

Abstract
Shennongjia Rhinopithecus roxellana (SNJ R. roxellana) is the smallest geographical population of R. roxellana. The phylogenetic relationships among its genera and species and the biogeographic processes leading to their current distribution are largely unclear. To address these issues, we resequenced and obtained a new, complete mitochondrial genome of SNJ R. roxellana by next‐generation sequencing and standard Sanger sequencing. We analyzed the gene composition, constructed a phylogenetic tree, inferred the divergence ages based on complete mitochondrial genome sequences, and analyzed the genetic divergence of 13 functional mtDNA genes. The phylogenetic tree and divergence ages showed that R. avunculus (the Tonkin snub‐nosed monkey) was the first to diverge from the Rhinopithecus genus ca. 2.47 million years ago (Ma). Rhinopithecus bieti and Rhinopithecus strykeri formed sister groups, and the second divergence from the Rhinopithecus genus occurred ca. 1.90 Ma. R. roxellana and R. brelichi diverged from the Rhinopithecus genus third, ca. 1.57 Ma. SNJ R. roxellana was the last to diverge within R. roxellana species in 0.08 Ma, and the most recent common ancestor of R. roxellana is 0.10 Ma. The analyses on gene composition showed SNJ R. roxellana was the newest geographic population of R. roxellana. The work will help to develop a more accurate protection policy for SNJ R. roxellana and facilitate further research on selection and adaptation of R. roxellana.
Keywords: divergence ages, mitochondrial genome (mtDNA), next‐generation sequencing, phylogenetic analyses, Rhinopithecus roxellana, Shennongjia Rhinopithecus roxellana
1. INTRODUCTION
The Rhinopithecus genus (snub‐nosed monkeys) has five species, including three species that only live in China, the Sichuan golden monkey (Rhinopithecus roxellana), the Guizhou golden monkey (Rhinopithecus brelichi), and the Yunnan golden monkey (Rhinopithecus bieti). One species lives in North Vietnam, the Tonkin snub‐nosed monkey (Rhinopithecus avunculus; Le & Covert, 2010; Yu, Wang, Ting, & Zhang, 2011). The Myanmar golden monkey (Rhinopithecus strykeri) was reported in Myanmar (Burma) but also exists in the contiguous forests in Nujiang Prefecture, China (Geissmann et al., 2011; Liedigk et al., 2012). Rhinopithecus roxellana was distributed widely in China during the Pleistocene; however, today, wild R. roxellana populations only exist in three isolated mountainous regions, including the Minshan and Qionglai mountains in Sichuan and Gansu provinces (SG), the Qinling mountains in Shaanxi province (QL), and the Shennongjia National Nature Reserve (SNJ) in Hubei province. Rhinopithecus roxellana is classified as an endangered species by the World Conservation Union and is protected at Level I by law of the People's Republic of China on the protection of wildlife (Le, 2015; Pan et al., 2009; Zhou et al., 2014). Due to the distinct appearance of R. roxellana, a beautiful golden coat and snub nose, and its rarity, R. roxellana is an endangered arboreal primate icon in China (Figure 1). R. roxellana has a population of approximately 22,000, including approximately 16,000 individuals from the SG population, approximately 5,500 individuals from the QL population, and approximately 1,000 individuals from the SNJ population (Chang, Liu, Yang, Li, & Vigilant, 2012; Yang et al., 2012).
Figure 1.

