

Abstract
IMPORTANCE
Tumor genetic sequencing identifies potentially targetable genetic alterations with therapeutic implications. Analysis has concentrated on detecting tumor-specific variants, but recognition of germline variants may prove valuable as well.
OBJECTIVE
To estimate the burden of germline variants identified through routine clinical tumor sequencing.
DESIGN, SETTING, AND PARTICIPANTS
Patients with advanced cancer diagnoses eligible for studies of targeted agents at Memorial Sloan Kettering Cancer Center are offered tumor-normal sequencing with MSK-IMPACT, a 341-gene panel. We surveyed the germline variants seen in 187 overlapping genes with Mendelian disease associations in 1566 patients who had undergone tumor profiling between March and October 2014.
MAIN OUTCOMES AND MEASURES
The number of presumed pathogenic germline variants (PPGVs) and variants of uncertain significance per person in 187 genes associated with single-gene disorders and the proportions of individuals with PPGVs in clinically relevant gene subsets, in genes consistent with known tumor phenotypes, and in genes with evidence of second somatic hits in their tumors.
RESULTS
The mean age of the 1566 patients was 58 years, and 54% were women. Presumed pathogenic germline variants in known Mendelian disease-associated genes were identified in 246 of 1566 patients (15.7%; 95% CI, 14.0%–17.6%), including 198 individuals with mutations in genes associated with cancer susceptibility. Germline findings in cancer susceptibility genes were concordant with the individual’s cancer type in only 81 of 198 cases (40.9%; 95% CI, 34.3%–47.9%). In individuals with PPGVs retained in the tumor, somatic alteration of the other allele was seen in 39 of 182 cases (21.4%; 95% CI, 16.1%–28.0%), of which 13 cases did not show a known correlation of the germline mutation and a known syndrome. Mutations in non–cancer-related Mendelian disease genes were seen in 55 of 1566 cases (3.5%; 95% CI, 27.1%–45.4%). Almost every individual had more than 1 variant of uncertain significance (1565 of 1566 patients; 99.9%; 95% CI, 99.6%–99.9%).
CONCLUSIONS AND RELEVANCE
Germline variants are common in individuals undergoing tumor-normal sequencing and may reveal otherwise unsuspected syndromic associations.
Cancer is in large part a genetic disease in the sense that acquired (somatic) mutations in tumor DNA contribute substantially to the malignant phenotype. Massively parallel (next-generation) sequencing makes it practical to define the spectrum of mutations in an individual cancer. Clinical researchers and practicing oncologists use tumor mutation profiling to guide treatment decisions and to determine patient eligibility for studies of targeted agents.1
The DNA of a patient’s cancer also contains the full range of that individual’s inherited (germline) genetic variation, unless it has been altered or lost through mutagenesis. It has been estimated that, on average, healthy individuals are carriers for 0.7 to 2.8 severe autosomal recessive pediatric disease-causing variants,2,3 and 1.2% to 3.4% of individuals will have high-penetrance, medically actionable mutations that substantially increase their risk for disease.4 The presence of germ-line variants can complicate the effort to identify targetable “driver” mutations in a patient’s cancer. For this reason, laboratories conducting somatic mutation profiling may sequence tumor and normal samples in parallel to identify those variants that are specific to the cancer and hence more likely to be related to the cancer phenotype.5
In some patients, a cancer-predisposing variant is present in the “normal” DNA, either inherited or as an early postzygotic event. The burden of germline variants differs by tumor type, although it can be substantial (eg, mutations of the Fanconi pathway genes can be seen in 20% of patients with ovarian cancer).6 Most germline susceptibility variants are not targetable, although there are exceptions (eg, PARP inhibitors in patients with BRCA1 and BRCA2 mutation-associated cancers). Efforts to characterize germline susceptibility are therefore usually ancillary to the primary purpose of clinical tumor mutation profiling. However, knowledge of these variants could guide the preventive care of the patient or the patient’s family, even if the knowledge does not influence the treatment of the patient’s cancer.7 Some groups, such as the American College of Medical Genetics (ACMG), have recommended a compulsory search for specific germline variants when tumor-normal pairs are used for somatic mutation profiling.8–10 This secondary analysis requires significant incremental effort to characterize the pathogenicity of detected germline variants. To understand the potential scope of such an effort, we identified and characterized germline variants in a series of 1566 oncology patients who underwent somatic mutation profiling at our institution.
