 | 45 | 25 | 30 |
| 10 k PEG-b-pDPB | 31.1 | 1.7 | N/A | 10 | 45 | 26 | 29 |
| 20 k PEG-b-pDPB | 55.2 | 1.5 | N/A | 20 | 47 | 28 | 25 |
Initially, varied molar percentages of 5 k PEG-b-pDPB and pD-b-pDPB polymers were titrated together to produce four mixed micelles: 100% pD-b-pDPB (100D/no PEG), 75% pD-b-pDPB/25% PEG-b-pDPB (75D/5 k PEG), 50% pD-b-pDPB/50% PEG-b-pDPB (50D/5 k PEG), and 25% pD-b-pDPB/75% PEG-b-pDPB (25D/5 k PEG). These four mixed micelles were formulated to determine the effect of the relative density of PEG and pDMAEMA in the corona. For a subset of experiments, 50/50 micelles were formulated with 10 k and 20 k PEG-containing PEG-b-pDPB polymers, rather than 5 k, forming 50D/10 k PEG and 50D/20 k PEG micelles, respectively. This mixed micelle approach provided a synthetically simple, robust, and well-defined method for synthesis of a small library of carriers with varied amount and length of PEG titrated into the pDMAEMA micelle corona. Because the mixed micelle method does not require inefficient, difficult to control chemical conjugations (e.g., as would be required for grafting of molecules to the surface of pre-made nanoparticles), it enables rapid completion of structure-function studies such as those carried out herein (see experimental outline in Scheme 1).
Scheme 1.

Experimental plan including measured outcomes.
3.2. Micelle characterization
The 100D/no PEG, 75D/5 k PEG, 50D/5 k PEG, and 25D/5 k PEG micelles all had hydrodynamic diameters of ~35 nm (Fig. 1A). However, micelle size increased for formulations with increased PEG molecular weight, ranging from 35 nm (50D/5 k PEG) to 65 nm (50D/20 k PEG) (Fig. 1B). Generally, these diameters are in a useful range for EPR-based tumor delivery and are large enough to avoid renal clearance and small enough to avoid rapid macrophage phagocytosis [39].
Fig. 1.

Chemicophysical characterization of mixed micelles. (A) Size and ζ potential of micelles containing variable ratios of PEG and pDMAEMA in the micelle corona, n = 3. (B) Size and ζ potential of 50D micelles containing varied PEG molecular weight (5, 10, or 20 k) in the micelle corona, n = 3. (C) Representative TEM images of mixed micelles.
The ζ potential of the mixed micelles correlated to the percent pD-b-pDPB in the 5 kDa PEG-containing mixed micelles: +17.6 mV (100D/no PEG), +11.2 mV (75D/5 k PEG), +4.6 mV (50D/5 k PEG), and +0.1 mV (25D/5 k PEG) (Fig. 1A). This trend suggests that, as expected, the addition of PEG in the micelle corona shielded the positive micelle surface charge caused by pDMAEMA. Similarly, an increase in PEG molecular weight in the 50D micelles, decreased the ζ potential for micelles containing 5 k, 10 k, and 20 k PEG-pDPB (+4.6 mV to +2.2 mV to +0.5 mV, respectively) (Fig. 1B). These data suggest that both relative percent and molecular weight of the PEG molecule can contribute to shielding of the micelle surface charge. These results are consistent with previous reports suggesting the addition of PEG on the surface of a nanoparticle results in a more-neutral ζ potential [40].
TEM images were used to visualize the size of the dehydrated mixed micelles (Fig. 1C) [33]. Diameter of the dehydrated micelles containing 5 k PEG-pDPB were consistently ~20 nm. The 50D micelles with the 10 k and 20 k PEG-pDPB had larger diameters of 25 nm and 40 nm, respectively; consistent with the relative size increases measured with DLS.
3.3. Cellular uptake and RNAi
Cellular uptake of Alexa488-labeled dsDNA mixed micelleplexes was evaluated by flow cytometry in MDA-MB-231 human mammary gland carcinoma cells (Fig. 2A and B). Regardless of mixed micelle stoichiometry, cellular uptake of the micelleplexes increased with treatment time. Cellular uptake inversely correlated with the percent PEG-b-pDPB and the molecular weight of PEG in the corona, especially at the 6h treatment time. Increasing the amount and molecular weight of the PEG decreased ζ potential, which was likely a significant contributor to the differences in rate and extent of cellular uptake and internalization. This result is consistent with the presumption that electrostatic interactions between cationic delivery systems and anionic proteoglycans on the cell surface promote internalization [41]. This relationship is especially apparent among the different molecular weight PEGs in the 50D formulations (Fig. 2B). The 50D/5 k PEG, 50D/10 k PEG, and 50D/20 k PEG micelleplexes resulted in significantly different cellular uptake at each of the treatment times. These results suggest that finely tuning both the stoichiometry and PEG molecular weight within the mixed micelles can be utilized to optimize the micelleplexes with an ideal combination of cellular uptake and gene silencing bioactivity.
Fig. 2.