The photograph of Rhinopithecu roxellana in Shennongjia National Nature Reserve. It would be used as the graphical table of contents
The phylogenetic relationships of the Rhinopithecus genus have been studied (Karanth, Singh, Collura, & Stewart, 2008; Li et al., 2010; Osterholz, Walter, & Roos, 2008; Perelman et al., 2011; Sterner, Raaum, Zhang, Stewart, & Disotell, 2006); however, debates on the origin of R. roxellana are still ongoing. Wang, Jiang, and Li (1998) found some morphological differences among the R. roxellana specimens from Sichuan‐Gansu (SG), Shanxi Qinling (QL), and Hubei Shennongjia (SNJ) and classified the SG, SNJ, and QL populations as R. roxellana ssp. roxellana, R. roxellana ssp. hubeiensis, and R. roxellana Qinlingensis, respectively (Li et al., 2007). Pan et al. (2009) proposed two hypotheses for the origin of R. roxellana. The mono‐origin hypothesis suggested that the QL and SNJ populations originated from the SG population, while the multiorigin hypothesis insisted that the SG population evolved from a fusion of the QL and SNJ populations. Nevertheless, neither of these two hypotheses can perfectly explain the evolutionary relationships among these three populations. The Shennongjia National Nature Reserve is the most eastern habitat of R. roxellana in China, and SNJ R. roxellana has the smallest R. roxellana population. To protect the SNJ R. roxellana population, it is particularly important to study the genetic evolution and the genetic diversity of SNJ R. roxellana. Analyses of the complete mitochondrial DNA control region suggested that the QL and SNJ populations originated from the SG population, but there was no gene flow between the SNJ and SG populations (Luo, Liu, Pan, Zhao, & Li, 2012). In contrast, the structure analyses of 16 microsatellite loci showed that the SNJ population consisted of two groups and that the SNJ population originated from the SG and QL populations (Chang, Liu, Yang, et al., 2012). Moreover, some studies showed SNJ population had the lowest diversity (Chang, Luo, Liu, et al., 2012; Chang, Liu, Yang, et al., 2012; Luo et al., 2012), other studied showed SNJ population had rich genetic diversity (He, Zhang, Peng, Li, & Li, 2010). However, until now the phylogenetic relationship and genetic diversity of SNJ R. roxellana is still unclear.
Mitochondrial DNA (mtDNA) has been widely used as a molecular marker in phylogenetic and phylogeographic studies of the Rhinopithecus genus and other primates because it is high mutation and substitution rates, low effective population size, high copy number, rare gene recombination, maternal transmission, and easy accessibility (Finstermeier et al., 2013; Kolleck et al., 2013; Liedigk et al., 2012; Yang et al., 2012; Yu et al., 2011; Zhang, 1998). Mitochondrial genes/fragments such as the D‐loop, Cytochrome b (Cytb), tRNA, and NADH gene have been used for phylogenetic studies on genetic divergence and phylogenetics of the R. roxellana species (Kolleck et al., 2013; Le, 2015; Luo et al., 2012; Sterner et al., 2006; Zhang, 1998). The complete mitochondrial genomes were used to analyze primate phylogenetic relationships and divergence date (Pozzi et al., 2014). Moreover, the complete mitochondrial genomes are effectively used as the molecular markers in molecular ecology, population genetics, conservation genetics, and evolutionary biology (Ekblom & Galindo, 2011; Morin et al., 2010; Yann & Juan, 2010). For example, complete mitochondrial genomes have been used to solve many evolutionary questions in humans (Green, Malaspinas, & Krause, 2008; Krause et al., 2010) and to investigate the evolutionary histories of endangered and enigmatic species, such as mammoths, brown bears, aurochs, Tasmanian tigers, and polar bears (Driscoll, Yamaguchi, & Bar, 2009; Edwards et al., 2010; Lindqvist et al., 2010; Miller et al., 2009, 2012; Morin et al., 2010).
In this study, we used the Illumina Hi‐seq2000 platform to resequence the complete mitochondrial genome of SNJ R. roxellana. Furthermore, we analyzed the mitochondrial genomic divergence and phylogenetic relationships within the R. roxellana species and the Rhinopithecus genus to determine whether R. roxellana is a monotypic or polytypic species and assess the divergence ages and evolutionary status of the SNJ population within R. roxellana.
2. MATERIALS AND METHODS
2.1. Ethics statement
The research complied with protocols approved by the Forestry Ministry of Hubei Province, China, and abided by the legal requirements of China. Permits to collect samples were provided by the Shennongjia National Nature Reserve, and the staff of the Shennongjia National Nature Reserve helped to collect samples and supported the study. The research was conducted according to animal care regulations and the principles of the American Society of Primatologists.