Methods
Participants
As part of an ongoing effort to identify patients who are eligible for studies of targeted agents, patients with advanced disease at Memorial Sloan Kettering Cancer Center are offered mutation profiling with a custom targeted capture panel (MSK-IMPACT).11 Individuals with specific disease types are offered profiling if they are potential candidates for a trial of a targeted therapy but are otherwise unselected. The 1566 participants in the present study are patients who underwent genetic profiling between March 2014 and October 2014 in the context of a study approved by the Memorial Sloan Kettering institutional review board (eTable 1 in the Supplement) (NCT01775072). All patients had provided their written informed consent for genetic profiling. An anonymized germ-line analysis was conducted under a waiver approved by the Memorial Sloan Kettering institutional review board. Overall, the mean age of the participants was 58 years, and there were 721 men (46%) and 845 women (54%).
At a Glance.
Targeted tumor sequencing with a panel of 341 genes and matched normal DNA in 1566 individuals with advanced malignant neoplasms revealed presumed pathogenic germline variants (PPGVs) in about 16% of individuals.
Most PPGVs (80.5%; 95% CI, 75.1%–85.0%) were in genes related to cancer susceptibility.
The PPGVs in genes previously designated as clinically actionable were seen in 5.0% (95% CI, 4.1%–6.2%) of individuals.
Most cancer-susceptibility PPGVs were retained in the tumor (91.9%; 95% CI, 87.3%–95.0%).
Almost all individuals carried germline variants of uncertain significance that will require significant curation to determine clinical significance.
Sequencing
The MSK-IMPACT assay was conducted as previously described.11 Briefly, DNA from formalin-fixed, paraffin-embedded tumors and patient matched-normal blood samples were extracted using using the EZ1 Advanced XL system (Qiagen) and sheared using the E220 instrument (Covaris). Sequencing libraries were prepared using the KAPAHTP protocol (KapaBiosystems) and the Biomek FX system (Beckman Coulter). Custom DNA probes targeting exons and selected introns of 341 genes, synthesized using the NimbleGen SeqCap EZ library custom oligo system, were used for hybridization capture and enrichment of target sequence (eTable 2 in the Supplement). Post-capture pooled libraries were sequenced on the Illumina HiSeq 2500 system as 2 × 100 base pairs paired-end reads.
Sequenced reads were trimmed for vestigial adapter sequence using Trim Galore! (Babraham Bioinformatics)12 and then aligned to the hg19 reference genome using BWA-MEM software (Burrows-Wheeler).13 Aligned reads were marked for polymerase chain reaction duplicates and base quality adjusted using GATK (Genome Analysis Toolkit)14 and Picard15 tools. Realignment was performed at indel sites with local assembly using ABRA (assembly-based realigner.16 Point mutations and short indels (<30 base pairs) were called using a combination of MuTect17 (run in single sample mode) and GATK HaplotypeCaller.14 Large deletions and duplications were identified as germline copy number aberrations using a custom copy number detection algorithm: coverage of targeted regions in tested samples were compared against coverage values from a set of reference blood normals, and thresholds for absolute-fold change were set to identify regions of single-copy gains (duplications, absolute-fold change >1.2) and losses (deletions, absolute-fold change less than −1.5).11 Large deletions and duplications were also detected as structural variants in cases where the aberration was caused by a genomic rearrangement and one end of the break point was located in a genomic region captured by the panel bait set. DELLY, version 0.3.3,18 was used to detect structural variants in germline samples, using an unmatched reference normal as a control.11
The mean coverage across tested samples was 371X. Probe sequences and concentrations were optimized to ensure maximally uniform and reproducible coverage across targets. As a result, more than 99% of exons were covered to greater than 20% of the median exon coverage for each sample (71X). To reduce false-positive findings, we included only variants with depth of unique sequencing coverage above 50X and variant allele frequencies above 25%. Variants not annotated as common population polymorphisms (<1% population frequency in the 1000 genomes and NHLBI ESP cohorts19) but present at frequencies greater than 5% in our cohort were flagged as possible systematic assay artifacts.