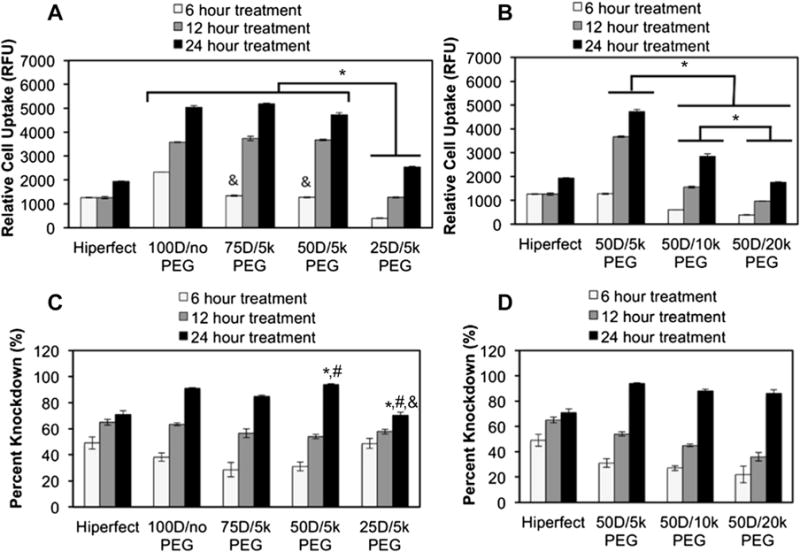
Cellular uptake and gene knockdown as a function of mixed micelle composition and cell treatment time. (A) Relative cellular uptake of mixed micelleplexes containing varied ratios of PEG and DMAEMA in the corona. * indicates significant difference (p < 0.05) and & indicates significant difference (p < 0.05) from 100D/no PEG. (B) Relative cell uptake of 50D micelleplexes containing varied molecular weight of PEG in the corona. *p < 0.05. (C) Percent knockdown of model gene luciferase achieved by luciferase siRNA delivered via mixed micelleplexes containing varied amounts of 5 k PEG on the corona. * indicates significance (p < 0.05) from 100D/no PEG treatment; # indicates significance (p < 0.05) from 75D/5 k PEG treatment; & indicates significance (p < 0.05) from 50D/5 k PEG treatment. (D) Percent knockdown of model gene luciferase using siRNA delivered in 50D mixed micelleplexes containing varied molecular weight of PEG in the corona.
RNAi bioactivity of the different micelleplexes was assessed in Luc-231 breast cancer cells based on protein-level knockdown activity of siRNA against luciferase (Fig. 2C and D). To ensure that the gene silencing results were not affected by cytotoxicity, luciferase signals were normalized to total cellular protein content for each sample. In these studies, gene knockdown correlated to treatment time, but in contrast to cellular uptake results, the gene knockdown did not show significant differences based on the percent or molecular weight of the PEG in the micelleplex corona (Fig. 2C and D). This implies that gene knockdown did not depend strongly on percent or molecular weight PEG. PEG molecular weight in the 50D mixed micelleplexes did not significantly influence gene knockdown (Fig. 2D), although cellular uptake was significantly different among these treatments (Fig. 2B). The lack of correlation between the uptake and the silencing bioactivity of the different mixed micelle formulations suggests that the amount of mixed micelle PEGylation may also have effects on intracellular siRNA bioavailability.
3.4. Cytotoxicity evaluation
Next, cytocompatibility was measured in Luc-3T3s to further screen the mixed micelles (Fig. 3). Cell viability after micelleplex treatment was not significantly different relative to untreated controls at 6 h and 12 h. The charge ratio and siRNA concentration was held constant for all micelleplex formulations; however, because the pD-b-pDPB is the sole contributor to the charge ratio calculation, the total amount of polymer is increased as the percent of pD-b-pDPB is decreased relative to PEG-b-pDPB in the micelleplex. The mild, but statistically significant, toxicity of the 25D/5 k PEG micelleplex after 24 h of treatment can likely be attributed to the higher total amount of polymer required to make this formulation with siRNA at a charge ratio of 6:1. While the 25D/5 k PEG micelle is favorable for its low ζ potential, it does require a higher polymer dose per siRNA. This was motivation to expand the current study to also consider larger molecular weight PEG molecules in order to achieve efficient charge shielding of the 50D formulation in a way that is both cytocompatible and produces potent gene silencing. As a result, further studies focused on the 50D formulations to compare 5 k, 10 k, and 20 k PEG.
Fig. 3.
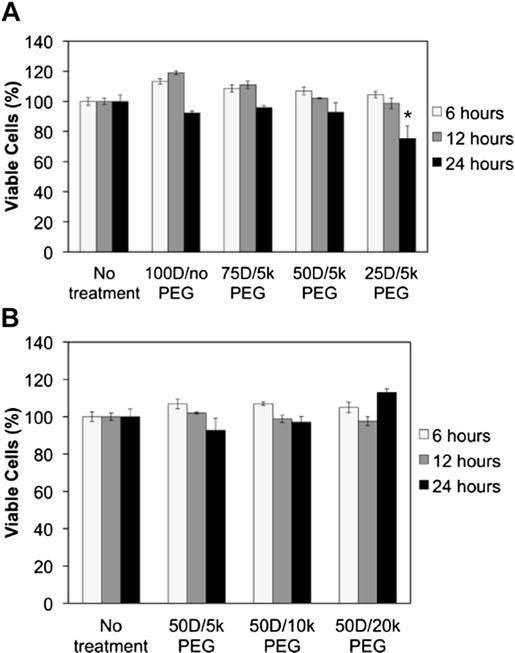
Cytotoxicity evaluation of mixed micelleplexes at different treatment times at siRNA dose of 100 nM. Relative number of viable NIH 3T3 cells after treatment with mixed micelleplexes containing (A) varied percent molar composition of PEG and (B) varied PEG molecular weight in the micelle corona. * represents a significant decrease from samples receiving no treatment (p < 0.05).