2.2. Sample information
For sequencing, we collected a frozen liver sample of a three‐month‐old male R. roxellana who was from Shennongjia National Nature Reserve in China and killed during a male takeover in August 2012. His corpse was immediately stored at −20°C after death.
2.3. DNA extraction, short‐read sequencing, de novo assembly and annotation
In the study, we resequenced the mitochondrial genome of SNJ R. roxellana. Firstly, we extracted pure liver mitochondria using a tissue mitochondria isolation kit (Beyotime Biotech Inst., Beijing, China), and then, mtDNA was extracted using the blood DNA kit (E.Z.N.A.; Omega, USA). The extract was prepared for both Sanger sequencing and next‐generation sequencing (NGS). One part of the mtDNA was used to construct a paired‐end sequencing library with an insertion size of approximately 500 base pairs (bp) and was sequenced by NGS with Illumina Hi‐seq2000. We prepared the libraries as follows: 200–500 ng DNA is fragmented by Covaris. The fragmented DNA is combined with End Repair Mix, incubate at 20°C for 30 min. Purify the end‐repaired DNA with QIAquick PCR Purification Kit (Qiagen) and then add A‐Tailing Mix, incubate at 37°C for 30 min. Combine the purified Adenylate 3′ Ends DNA, Adapter and Ligation Mix, incubate the ligation reaction at 20°C for 15 min. Adapter‐ligated DNA is selected by running a 2% agarose gel to recover the target fragments. Purify the gel with QIAquick Gel Extraction kit (Qiagen). Several rounds of PCR amplification with PCR Primer Cocktail and PCR Master Mix are performed to enrich the Adapter‐ligated DNA fragments. Then, the PCR products are selected by running another 2% agarose gel to recover the target fragments. Purify the gel with QIAquick Gel Extraction kit (Qiagen). The remainder mtDNA was used to perform a PCR amplification by the primers MT(15,928–16,543) and Mt(7–535) (Table 1) and general sequencing to link the gaps between scaffolds. The PCR products were preliminarily confirmed on a 1.0% agarose gel, sequenced directly in two reactions with forward and reverse primers by Sanger sequencing and then spliced with contigs or scaffolds using the ContigExpress software package.
Table 1.
The information on the primers
| Sequence (5′–>3′) | Tm | Product length | |
|---|---|---|---|
| MT(15,928–16,543) forward primer | ATTTAGTCTGGCTTTTGAAG | 60.46 | 615 bp |
| Reverse primer | GATAACAGCGCAATCCTATTC | 59.48 | |
| Mt(7–535) forward primer | ATCGACATAGGGTTTACGA | 59.01 | 528 bp |
| Reverse primer | CTTAAAACCTTCAACCTCC | 60.14 |
We used SOAP de novo to assemble the short reads from NGS data (http://soap.genomics.org.cn). In order to successfully de novo assemble the short reads, we filtered the low‐quality reads (When the quality value of a base was less than or equail 7, we defined the base as low‐quality base. If there was more than 10% low‐quality base in a read, the read was referred as low‐quality read.), and used the high‐quality clear data, and used –K, –R, –F, and –u as the program parameters in the SOAP to finish assembly. Each read that was identified as mtDNA was aligned to mtDB. These alignments were then merged, and each alignment column was examined to determine the majority base, yielding large‐assembled contigs or scaffolds of mtDNA. To evaluate accuracy and completeness, the assembled mitochondrial genome sequence homology analysis was conducted using online BlastN in GenBank. Mitochondrial genomes were annotated in reference to the R. roxellana mtDNA sequence (JQ821835.1). If the reading frame of protein‐coding genes was disrupted, the original read assembly was revised and corrected manually.