Variant Curation
Sequence data from tumor-normal pairs were linked through a unique study identifier and then irretrievably disconnected from personal identifiers through anonymization procedures approved by the institutional review board before variant curation. Curation of single-nucleotide variants (SNVs) and indels was performed by Ingenuity Variant Analysis software, version 3.1.20141014, a decision support product that uses an automated algorithm to categorize variants as pathogenic, likely pathogenic, uncertain significance, likely benign, and benign based on the 2015 ACMG guidelines,20 followed by manual confirmation of pathogenic and likely pathogenic variants according to the ACMG pathogenic criteria.20 Of the 341 genes evaluated on the panel, 187 were associated with Mendelian disease in the Online Mendelian Inheritance in Man database (http://omim.org) (OMIM genes) (eTable 4 in the Supplement). Various modes of inheritance are represented within the OMIM genes ranging from autosomal dominant (127 genes), autosomal recessive (27 genes), X-linked recessive (8 genes), X-linked dominant (4 genes), mixed patterns of inheritance (17 genes), and those frequently associated with somatic mosaicism (4 genes). Of these 187 genes evaluated, 93 (49.7%) were associated with a susceptibility to cancer affecting 1 or more organs (eTable 4 in the Supplement). Included on the panel were 26 genes that are also present on the list of genes recommended for secondary analysis by the ACMG (ACMG genes)8 (eTable 3 and eTable 4 in the Supplement).
Tumor Analysis
Pathogenic and likely pathogenic variants were considered together as presumed pathogenic germline variants (PPGVs), which were assessed in the corresponding anonymized tumor samples. Estimations of retention, loss of heterozygosity (LOH), loss of the PPGV, and somatic mosaicism were based on assumptions regarding high tumor purity and diploid tumor genome ploidy, which may have underestimated the frequency of LOH. The classification of LOH was applied when the germline PPGV allele frequency (GAF) was between 0.3 and 0.7 and its frequency in the tumor, labeled tumor allele frequency (TAF), was greater than 0.7, indicating retention of the germline PPGV and loss of the wild-type allele. Variants were considered lost in the tumor if the GAF was between 0.3 and 0.7 and the TAF was less than 0.3. Variants were considered possibly somatic mosaic if the GAF and TAF were both less than 0.3.
Statistical Analysis
Patient demographics were summarized by tumor type. Age was described by mean, and sex was described by frequency and percentage. With 1566 cases, we could estimate various proportions of interest to within at most ±2.5%. This calculation used the Wilson method.21 The actual 95% confidence intervals after observing the data were computed by Wilson score method as well. All statistical analyses were conducted in R, version 3.1.1. Package “PropCIs” was used to compute the Wilson score confidence intervals.
Results
Considering all 187 genes with genetic disease associations in OMIM (eTable 4 in the Supplement), we found a mean of 63.3 (median, 63; mode, 62; range 29–92) protein-coding variants per individual. Although most of these variants were classified as benign or likely benign, 246 of the 1566 participants (15.7%; 95% CI, 14.0%–17.6%) had at least 1 pathogenic or likely pathogenic protein-coding variant in 1 of the genes in the OMIM gene list (Figure 1). There were 2 participants (0.1%; 95% CI, 0.04%–0.46%) who carried 3 PPGVs and 20 (1.3%; 95% CI, 0.83%–1.96%) with 2 PPGVs. Most PPGVs were single-nucleotide variants or small insertions or deletions (eTable 5 in the Supplement). Assessment of germline copy number variation identified 10 presumed pathogenic whole or partial gene deletion or duplication events (0.64% of individuals; 95% CI, 0.35%–1.17%; 3.7% of PPGVs [10 of 270]; 95% CI, 2.02%–6.68%).