3.5. Mechanistic studies of intracellular bioavailability
RNAi activity depends upon siRNA loading into the RNA-induced silencing complex (RISC), which occurs in the cell cytoplasm [42]. Two of the primary barriers against intracellular bioavailability in this context are siRNA release from its carrier and escape from entrapment within endolysosomal vesicles. It was hypothesized that, despite lower rate of cellular internalization due to decreased ζ potential, micelleplexes with higher percent and/or larger molecular weight PEG in the corona achieve similar gene silencing bioactivity due to greater intracellular bioavailability. This hypothesis was motivated by our initial cell internalization and gene silencing observations and fits with the concept that incorporation of larger molecular weight PEG in the micelleplex corona may lead to reduced valency of the pDMAEMA/siRNA electrostatic interactions and thus enable more efficient intracellular siRNA release.
To test this hypothesis, micelleplexes were first exposed to the polyanion heparin, and siRNA unpackaging was measured using RiboGreen (Fig. 4A). Micelleplexes with larger molecular weight PEG in the corona more rapidly and completely released siRNA over 120 min. This result is consistent with the hypothesis that the micelleplexes with higher molecular weight PEG in the corona release siRNA more efficiently intracellularly, resulting in improved siRNA intracellular bioavailability and gene silencing.
Fig. 4.
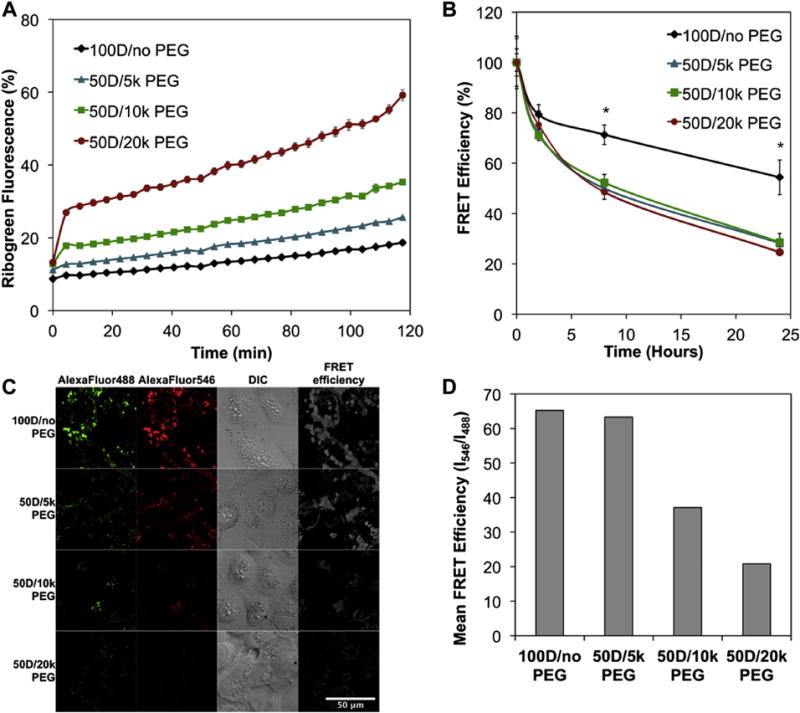
Micelleplex incorporation of PEG-pDPB increases siRNA intracellular unpackaging and is dependent on PEG molecular weight. (A) Kinetics of siRNA decomplexation from micelles after exposure to 250 U/L heparin at siRNA concentration of 100 nM, n = 3. (B) In vitro kinetics of siRNA intracellular unpackaging from mixed micelles following a 4 h treatment with 100 nM siRNA, n = 3. (C) Representative FRET microscopy images following a 4 h treatment at siRNA concentration of 100 nM (D) Quantitative evaluation of FRET efficiency calculated as I546/I488 for pixels with non-zero fluorescence intensity over a 425 μm × 425 μm field of view (*p < 0.05).
To confirm that siRNA is released more efficiently intracellularly in cells when larger molecular weight PEG polymers compose the micelleplex, in vitro FRET-based flow cytometry measurements were used to assess intracellular release of cargo from 50D mixed micelles containing 5 k, 10 k, or 20 k PEG (Fig. 4B). FRET signal is higher when the pair of fluorescent molecules is closer together, and a decrease in FRET efficiency indicates the two molecules are further apart from each other. In this study, decrease in FRET efficiency indicated carrier release of two co-loaded dsDNA molecules labeled with the FRET pair Alexa Fluor 488 and Alex Fluor 546. These FRET studies revealed that mixed micelles containing PEG-b-pDPB showed significantly greater intracellular unpacking, based on loss in FRET efficiency, relative to 100D micelles that do not contain PEG. No significant effect of PEG molecular weight was seen.
To observe changes in intracellular FRET, confocal microscopy imaging was also utilized to qualitatively and quantitatively assess intracellular unpackaging (Fig. 4C and D). In FRET confocal images, the ratio of green (donor) relative to red (in web version) (acceptor) fluorescent signal was visibly higher as the PEG molecular weight was increased (Fig. 4C). FRET efficiency was quantified for each treatment group and indicated that the level of siRNA intracellular release over 4 h of treatment correlated to the molecular weight of the PEG used on the 50D mixed micelle formulation (Fig. 4D). These microscopy studies confirmed the impact of mixed micelle formulation on intracellular siRNA release, and this measurement was more sensitive to the effect of the molecular weight of PEG in the corona than flow cytometry. Taken together, these data indicate that the intracellular siRNA bioavailability of siRNA was higher in the mixed micelles with larger molecular weight PEG at least partially due to improved intracellular unpackaging.