2.4. Genetic divergence
To estimate the genetic divergence of the SNJ R. roxellana in base composition, we performed the genomic analyses including base composition, base substitution, amino acid replacement with respect to reference mtDNA sequence, and the number of diversity base and the gaps to R. brelichi mtDNA sequence using the complete mitochondrial sequence data. Hence, alignment between SNJ R. roxellana mtDNA and QL R. roxellana mtDNA (reference sequence) was conducted. Alignment between QL R. roxellana mtDNA and R. brelichi mtDNA, alignment between SG R. roxellana mtDNA and R. brelichi mtDNA, alignment between SNJ R. roxellana mtDNA and R. brelichi mtDNA were performed. All pairs’ alignment was conducted using online BlastN in GenBank, and amino acid replacement was analyzed according to gene annotation in GenBank. We further analyzed the genetic divergence between SNJ R. roxellana and reference mtDNA based on 13 functional mtDNA genes.
2.5. Phylogenetic reconstruction of genus Rhinopithecus
To assess the phylogenetic position of SNJ R. roxellana among the Rhinopithecus genus and the relationships within the R. roxellana species, multiple alignments and phylogenetic analyses were conducted using the neighbor‐joining (NJ), maximum‐likelihood (ML), and Bayesian inference (BI) methods. NJ and ML phylogenetic trees were constructed using the MEGA6.0 program (Kimura, 1980; Tamura, Stecher, Peterson, Filipski, & Kumar, 2013). The Kimura 2‐parameter nucleotide model was employed to delete pairwise gaps in the NJ analysis. The reliability of tree topologies was evaluated with 10,000 bootstrap replicates in the NJ analysis and 1,000 bootstrap replicates in the ML analysis. The appropriate DNA substitution model was identified by the lowest Bayesian information criterion scores and calculated using Modeltest version 3.06. BI phylogenetic trees were constructed using BEAST Software (Drummond, Suchard, Xie, & Rambaut, 2012). The Markov chain Monte Carlo algorithm was run for 2 × 1,000,000 generations with four incrementally heated chains. The analyses started with random trees and were sampled every 1 × 1,000 generations. The first 5 × 10,000 trees were treated as burnt in and discarded. The Bayesian consensus tree was constructed using the remaining trees. Internodes with posterior probabilities of 95% were considered statistically significant. In all analyses, positions containing gaps and missing data were eliminated in the phylogenetic tree construction. In this study, eleven complete mtDNA sequences of seven Rhinopithecus, one Pygathrix nigripes, one Nasalis larvatus, Trachypithecus germaini, and Presbytis melalophos were retrieved from GenBank Database (Table 2); the latter four species were used as outgroups.
Table 2.
The information on sequences constructing phylogenetic tree
| Acc. no. | Species and sequence information | Abbreviation |
|---|---|---|
| KM504390.1 | SNJ Rhinopithecu roxellana mitochondrion, complete genome | SNJ R. roxellana |
| DQ355300.1 | SG Pygathrix roxellana mitochondrion, complete genome | SG R. roxellana |
| JQ821835.1 | QL R. roxellana mitochondrion, complete genome | QL R. roxellana |
| JQ821836.1 | Rhinopithecus brelichi mitochondrion, complete genome | R. brelichi |
| HM125579.1 | Rhinopithecus bieti mitochondrion, complete genome | R. bieti |
| JQ821838.1 | Rhinopithecus strykeri mitochondrion, complete genome | R. strykeri |
| HM125578.1 | Rhinopithecus avunculus mitochondrion, complete genome | R. avunculus |
| JF293094.1 | Nasalis larvatus mitochondrion, complete genome | N. larvatus |
| JQ821840.1 | Pygathrix nigripes mitochondrion, complete genome | P. nigripes |
| HQ149047.1 | Trachypithecus germaini mitochondrion, complete genome | T. germaini |
| DQ355299.1 | Presbytis melalophos mitochondrion, complete genome | P. melalophos |
2.6. Divergence age analysis