Figure 1. Individuals With at Least 1 Presumed Pathogenic Germline Variant in OMIM Genes, Including the Cancer and ACMG Subsets.
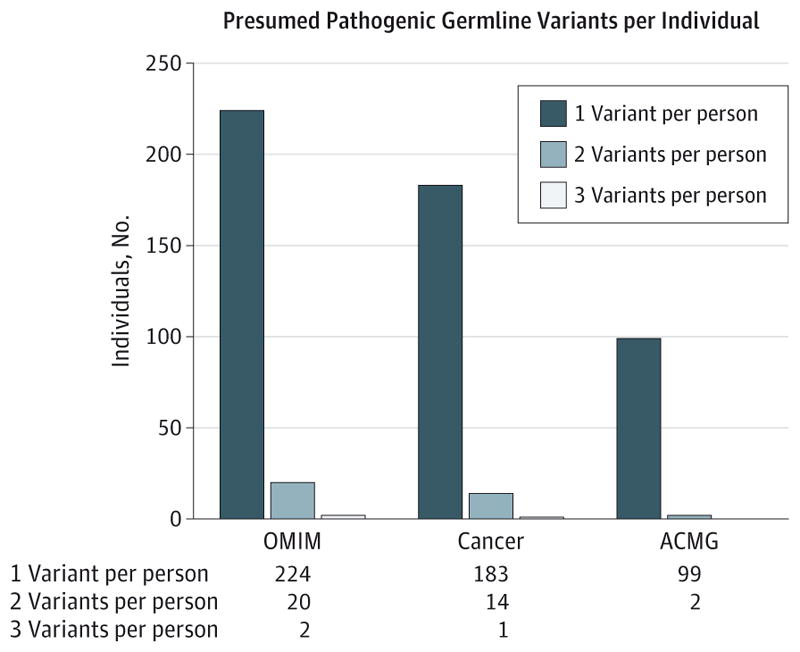
The number of genes in the entire Online Mendelian Inheritance in Man (OMIM) subset is 187 (http://omim.org), which includes the Cancer subset of 93 genes and the partially overlapping American College of Medical Genetics (ACMG) subset of 26 genes8 (eTable 4 in the Supplement).
In the 1566 individuals studied, 1565 (99.9%; 95% CI, 99.6%–99.9%) carried 1 or more variants of uncertain significance in a gene associated with a disease. The median number of variants of uncertain significance per individual was 6, mode 5 (range 0–20). Most individuals (1546 [98.7%; 95% CI, 98.0%–99.2%]) carried more than 1 variant of uncertain significance (19 with 1, 49 with 2, 126 with 3, 176 with 4, 244 with 5 [mode], and 951 with 6 or more). Variants of uncertain significance were seen in almost all genes (eTable 6 in the Supplement).
The identified PPGVs were grouped by potential for phenotypic expression in the patient by consideration of the predetermined subcategories of the OMIM genes (Cancer and ACMG; eTable 4 in the Supplement), mode of inheritance, known penetrance for a disease phenotype, and presence on the ACMG list or beyond, with consideration of cancer-related, non–cancer-related, and likely somatic mosaicism (Figure 2). Most PPGVs (198 of 246 individuals, 80.5%; 95% CI, 75.1%–85.0%) were found in genes known to be associated with cancer-susceptibility syndromes. The most commonly mutated cancer susceptibility genes were BRCA2 (n = 31), CHEK2 (n = 23), MUTYH (n = 23), and BRCA1 (n = 21). Other genes within which there were 4 or more PPGVs identified were ATM (n = 9), BRIP1 (n = 9), CDH1 (n = 7), ERCC2 (n = 7), RET (n = 6), SDHA (n = 6), MSH6 (n = 5), PALB2 (n = 5), NF1 (n = 4), TP53 (n = 4), MSH2 (n = 4), BAP1 (n = 4), and RECQL4 (n = 4). An additional 42 PPGVs were found in 26 other cancer-susceptibility genes (eTable 6 in the Supplement). All variants were heterozygous and monoallelic except in one case of a participant in the sixth decade of life with small bowel cancer who had compound heterozygous mutations in MUTYH.