Utilization of FRET to quantitatively measure nanocarrier stability and drug release is increasingly being used [34,35,43–47]. Here, FRET provided, to our knowledge, the first direct measurement of the effect of PEG in mixed micelles on intracellular release and bioavailability of siRNA. These results are also in agreement with other studies assessing the effects of PEG on other (non-mixed micelle) systems. Previous (non-FRET-based) studies showed that grafting of PEG to PEI improved both RNase resistance and intracellular unpackaging of PEI-siRNA polyplexes [15]. In addition, a recent, FRET-based study showed that a triblock polymer-based tri-layer micelle comprising (1) PEG, (2) cationic poly(L-lysine), and (3) hydrophobic poly{N-[N-(2-aminoethyl)-2-aminoethyl]aspartamide} bearing a hydrophobic dimethoxy nitrobenzyl ester in the side chain showed superior intracellular siRNA release relative to the analogous bi-layer micelle that did not contain the outer PEG shell [44]. These studies support the effect of PEG on nanocarrier siRNA release, as detected in our mixed micelles studies. However, the PEG functionality was spatially distinct from the cationic siRNA packaging component in each of these studies, and, though they have not been directly compared, the mixed corona utilized with the current approach may better reduce the charge density within the siRNA loading shell and enhance intracellular release relative to these previous systems.
Another advantage of the current design is that it provides an efficient endosomal escape mechanism based on the pDPB micelle core, which provides active pH-dependent membrane disruptive behavior as previously confirmed in polyplexes [19], micelles [20], and PEGylated triblock micelles [33]. The expected pH-dependent membrane disruptive activity of the mixed micelles was confirmed using a red blood cell hemolysis assay [48] (Figure S1). The effect of PEG molecular weight in 50D mixed micelles on intracellular trafficking and endosomal escape was further explored using confocal microscopy (Fig. 5). Colocalization of AlexaFluor488-labeled mixed micelle treatments and Lysotracker staining (marks late endosomes/lysosomes) was quantified to determine the level of endosomal escape using a previously established protocol [35] (Fig. 5A and B). Mixed micelles containing PEG (50D/5 k PEG, 50D/10 k PEG, and 50D/20 k PEG) showed significantly lower Lysotracker colocalization relative to 100D/no PEG micelles. While mixed micelles had an increased capacity to escape the endosome, no significant dependence on PEG length on the corona was observed.
Fig. 5.

The 50D mixed micelleplexes improved siRNA endosomal escape relative to 100D micelleplexes. (A) Representative confocal microscopy images showing decreased colocalization of Lysotracker and AlexaFluor488-labeled cargo for 50D/10 k PEG versus 100D/no PEG micelles following 24 h of treatment (50D/5 k PEG and 50D/20 k PEG treatment images are in Supplementary Fig. 2). (B) Quantification of colocalization (percent AlexaFluor488 overlapping with Lysotracker) demonstrates decreased endosomal localization of all 50D formulations relative to 100D micelleplexes (p < 0.05, n = 4).
Two factors likely contribute to the increased endosomal escape of the mixed micelles. Increased intracellular unpackaging of the mixed micelles, as described above, may enable more rapid diffusion from endolysosomal compartments once they are partially disrupted. Another possible explanation is that membrane disruptive function is dependent on local pDPB concentration. Because cargo is electrostatically condensed to positively-charged pDMAEMA in the polymers, an equivalent dose of siRNA contains approximately twice as much of the pDPB component for the 50% PEG mixed micelleplexes as for the 100D/no PEG micelles. Thus, the mixed micelle formulations may increase the endosomal concentration of the pDPB block and result in improved endosomal escape.
3.6. Micelle interactions with whole blood components
To predict in vivo translatability based on ability to overcome systemic delivery barriers, mixed micelle inertness was measured upon exposure to human whole blood. Upon intravenous injection, cationic micelles are pre-disposed to aggregate due to interactions with blood serum proteins and red blood cells [49], which affect organ distribution and pharmacokinetics [50] and can cause a toxicity due to emboli within lung capillaries [10]. To model this phenomenon, Cy5-labeled dsDNA micelleplexes were incubated in fresh, anticoagulated whole human blood, and the percentage of micelles remaining in the plasma was quantified (Fig. 6A) after incubation and a centrifugation step intended to separate out red blood cells and any micelle aggregates. Increased molecular weight PEG in the 50D mixed micelles correlated with improved retention in the plasma supernatant, 53% for the 50D/5 k PEG and 98% for the 50D/20 k PEG. This trend is likely due to the higher charge shielding and decreased ζ potential achieved by incorporation of larger molecular weight PEG. This ex vivo screening suggests that micelles with the largest PEG would be the most compatible for in vivo studies.
Fig. 6.