To estimate R. roxellana divergence ages, we inferred the evolutionary history based on complete mtDNA, employing a complete molecular clock approach as implemented in MEGA6 (Tamura et al., 2013). In order to ensure a relatively accurate genetic divergence time, we first analyzed the information site and genetic distance of all sequences and then used the NJ and ML methods to infer the evolutionary history (Tamura, Nei, & Kumar, 2004; Tamura et al., 2012). All positions containing gaps and missing data were eliminated, and evolutionary analyses were conducted in MEGA6. In the NJ method, the evolutionary distances are computed using the ML method (Felsenstein, 1985) and are measured as the number of base substitutions per site. The percentage of replicate trees in which associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches (Tamura et al., 2012). The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances that are used to infer the phylogenetic tree. Divergence times for all branching points in the topology were calculated with the RelTime method (Tamure et al., 2012) using the branch lengths contained in the inferred tree. In the ML method, initial tree(s) for the heuristic search were obtained automatically by applying the NJ and BioNJ algorithms to a matrix of pairwise distances estimated to use the maximum composite likelihood approach and then selecting the topology with the superior log likelihood value. The timetree that is shown was also generated using the RelTime method (Tamure et al., 2012). Divergence times for all branching points in the topology were calculated using the ML method based on the Tamura–Nei model (Tamure et al., 2012). Bars around each node represent 95% confidence intervals, which were computed using the method described by Tamure et al (2012). The estimated log likelihood value of the topology is shown. The tree is drawn to scale, with branch lengths measured in the relative number of substitutions per site. We selected the proposed split for the divergence between SG R. roxellana (DQ355300.1) and P. nigripes of 6.57 (6.69–6.45) million year ago (MYA, Ma) as the calibration points in two models (Perelman et al., 2011).
3. RESULTS AND ANALYSIS
3.1. Assembly and annotation on mtDNA of SNJ R. roxellana
We resequenced the mtDNA of SNJ R. roxellana and obtained 217.09 Mb of raw data and 226.95 Mb of raw data from Illumina‐Pipeline. The sequencing depth was 13,568× and 14,184.4×, which was calculated on a base of 16 Kb. After filtering, we got 192.14 Mb of clean data and 200.28 Mb of clean data with coverage of 12,008.8× and 12,517.5×, respectively. Finally, two scaffolds were de novo assembled; one was named N50 (Scaffold 1) with a length of 15,768 bp, and the other was named N90 (Scaffold 2) with the length of 848 bp. The total length was 16,616 bp, but these two scaffolds could not align into a complete genome. In order to link the gap between Scaffolds 1 and Scaffold 2, we designed primers MT(15,928–16,543) and Mt(7–535) according to R. roxellana reference mtDNA (GenBank acc. no.: JQ821835.1) to amplify the remaining sequence segment. We got 615‐bp and 528‐bp fragments by PCR amplification and Sanger sequencing. Finally, we got the complete mtDNA genome of the SNJ R. roxellana with 16,552 bp by merging sequences with overlapping alignment. The sequence was deposited in GenBank (acc. no.: KM504390.1). The results of the homologous alignment showed that the value of query coverage and identification between the mtDNA sequences of SNJ R. roxellana and either R. roxellana geographic population (GenBank acc. no: DQ355300.1, SG and JQ821835.1, QL) was 99% and 100%, respectively. These results confirmed the correctness and completeness of the SNJ R. roxellana mtDNA sequence assembly. Finally, the same genes as the R. roxellana mtDNA sequence (JQ821835.1) were annotated in the complete mtDNA genome of SNJ R. roxellana, including 13 protein‐coding genes (ATP6, ATP8, COI‐III, ND1‐6 and 4L, and Cytb), 22 transfer RNA genes (tRNAs), 2 ribosomal RNA genes (12S and 16SrRNA), and two noncoding regions.