Figure 2. Total Presumed Pathogenic Germline Variants (PPGVs) Identified in 61 Genes.

Results of germline analysis of 187 Mendelian disease-associated genes from a 341-gene panel (MSK-IMPACT11) in 246 of 1566 individuals. AD indicates autosomal dominant (127 genes); AR, autosomal recessive (27 genes); CX, mixed patterns of inheritance (17 genes); SM, frequently associated with somatic mosaicism (4 genes); XD, X-linked dominant (4 genes); and XR, X-linked recessive (8 genes).
In 55 of the 1566 participants (3.5%; 95% CI, 2.7%–4.5%), 56 PPGVs were noted in 18 genes associated with Mendelian conditions but not known to cause cancer susceptibility. Of these, 27 PPGVs were seen in 5 genes known to be somatically mutated in hematologic clonal expansions, myelodysplasia, or related conditions (DNMT3A, MPL, ASXL1, JAK2, and BCOR) and may represent postzygotic clonal hematopoiesis rather than constitutional mutation22–25 (eTables 7–9 in the Supplement). Variants indicating a carrier state for an autosomal recessive disorder were noted in 125 individuals (8.0%; 95% CI, 6.7%–9.4%). A total of 209 variants linked to autosomal dominant genetic disease were seen in 192 individuals. The anonymized design of this study precluded genotype-phenotype correlation for diagnoses other than cancer. One X-linked dominant variant was seen in a man.
There were 26 genes included in the panel that ACMG has indicated are of sufficient clinical utility to mandate secondary analysis in the course of clinical tumor-normal sequencing (Figure 3).8–10 There were 101 individuals (6.4%; 95% CI,5.3%–7.8%) with at least 1 PPGV in an ACMG-endorsed gene, including 2 individuals with 2 variants (eTable 5 in the Supplement). If monoallelic germline MUTYH variants are excluded from analysis of the ACMG subset, there were a total of 80 PP-GVs in 79 individuals (5.0%; 95% CI, 4.1%–6.2%) (eTable 9 in the Supplement).
Figure 3. Aggregate Data Showing Presumed Pathogenic Germline Variants (PPGVs) in ACMG Genes by Tumor Type.

Phenotypes are designated as expected to be secondary to pathogenic PPGVs based on common presentations of the known syndromes and gene associations (dark beige) or unexpected (light beige). ACMG indicates American College of Medical Genetics (ACMG).20
There were PPGVs observed in individuals with cancers other than those typically associated with the gene containing the PPGV (eTable 7 in the Supplement). Only 81 of 198 participants (40.9%; 95% CI, 34.3%–47.9%) with PPGVs in cancer-susceptibility genes had diagnoses considered to be associated with the mutated gene. For example, only 28 of 52 PPGVs (53.8%; 95% CI, 40.5%–66.7%) in BRCA1 and BRCA2 were observed in individuals with breast, ovary, prostate, or pancreas cancer. The percentage of PPGVs in ACMG genes associated with unexpected phenotypes varied per tumor (Figure 3), indicating that genotype-phenotype correlations with breast, colon, and prostate cancer and other rarer tumors such as gastrointestinal stromal tumor, adrenocortical carcinoma, renal cell carcinoma, and cancer of the small bowel were more expected than those for other cancers (eg, non-small-cell lung cancer). Within the cancer-susceptibility genes, the PPGVs were retained in the tumor in 182 of 198 participants (91.9%; 95% CI, 87.3%–95.0%), and there was LOH or mutation of the other allele in 39 of these 182 (21.4%; 95% CI, 16.1%–28.0%). Of the 39 cases with retention of the PPGVs in the tumor and apparent inactivation of the other allele, 13 (33.3%; 95% CI, 20.6%–49.0%) were associated with unexpected phenotypes. For example, 3 cases of stomach adenocarcinoma, 1 neuroendocrine tumor, and 1 sarcoma were associated with BRCA1 or BRCA2 PPGVs (eTables 8 and 9 in the Supplement).