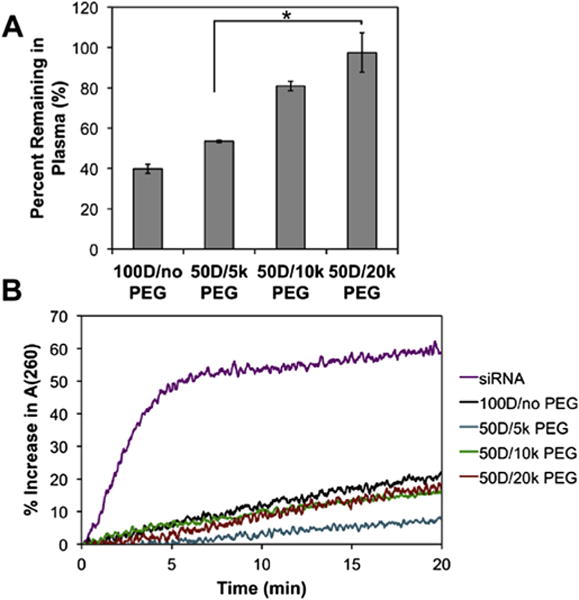
Characterization of mixed micelleplex blood inertness and protection of siRNA against nuclease degradation (A) Blood inertness of mixed micelles indicates that increased PEG molecular weight in the corona reduces aggregation or adsorption to blood components. (B) siRNA protection of mixed micelleplexes against nucleases indicates the all micelles effectively protect cargo against nucleases.
3.7. Micelle protection of siRNA from RNase degradation
Another barrier to siRNA bioactivity in vivo is degradation and inactivation of siRNA by nucleases. Here, siRNA protection by the mixed micelles was characterized by the hyperchromic effect during exposure to a Riboshredder RNAse blend (Fig. 6B). All of the mixed micelle formulations effectively protected the siRNA against degradation in comparison with siRNA alone. After 20 min, free siRNA exposed to the Riboshredder showed a 60% increase in A(260). The siRNA formulated with the 100D/no PEG micelle exposed to Riboshredder resulted in the least protection among micelles, with an increase in A(260) of 20%, while all of the mixed micelles effectively protected the siRNA from nuclease degradation. These data suggest that inclusion of PEG in the corona may contribute to siRNA nuclease protection, which is in agreement with prior PEG-PEI polyplex studies [15].
3.8. In vivo blood circulation half-life and biodistribution
The desirable in vitro properties observed for the 50D mixed micelles justified initial characterization in vivo. Extended blood circulation half-life is critical for effective tumor accumulation of nanomaterials by the EPR effect [51,52]. Previous studies using PEGylated, cationic polymeric siRNA carriers achieve blood circulation half-life values of <5 min [12,53]. Here, blood circulation half-life (t1/2) was determined by collecting blood at known times, and fitting the data to a one-variable exponential model that assumes starting dose (t = 0) in serum is 100%, (Fig. 7A and B). Fitting the data to this model yields significantly different blood circulation half-lives of 4.6 min for 50D/5 k PEG, 7.5 min for 50D/10 k PEG, and 17.7 min for 50D/20 k PEG (Fig. 7B). These data demonstrate a strong dependence of blood circulation half-life on the molecular weight of the PEG within the 50D micelles. These data also imply that simply altering PEG molecular weight in the corona can modulate in vivo half-life and function of otherwise structurally similar mixed micelles. Previous studies indicate that an increased blood circulation half-life of nanoparticles correlates to an improved tumor biodistribution via the EPR effect following intravenous injection [54–56].
Fig. 7.

Effect of PEG molecular weight in corona of 50D mixed micelleplexes on in vivo blood circulation half-life. (A) Concentration of 50D mixed micelleplexes present in blood serum over time, n = 5. (B) Blood circulation half-life time values of 50D micelleplexes with varied PEG molecular weight in corona. * represents significance (p < 0.05).
Biodistribution of the micelleplexes was also more comprehensively evaluated in the mice, postmortem. Fluorescent imaging and quantification of organ biodistribution (Fig. 8) confirmed that, as expected, a large percentage of the nanocarriers accumulated within the liver for all 50D mixed micelles. The 50D/5 k PEG micelles resulted in a 2-fold higher fluorescence in the lungs compared to the 50D/10 k PEG micelles and 4-fold higher than the 50D/20 k PEG micelles. The higher lung accumulation of the 50D/5 k PEG micelles may be due to lodging of aggregates within lung capillaries, consistent with the ex vivo whole blood inertness data in Fig. 2. The decreased mixed micelle lung biodistribution achieved with larger molecular weight PEG would also be predicted to decrease the potential for longer-term carrier toxicity in vivo. Similarly, increasing PEG molecular weight also affected bio-distribution in the kidneys. The 50D/5 k PEG micelleplexes had significantly increased distribution in the kidneys relative to the 50D/10 k PEG and the 50D/20 k PEG micelleplexes. This would also imply reduced stability of the 50D/5 k PEG micelles within the circulation, which results in rapid renal clearance of free siRNA molecules [12]. Tissue distribution was as expected for the 50D/20 k PEG micelleplexes, and their extended circulation time suggests that they may be ideal for EPR-based tumor delivery in vivo.
Fig. 8.

In vivo tissue distribution of 50D micelleplexes containing varied PEG molecular weights in the micelle corona 20 min after injection into BALB/c mice. (A) Total radiant flux and (B) representative images of the 50D micelleplexes in 5 main organs, n = 8. * represents significance (p < 0.05).