3.2. Genetic divergence
Alignment of the consensus SNJ R. roxellana mtDNA sequence with the R. roxellana mtDNA reference sequence (GenBank acc. no.: JQ821835.1) exhibited a total of 73 variable nucleotide positions, consisting of 62 transitions (C‐to‐T = 36, 12 A‐to‐G = 26), six transversions (C‐to‐A = 3, C‐to‐G = 2, T‐to‐A = 1), and five in dels (Table 3). These results confirmed the strong transitional bias (transitions > transversions) in primate mtDNA sequences as reported in the previous study (Zhang, 1998). A total of 19 of these 68 nucleotide substitutions (27.94%) occurred within the noncoding control region of the mtDNA genome. Of the remaining 49 substitutions (72.06%), 37 (54.41%, mean = 4.53%) occurred in 12 of the 13 mitochondrial protein‐coding genes, five (7.35%, mean = 0.33%) occurred in the 22 mitochondrial tRNA genes, and seven (10.29%, mean = 5.15%) occurred in the two mitochondrial rRNA genes (12S and 16S). All the results indicate that the noncoding control region appears to be the region with the fastest evolution speed, with the protein‐encoding genes ranked second and the tRNAs exhibiting slow evolution. Next, we compared the substitutions of 13 protein‐coding genes in SNJ R. roxellana with those in the reference R. roxellana. Only one gene (ATP8) did not show substitutions, and four genes did not have nonsynonymous substitutions. Substitutions of the two protein‐encoding genes (ND1 and ND5) resulted in two amino acids replacement, and substitutions of six protein‐encoding genes led to one amino acid replacement (Table 3). The effect of natural selection during protein evolution can be expressed using the ratio of non‐synonymous mutation (dN) and synonymous mutation (dS). Low dN/dS (<1), high dN/dS (>1), and dN/dS (=1) signify purifying selection, positive selection, and neutral selection, respectively. Overall, ND1 showed positive selection, ND2, COXI, and COXII displayed neutral selection, and the others exhibited purifying selection. The base composition of SNJ R. roxellana mtDNA was different from that of SG R. roxellana and QL R. roxellana mtDNA, which occurred between A and G base pairs (Table 4). In addition, the genetic divergence between R. roxellana and R. brelichi demonstrated that SNJ R. roxellana had the highest number (766) of divergence bases and fewer gaps (11; Table 5).
Table 3.
Number of synonymous and nonsynonymous substitutions and amino acid replacement
| Gene | Synonymous | Nonsynonymous | Amino acid replacement | dN/dS |
|---|---|---|---|---|
| ND1 | 1 | 2 | T‐M/A‐T | 2 |
| ND2 | 1 | 1 | T‐A | 1 |
| COXI | 1 | 1 | I‐V | 1 |
| COXII | 1 | 1 | A‐G | 1 |
| ATP8 | 0 | 0 | — | — |
| ATP6 | 2 | 0 | — | — |
| COXIII | 2 | 1 | Y‐H | 0.5 |
| ND3 | 2 | 0 | — | — |
| ND4L | 1 | 0 | — | — |
| ND4 | 5 | 1 | T‐A | 0.2 |
| ND5 | 6 | 2 | L‐F/V‐A | 0.333333 |
| ND6 | 1 | 0 | — | — |
| CTYb | 4 | 1 | N‐S | 0.25 |
COXI–III, cytochrome c oxidase subunit 1–3; ATP6 and ATP8, two subunits of ATP synthase; ND1–6 and 4L, NADH dehydrogenase subunits 1–6 and 4L; Cytb, cytochrome b.
Table 4.
The difference in base pairs of mtDNA of Rhinopithecu roxellana mtDNA
| T(U) | C | A | G | Total | |
|---|---|---|---|---|---|
| SNJ R. roxellana | 30.4 | 25.9 | 32.1 | 11.6 | 16,552 |
| SG R. roxellana | 30.4 | 25.9 | 32 | 11.7 | 16,549 |
| Ql R. roxellana | 30.4 | 25.9 | 32 | 11.7 | 16,551 |
Table 5.
Analyses of genetic divergence of Rhinopithecu roxellana mtDNA
| Alignment sequences | Divergence base number (%) | Gap number |
|---|---|---|
| DQ355300.1–JQ821836.1 | 753 (4.55) | 12 |
| JQ821835.1–JQ821836.1 | 756 (4.57) | 12 |
| KM504390.1–JQ821836.1 | 766 (4.63) | 11 |
3.3. Phylogenetic analyses
In this study, after all the positions containing gaps and missing data were eliminated, 16,460 positions were used to construct the phylogenetic tree. Regardless of the phylogenetic method employed, the topology structure of three phylogenetic trees was highly congruent, so we only present the NJ tree here (Figure 2). All analyses indicated that the Rhinopithecus genus possessed a monophyletic origin and R. avunculus diverged first, R. bieti and R. strykeri formed sister taxa and diverged second, and R. roxellana and R. brelichi formed sister taxa and diverged last. The phylogenetic tree shows R. roxellana has a monophyletic origin and SNJ R. roxellana diverged at the latest among R. roxellana geographical populations.