Other examples of unexpected PPGVs that were also retained in the tumor included a truncating SDHD PPGV in a premenopausal woman with breast cancer, although we do not know if the variant was paternally inherited. Three unique protein-truncating mutations and a partial gene duplication in BAP1, associated with malignant mesothelioma, uveal melanoma, and cutaneous melanoma (OMIM 614327), were identified in 4 individuals, 1 each with high-grade serous ovarian cancer, hepatocellular carcinoma, stomach adenocarcinoma, and early-onset prostate cancer. Of note, the ovarian cancer harbored a second somatic splice site mutation. The full spectrum of BAP1-associated tumors is yet to be elucidated.
Loss of function PPGVs in SDHA, a gene associated with hereditary paraganglioma-pheochromocytoma syndrome (OMIM 600857), were seen in 6 individuals, with only a gastrointestinal stromal tumor having previously been associated. A PPGV in RET was seen in an individual with colon cancer, which was further unexpectedly associated with LOH, although a second hit has been reported with metastatic medullary thyroid carcinoma.26 Furthermore, 2 cases (breast and thyroid cancer) occurred in association with a known founder PPGV in MUTYH and showed LOH in the tumors, raising the possibility for the tumors to develop an increased mutational load secondary to aberrant base-excision repair, and potentially relevant to the consideration of therapeutic strategies targeting tumor immunogenicity (eTable 8 in the Supplement).27 The PPGVs in non–cancer-susceptibility genes were only retained in the tumor in 34 of 55 participants (61.8%; 95% CI, 48.6%–73.5%). Loss of heterozygosity for the other allele was observed in 2 of these 34 (5.9%; 95% CI, 1.6%–19.1%) (eTable 8 in the Supplement).
Discussion
Somatic mutation profiling using massively parallel next-generation sequencing is one of the most prevalent applications of precision medicine. In the course of analyzing tumor DNA (without matched normal DNA), sequencing can identify constitutional (germline) DNA variations that are associated with disease or susceptibility to disease as well as carrier states for Mendelian disorders. Many centers use matched tumor-normal sequencing to facilitate more accurate calling of somatic mutations by using the normal DNA to exclude germline variants from the tumor calls.5 In the present analysis of 1566 consecutive patients undergoing tumor-normal sequencing with a custom 341-gene panel, nearly 16% of patients carried germline pathogenic or likely pathogenic variants in a gene linked to an inherited human disease. Unsurprisingly, as this was an ascertainment from patients with cancer sequenced using a cancer-specific panel, most of these variants were in cancer-susceptibility genes. Overall,12.6% of patients in this series carried a mutation in a cancer-susceptibility gene, a higher prevalence than the 3% recently described in a cohort of patients with apparently sporadic cancers.5 The higher prevalence of mutations in cancer-predisposing genes observed here may be related to differences in the number of genes contained on the panel and the diagnoses of the patients included in this report. Breast cancer was the most common diagnosis in our cohort, and BRCA1 and BRCA2 mutations were the most prevalent germline mutations, with 3 Ashkenazi Jewish mutations accounting for almost half of the BRCA1 and BRCA2 PPGVs. This illustrates that the anticipated prevalence of mutations may vary among ascertainments as a result of population characteristics.
When tumors are analyzed without a matched normal comparison sequence, germline variants will be detected in the tumors themselves. This complicates interpretation because it may be difficult to differentiate germline from somatic variants in a tumor-only sequence. Sequencing strategies that do not use matched normal analyses will therefore have to contend with the germline findings we describe, as the PPGV is retained in the tumor more than 90% of the time.