4. Conclusions
The present study identified a surface charge neutral micelle that simultaneously overcomes both systemic and intracellular siRNA delivery barriers. This formulation (50D/20 k PEG) was identified from screening a series of mixed micelleplexes made with varied molar percentages of PEG and different molecular weight PEG in the corona. Higher PEG percent composition and molecular weight PEG in the corona of the mixed micelles resulted in decreased ζ potential and decreased cellular uptake in vitro. In spite of the lower cellular uptake, mixed micelleplexes with higher molar percentages and higher molecular weight PEG in the corona achieved similar gene knockdown. A series of quantitative FRET and confocal microscopy studies confirmed that this is due to higher efficiency of intracellular siRNA unpackaging and endosomal escape in mixed micelles with larger molecular weight PEG. Ex vivo and in vivo analyses further validated that, based on hemocompatibility and blood circulation half-life measurements, the 50D/20 k PEG mixed micelles are also ideally suited to overcome systemic delivery barriers. By applying a systematic, mixed micelle approach and quantitative FRET methods, these studies provide new structure-function insights for formulations intended to overcome both systemic and intracellular delivery barriers.
Supplementary Material
Acknowledgments
This research was supported by a grant from the Department of Defense Congressionally Directed Medical Research Program (W81XWH-10-1-0446). Fluorescence and bioluminescence readings were performed through the Vanderbilt University Institute for Imaging Science (VUIIS). DLS, spectrofluorometry, and TEM were conducted through the use of core facilities of the Vanderbilt Institute of Nanoscale Sciences and Engineering (VINSE) (supported by NSF EPS grant 1004083). Confocal microscopy was performed in the VUMC Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, HD15052, DK59637 and EY08126).
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.biomaterials.2014.10.036.
References
- 1.Meister G, Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–9. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 2.Paul CP, Good PD, Winer I, Engelke DR. Effective expression of small interfering RNA in human cells. Nat Biotechnol. 2002;20:505–8. doi: 10.1038/nbt0502-505. [DOI] [PubMed] [Google Scholar]
- 3.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129–38. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanasty RL, Whitehead KA, Vegas AJ, Anderson DG. Action and reaction: the biological response to siRNA and its delivery vehicles. Mol Ther. 2012;20:513–24. doi: 10.1038/mt.2011.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Urban-Klein B, Werth S, Abuharbeid S, Czubayko F, Aigner A. RNAi-mediated gene-targeting through systemic application of polyethylenimine (PEI)-complexed siRNA in vivo. Gene Ther. 2005;12:461–6. doi: 10.1038/sj.gt.3302425. [DOI] [PubMed] [Google Scholar]
- 6.Wightman L, Kircheis R, Rössler V, Carotta S, Ruzicka R, Kursa M, et al. Different behavior of branched and linear polyethylenimine for gene delivery in vitro and in vivo. J Gene Med. 2001;3:362–72. doi: 10.1002/jgm.187. [DOI] [PubMed] [Google Scholar]
- 7.Shrestha R, Elsabahy M, Florez-Malaver S, Samarajeewa S, Wooley KL. Endosomal escape and siRNA delivery with cationic shell crosslinked knedel-like nanoparticles with tunable buffering capacities. Biomaterials. 2012;33:8557–68. doi: 10.1016/j.biomaterials.2012.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reischl D, Zimmer A. Drug delivery of siRNA therapeutics: potentials and limits of nanosystems. Nanomedicine Nanotech Biol Med. 2009;5:8–20. doi: 10.1016/j.nano.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Dokka S, Toledo D, Shi X, Castranova V, Rojanasakul Y. Oxygen radical-mediated pulmonary toxicity induced by some cationic liposomes. Pharm Res. 2000;17:521–5. doi: 10.1023/a:1007504613351. [DOI] [PubMed] [Google Scholar]
- 10.Audouy SAL, de Leij LFMH, Hoekstra D, Molema G. In vivo characteristics of cationic liposomes as delivery vectors for gene therapy. Pharm Res. 2002;19:1599–605. doi: 10.1023/a:1020989709019. [DOI] [PubMed] [Google Scholar]
- 11.Brenner BM, Hostetter TH, Humes HD. Glomerular permselectivity: barrier function based on discrimination of molecular size and charge. Am J Physiol Renal Physiol. 1978;234:F455–60. doi: 10.1152/ajprenal.1978.234.6.F455. [DOI] [PubMed] [Google Scholar]
- 12.Zuckerman JE, Choi CHJ, Han H, Davis ME. Polycation-siRNA nanoparticles can disassemble at the kidney glomerular basement membrane. Proc Natl Acad Sci U S A. 2012;109:3137–42. doi: 10.1073/pnas.1200718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Godbey WT, Wu KK, Mikos AG. Tracking the intracellular path of poly(-ethylenimine)/DNA complexes for gene delivery. Proc Natl Acad Sci U S A. 1999;96:5177–81. doi: 10.1073/pnas.96.9.5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao Z-X, Ho Y-C, Chen H-L, Peng S-F, Hsiao C-W, Sung H-W. Enhancement of efficiencies of the cellular uptake and gene silencing of chitosan/siRNA complexes via the inclusion of a negatively charged poly(γ-glutamic acid) Bio-materials. 2010;31:8780–8. doi: 10.1016/j.biomaterials.2010.07.086. [DOI] [PubMed] [Google Scholar]