Figure 2.
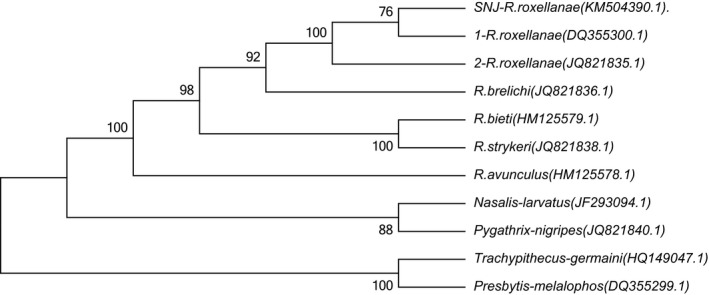
Evolutionary relationships of Rhinopithecus genus was inferred using the neighbor‐joining method. The evolutionary distances were computed using the maximum composite likelihood method and are shown as the number of base substitutions per site
3.4. Divergence age analysis
Divergence times for all branching points in the topology were calculated with the RelTime method, and all branching points in the topology were calculated using the NJ method and the ML method based on the Tamura–Nei model. Codon positions included 1st + 2nd + 3rd, using the branch lengths contained in the inferred tree. In the NJ methods, the optimal tree with the sum of branch length was 0.47216046. In the ML methods, the estimated log likelihood value of the topology was −57,223.6675. In the NJ method, the divergence time of Rhinopithecus genus was from ca. 2.23 to 2.64 Ma, R. roxellana and R. brelichi diverged in ca. 2.23 Ma, R. bieti and R. strykeri diverged in ca. 0.89 Ma, R. avunculus separated from genus Rhinopithecus in ca. 2.64 Ma, and divergence time of SNJ R. roxellana was ca. 0.14 Ma. The most recent common ancestor of R. roxellana lived ~0.16 Ma (Figure 3). In the ML method, the divergence time of genus Rhinopithecus was from ca. 1.57–2.47 Ma, R. roxellana and R. brelichi diverged in ca. 1.57 Ma, R. bieti and R. strykeri diverged in ca. 0.68 Ma, R. avunculus separated from genus Rhinopithecus in ca. 2.47 Ma, and divergence time of SNJ R. roxellana was ca. 0.08 Ma. The most recent common ancestor of R. roxellana lived approximately 0.10 Ma (Figure 4).
Figure 3.
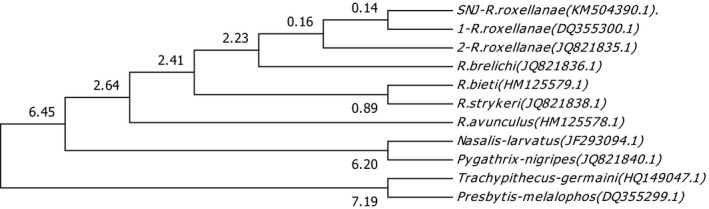
Evolutionary relationships of timetree. The evolutionary history was inferred using the neighbor‐joining method. Divergence times were showed in the branch in the topology tree
Figure 4.