The presence of potentially significant germline mutations in over 10% of patients has important implications for clinical centers considering routine reporting of potentially pathogenic variants detected after sequencing tumor-normal pairs. These findings indicate that it will not be uncommon to detect unexpected actionable variants. Even restricting reporting to the ACMG-endorsed gene set would identify potentially actionable mutations in at least 5% of our patients. Those medical centers offering testing must establish appropriate pretest education tools to inform patients about the potential for identifying inherited susceptibility or previously undiagnosed genetic disease and consider mechanisms for eliciting patient preferences regarding the communication of such results. Medical centers offering such genomic analysis will also need to develop appropriate posttest result communication protocols, as patients currently undergoing tumor mutation profiling may be difficult to engage in traditional posttest genetic counseling owing to their advanced disease. The clinical cancer genetics community will be challenged to establish best practices for communicating results to family members, particularly if a patient with advanced disease who is undergoing tumor-normal sequencing dies before receiving his or her test results.28
The present study illustrates the potential burden of routine germline analysis for laboratories and cancer care clinicians engaged in tumor-normal analysis on identified samples. Although only 5% to 16% of patients carry potentially pathogenic variants, numerous benign and likely benign variants were detected in nearly all patients, as were rare missense mutations of uncertain significance. In our experience, over 60% of patients carried 6 or more variants of uncertain significance in genes linked to genetic disease. Each of these variants requires careful curation to determine pathogenicity, a time-consuming process. Automated decision-support tools are being developed by both academic centers and commercial enterprises, but these efforts are still fairly rudimentary. Based on the experience reported here, reporting laboratories will require substantial resources for manual variant curation to ensure the quality of their reports.
The present report demonstrates the potential value of a broad germline sequencing approach in the context of tumor-normal analysis. In 59% of individuals with a potentially pathogenic variant in a cancer-susceptibility gene, their cancer type was not known to be associated with that gene. Finding unexpected mutations creates an opportunity to institute beneficial preventive measures for the patient’s family, and possibly to use specific targeted treatments (such as PARP inhibitors) for the patient. The significant prevalence of such unexpected germline mutations suggests that cancer susceptibility due to rare variation may be more common than previously believed. Furthermore, the observation that cancer-susceptibility PPGVs were commonly retained in the tumor and that the wild-type allele appeared to be lost more frequently than in noncancer disease genes suggests that the spectrum of cancer risk linked to these variants may be broader than currently believed. This hypothesis requires further investigation in appropriate laboratory and clinical studies.
Conclusions
Tumor-normal sequencing in a consecutive series of 1566 patients undergoing somatic tumor mutation profiling identified potentially pathogenic mutations in the normal sample of a substantial proportion of patients, including variants in genes not known to be associated with inherited predisposition to the tumor types of the patients studied. Variants of uncertain significance were common, and most patients had multiple such variants, which will require substantial effort to curate. These findings have important implications for those considering routine reporting of germline results from somatic mutation profiling using tumor-normal pairs.
Supplementary Material
Acknowledgments
Funding/Support: This work was supported by a gift from Mindy and Jonathan Gray and by The Robert and Kate Niehaus Center for Inherited Cancer Genomics. Dr Schrader is supported by the Canadian Institutes of Health Research, Michael Smith Foundation for Health Research, and British Columbia Cancer Foundation. Dr Robson is supported by the Breast Cancer Research Foundation.
Footnotes
Correction: This article was corrected on February 11, 2016, to add the middle initial to an author’s name.
Conflict of Interest Disclosures: None reported.
Previous Presentation: This research was presented at the Canadian Cancer Research Conference; November 10, 2015; Montreal, Canada.
Additional Contributions: We thank the members of the Molecular Diagnostics Service in the Department of Pathology, Memorial Sloan Kettering Cancer Center, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology for their role in conducting the MSK-IMPACT genetic sequencing analyses.
Author Contributions: Drs Schrader and Cheng contributed equally to this work; both had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Schrader, Cheng, Joseph, Benayed, Solit, Hyman, Klimstra, Offit, Berger, Robson.
Acquisition, analysis, or interpretation of data: Schrader, Cheng, Joseph, Prasad, Walsh, Zehir, Ni, Thomas, Ashraf, Lincoln, Arcila, Stadler, Solit, Hyman, Zhang, Ladanyi, Offit, Berger, Robson.
Drafting of the manuscript: Schrader, Cheng, Joseph, Stadler, Solit, Hyman, Offit, Robson.
Critical revision of the manuscript for important intellectual content: Schrader, Joseph, Prasad, Walsh, Zehir, Ni, Thomas, Benayed, Ashraf, Lincoln, Arcila, Stadler, Solit, Zhang, Klimstra, Ladanyi, Offit,
Berger, Robson.