- 15.Mao S, Neu M, Germershaus O, Merkel O, Sitterberg J, Bakowsky U, et al. Influence of polyethylene glycol chain length on the physicochemical and biological properties of poly(ethylene imine)-graft-poly(ethylene glycol) block copolymer/SiRNA polyplexes. Bioconjug Chem. 2006;17:1209–18. doi: 10.1021/bc060129j. [DOI] [PubMed] [Google Scholar]
- 16.Smith AE, Sizovs A, Grandinetti G, Xue L, Reineke TM. Diblock glycopolymers promote colloidal stability of polyplexes and effective pDNA and siRNA delivery under physiological salt and serum conditions. Biomacromolecules. 2011;12:3015–22. doi: 10.1021/bm200643c. [DOI] [PubMed] [Google Scholar]
- 17.Green MD, Foster AA, Greco CT, Roy R, Lehr RM, Epps TH, et al. Catch and release: photocleavable cationic diblock copolymers as a potential platform for nucleic acid delivery. Polym Chem. 2014;5:5535–41. doi: 10.1039/C4PY00638K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duvall CL, Convertine AJ, Benoit DSW, Hoffman AS, Stayton PS. Intracellular delivery of a proapoptotic peptide via conjugation to a RAFT synthesized endosomolytic polymer. Mol Pharm. 2010;7:468–76. doi: 10.1021/mp9002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Convertine AJ, Benoit DSW, Duvall CL, Hoffman AS, Stayton PS. Development of a novel endosomolytic diblock copolymer for siRNA delivery. J Control Release. 2009;133:221–9. doi: 10.1016/j.jconrel.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Convertine AJ, Diab C, Prieve M, Paschal A, Hoffman AS, Johnson PH, et al. pH-responsive polymeric micelle carriers for siRNA drugs. Biomacromolecules. 2010;11:2904–11. doi: 10.1021/bm100652w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson CE, Kim AJ, Adolph EJ, Gupta MK, Yu F, Hocking KM, et al. Tunable delivery of siRNA from a biodegradable scaffold to promote angiogenesis in vivo. Adv Mater. 2014;26:607–14. 506. doi: 10.1002/adma.201303520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ko YT, Kale A, Hartner WC, Papahadjopoulos-Sternberg B, Torchilin VP. Self-assembling micelle-like nanoparticles based on phospholipid–polyethyleneimine conjugates for systemic gene delivery. J Control Release. 2009;133:132–8. doi: 10.1016/j.jconrel.2008.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burke RS, Pun SH. Extracellular barriers to in vivo PEI and PEGylated PEI polyplex-mediated gene delivery to the liver. Bioconjug Chem. 2008;19:693–704. doi: 10.1021/bc700388u. [DOI] [PubMed] [Google Scholar]
- 24.Maldiney T, Richard C, Seguin J, Wattier N, Bessodes M, Scherman D. Effect of core diameter, surface coating, and PEG chain length on the biodistribution of persistent luminescence nanoparticles in mice. ACS Nano. 2011;5:854–62. doi: 10.1021/nn101937h. [DOI] [PubMed] [Google Scholar]
- 25.Mosqueira VCF, Legrand P, Morgat J-L, Vert M, Mysiakine E, Gref R, et al. Biodistribution of long-circulating PEG-grafted nanocapsules in mice: effects of PEG chain length and density. Pharm Res. 2001;18:1411–9. doi: 10.1023/a:1012248721523. [DOI] [PubMed] [Google Scholar]
- 26.Khargharia S, Kizzire K, Ericson MD, Baumhover NJ, Rice KG. PEG length and chemical linkage controls polyacridine peptide DNA polyplex pharmacokinetics, biodistribution, metabolic stability and in vivo gene expression. J Control Release. 2013;170:325–33. doi: 10.1016/j.jconrel.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dash PR, Read ML, Barrett LB, Wolfert MA, Seymour LW. Factors affecting blood clearance and in vivo distribution of polyelectrolyte complexes for gene delivery. Gene Ther. 1999;6:643–50. doi: 10.1038/sj.gt.3300843. [DOI] [PubMed] [Google Scholar]
- 28.Adolph EJ, Nelson CE, Werfel TA, Guo R, Davidson J, Guelcher SA, et al. Enhanced performance of plasmid DNA polyplexes stabilized by a combination of core hydrophobicity and surface PEGylation. J Mater Chem B. 2014 doi: 10.1039/C4TB00352G. http://dx.doi.org/10.1039/C4TB00352G. [DOI] [PMC free article] [PubMed]
- 29.Kim N, Jiang D, Jacobi AM, Lennox KA, Rose SD, Behlke MA, et al. Synthesis and characterization of mannosylated pegylated polyethylenimine as a carrier for siRNA. Int J Pharm. 2012;427:123–33. doi: 10.1016/j.ijpharm.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Omedes Pujol M, Coleman DJL, Allen CD, Heidenreich O, Fulton DA. Determination of key structure–activity relationships in siRNA delivery with a mixed micelle system. J Control Release. 2013;172:939–45. doi: 10.1016/j.jconrel.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maeda H, Matsumura Y, Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136–51. doi: 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 32.Nelson CE, Gupta MK, Adolph EJ, Shannon JM, Guelcher SA, Duvall CL. Sustained local delivery of siRNA from an injectable scaffold. Biomaterials. 2012;33:1154–61. doi: 10.1016/j.biomaterials.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, Yu SS, Miteva M, Nelson CE, Werfel T, Giorgio TD, et al. Matrix metalloproteinase responsive, proximity-activated polymeric nanoparticles for siRNA delivery. Adv Funct Mater. 2013;23:3040–52. doi: 10.1002/adfm.201202215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alabi CA, Love KT, Sahay G, Stutzman T, Young WT, Langer R, et al. FRET-labeled siRNA probes for tracking assembly and disassembly of siRNA nano-complexes. ACS Nano. 2012;6:6133–41. doi: 10.1021/nn3013838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson CE, Kintzing JR, Hanna A, Shannon JM, Gupta MK, Duvall CL. Balancing cationic and hydrophobic content of PEGylated siRNA polyplexes enhances endosome escape, stability, blood circulation time, and bioactivity in vivo. ACS Nano. 2013;7:8870–80. doi: 10.1021/nn403325f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–32. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 37.Kirkland-York S, Zhang Y, Smith AE, York AW, Huang F, McCormick CL. Tailored design of Au nanoparticle-siRNA carriers utilizing reversible addition-fragmentation chain transfer polymers. Biomacromolecules. 2010;11:1052–9. doi: 10.1021/bm100020x. [DOI] [PubMed] [Google Scholar]
- 38.Yu SS, Lau CM, Barham WJ, Onishko HM, Nelson CE, Li H, et al. Macrophage-specific RNA interference targeting via “click”, mannosylated polymeric micelles. Mol Pharm. 2013;10:975–87. doi: 10.1021/mp300434e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505–15. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gref R, Lück M, Quellec P, Marchand M, Dellacherie E, Harnisch S, et al. “Stealth” corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf B Biointerfaces. 2000;18:301–13. doi: 10.1016/s0927-7765(99)00156-3. [DOI] [PubMed] [Google Scholar]
- 41.Mislick KA, Baldeschwieler JD. Evidence for the role of proteoglycans in cation-mediated gene transfer. Proc Natl Acad Sci U S A. 1996;93:12349–54. doi: 10.1073/pnas.93.22.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohrt T, Mütze J, Staroske W, Weinmann L, Höck J, Crell K, et al. Fluorescence correlation spectroscopy and fluorescence cross-correlation spectroscopy reveal the cytoplasmic origination of loaded nuclear RISC in vivo in human cells. Nucleic Acids Res. 2008;36:6439–49. doi: 10.1093/nar/gkn693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta MK, Meyer TA, Nelson CE, Duvall CL. Poly(PS-b-DMA) micelles for reactive oxygen species triggered drug release. J Control Release. 2012;162:591–8. doi: 10.1016/j.jconrel.2012.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim HJ, Miyata K, Nomoto T, Zheng M, Kim A, Liu X, et al. siRNA delivery from triblock copolymer micelles with spatially-ordered compartments of PEG shell, siRNA-loaded intermediate layer, and hydrophobic core. Biomaterials. 2014;35:4548–56. doi: 10.1016/j.biomaterials.2014.02.016. [DOI] [PubMed] [Google Scholar]
- 45.Chen H, Kim S, Li L, Wang S, Park K, Cheng J-X. Release of hydrophobic molecules from polymer micelles into cell membranes revealed by forster resonance energy transfer imaging. Proc Natl Acad Sci U S A. 2008;105:6596–601. doi: 10.1073/pnas.0707046105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee H, Kim I-K, Park TG. Intracellular trafficking and unpacking of siRNA/quantum dot-PEI complexes modified with and without cell penetrating peptide: confocal and flow cytometric FRET analysis. Bioconjug Chem. 2010;21:289–95. doi: 10.1021/bc900342p. [DOI] [PubMed] [Google Scholar]
- 47.Morton SW, Zhao X, Quadir MA, Hammond PT. FRET-enabled biological characterization of polymeric micelles. Biomaterials. 2014;35:3489–96. doi: 10.1016/j.biomaterials.2014.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evans BC, Nelson CE, Yu SS, Beavers KR, Kim AJ, Li H, et al. Ex vivo red blood cell hemolysis assay for the evaluation of pH-responsive endosomolytic agents for cytosolic delivery of biomacromolecular drugs. J Vis Exp. 2013;73:e50166. doi: 10.3791/50166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buyens K, De Smedt SC, Braeckmans K, Demeester J, Peeters L, van Grunsven LA, et al. Liposome based systems for systemic siRNA delivery: stability in blood sets the requirements for optimal carrier design. J Control Release. 2012;158:362–70. doi: 10.1016/j.jconrel.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 50.Furgeson DY, Dobrovolskaia MA, Aggarwal P, Hall JB, McLeland CB, Dobrovolskaia MA, et al. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv Drug Deliv Rev. 2009;61:428–37. doi: 10.1016/j.addr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maeda H, Matsumura Y, Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63:131–5. doi: 10.1016/j.addr.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 52.Kwon G, Suwa S, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Enhanced tumor accumulation and prolonged circulation times of micelle-forming poly (ethylene oxide-aspartate) block copolymer-adriamycin conjugates. J Control Release. 1994;29:17–23. [Google Scholar]
- 53.Naeye B, Deschout H, Caveliers V, Descamps B, Braeckmans K, Vanhove C, et al. In vivo disassembly of IV administered siRNA matrix nanoparticles at the renal filtration barrier. Biomaterials. 2013;34:2350–8. doi: 10.1016/j.biomaterials.2012.11.058. [DOI] [PubMed] [Google Scholar]
- 54.Li S-D, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. J Control Release. 2010;145:178–81. doi: 10.1016/j.jconrel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gabizon AA. Liposome circulation time and tumor targeting: implications for cancer chemotherapy. Adv Drug Deliv Rev. 1995;16:285–94. [Google Scholar]
- 56.Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K. Preparation and biological characterization of polymeric micelle drug carriers with intracellular pH-triggered drug release property: tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy. Bioconjug Chem. 2004;16:122–30. doi: 10.1021/bc0498166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.