Statistical analysis: Cheng, Joseph, Ni, Berger.
Obtained funding: Solit, Offit.
Administrative, technical, or material support: Joseph, Thomas, Ashraf, Lincoln, Arcila, Solit, Hyman, Zhang, Klimstra, Offit, Berger.
Study supervision: Schrader, Joseph, Arcila, Solit, Offit, Berger, Robson.
Institutional review board regulatory compliance: Lincoln.
Role of the Funder/Sponsor: The funding institutions had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
References
- 1.Hyman DM, Solit DB, Arcila ME, et al. Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug Discov Today. doi: 10.1016/j.drudis.2015.08.005. S1359-6446(15)00321-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bell CJ, Dinwiddie DL, Miller NA, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3(65):65ra4. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tabor HK, Auer PL, Jamal SM, et al. NHLBI Exome Sequencing Project. Pathogenic variants for Mendelian and complex traits in exomes of 6,517 European and African Americans: implications for the return of incidental results. Am J Hum Genet. 2014;95(2):183–193. doi: 10.1016/j.ajhg.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dorschner MO, Amendola LM, Turner EH, et al. National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project. Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am J Hum Genet. 2013;93(4):631–640. doi: 10.1016/j.ajhg.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones S, Anagnostou V, Lytle K, et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med. 2015;7(283):283ra53. doi: 10.1126/scitranslmed.aaa7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanchi KL, Johnson KJ, Lu C, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat Commun. 2014;5:3156. doi: 10.1038/ncomms4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stadler ZK, Schrader KA, Vijai J, Robson ME, Offit K. Cancer genomics and inherited risk. J Clin Oncol. 2014;32(7):687–698. doi: 10.1200/JCO.2013.49.7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green RC, Berg JS, Grody WW, et al. American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15(7):565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bombard Y, Robson M, Offit K. Revealing the incidentalome when targeting the tumor genome. JAMA. 2013;310(8):795–796. doi: 10.1001/jama.2013.276573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parsons DW, Roy A, Plon SE, Roychowdhury S, Chinnaiyan AM. Clinical tumor sequencing: an incidental casualty of the American College of Medical Genetics and Genomics recommendations for reporting of incidental findings. J Clin Oncol. 2014;32(21):2203–2205. doi: 10.1200/JCO.2013.54.8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17(3):251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Babraham Bioinformatics. [Accessed October 1, 2013];Taking appropriate QC measures for RRBS-type or other -Seq applications with Trim Galore! http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
- 13.Li H. [Accessed October 1, 2013];Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. http://arxiv.org/abs/1303.3997.
- 14.McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. [Accessed November 20, 2014];Picard. http://broadinstitute.github.io/picard/
- 16.Mose LE, Wilkerson MD, Hayes DN, Perou CM, Parker JS. ABRA: improved coding indel detection via assembly-based realignment. Bioinformatics. 2014;30(19):2813–2815. doi: 10.1093/bioinformatics/btu376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28(18):i333–i339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.NHLBI Exome Sequencing Project (ESP) [Accessed June 15, 2014];Exome Variant Server. http://evs.gs.washington.edu/EVS/
- 20.Richards S, Aziz N, Bale S, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson EB. Probable inference, the law of succession, and statistical inference. J Am Stat Assoc. 1927;22(158):209–212. [Google Scholar]
- 22.Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kilpivaara O, Mukherjee S, Schram AM, et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat Genet. 2009;41(4):455–459. doi: 10.1038/ng.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quadro L, Fattoruso O, Cosma MP, et al. Loss of heterozygosity at the RET protooncogene locus in a case of multiple endocrine neoplasia type 2A. J Clin Endocrinol Metab. 2001;86(1):239–244. doi: 10.1210/jcem.86.1.7144. [DOI] [PubMed] [Google Scholar]
- 27.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bombard Y, Offit K, Robson ME. Risks to relatives in genomic research: a duty to warn? Am J Bioeth. 2012;12(10):12–14. doi: 10.1080/15265161.2012.699157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.