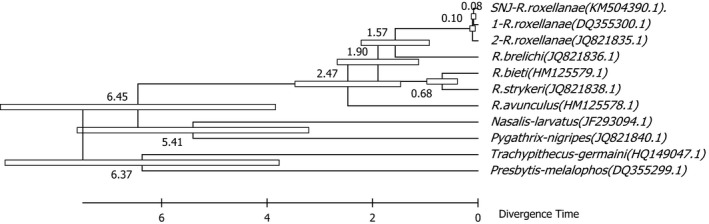
Molecular phylogenetic analysis was conducted by maximum‐likelihood method (timetree). Divergence times were showed on the branch in the topology tree
4. DISCUSSION
4.1. Evolutionary relationship of geographic groups of R. roxellana
The phylogenetic relationships of the Rhinopithecus genus have been studied (Chang, Liu, Yang, et al., 2012; Karanth et al., 2008; Li et al., 2010; Osterholz et al., 2008; Yang et al., 2012); however, the evolutionary relationships of R. roxellana are still in dispute (Pan et al., 2009). Some researchers insisted that the QL and SNJ populations originated from the SG population (Chang, Liu, Yang, et al., 2012; Li et al., 2007; Luo et al., 2012), while others believed that the SG population was a heterozygosis of the QL and SNJ populations (Chang, Luo, Liu, et al., 2012; Liedigk et al., 2012). The Shennongjia National Nature Reserve is the most eastern habitat of R. roxellana in China, and SNJ R. roxellana has the smallest population (Pan et al., 2009). Analyses of the complete mtDNA control region suggested that the QL and SNJ populations originated from the SG population, but there was no gene flow between the SNJ and SG populations (Luo et al., 2012). In contrast, the structure analyses of 16 microsatellite loci showed that the SNJ population consisted of two groups and that the SNJ population originated from the SG and QL populations (Chang, Liu, Yang, et al., 2012). Moreover, some studies showed SNJ population had the lowest diversity (Chang, Luo, Liu, et al., 2012; Chang, Liu, Yang, et al., 2012; Luo et al., 2012), another study showed SNJ population had rich genetic diversity (He et al., 2010). Therefore, the origin of R. roxellana is controversial in previous studies because of different genetic markers. In this study, we used three complete mitochondrial genome sequences, instead of the complete mtDNA control region (D‐loop), to assess the origin of R. roxellana by analyzing the genetic divergence,divergence ages and constructing the phylogenetic tree. The result showed SNJ R. roxellana was well clustered together with other two R. roxellana in the phylogenetic tree, and SNJ R. roxellana diverged at the latest among R. roxellana geographical populations. Although SNJ R. roxellana had the richest genetic divergence in gene composition, and SNJ R. roxellana was well clustered together with other two R. roxellana in the phylogenetic tree, the bootstrap value (76) of phylogenetic tree between SNJ R. roxellana and other two R. roxellana was low. These results indicated SNJ R. roxellana was a newest rapid evolutionary geographic population of R. roxellana, which was similar with recently report (Zhou et al., 2016). The result, to some extent, could explain why the early results were controversial.
4.2. The divergence ages of geographic groups of R. roxellana
The divergence ages of endangered species motivate us to determine their protected status and inform protection policy. The early study on the base of autosomal genome clusters showed the divergence ages between the northern species (R. roxellana and R. brelichi) and the “Himalayan” species (R. bieti and R. strykeri) was 1.60 Ma, and the divergence ages between R. roxellana and R. brelichi was 1.69 Ma (Zhou et al., 2014). But the result on the base of mitochondrial haplotype clusters showed the divergence ages of geographic of R. roxellana were 1.17 ± 0.70 and 0.53 ± 0.30 Ma (Chang, Luo, Liu, et al., 2012). In the study, although the evolutionary tendency of the Rhinopithecus genus and R. roxellana was consistent in the NJ and ML methods, the variation in divergence times was greater in ML than in NJ, and the divergence times occurred later in ML than in NJ. In the NJ method, the divergence ages between the northern species and the “Himalayan” species were 2.41 Ma, and the divergence ages between R. roxellana and R. brelichi were 2.23 Ma, and the divergence time of the R. roxellana was from ca. 0.14–1.16 Ma, and that of SNJ R. roxellana was ca. 0.14 Ma. In the ML method, the divergence ages between the northern species and the “Himalayan” species were 1.9 Ma, and the divergence ages between R. roxellana and R. brelichi were 1.57 Ma, and the divergence time of the R. roxellana was from ca. 0.08–0.1 Ma, and that of SNJ R. roxellana was ca. 0.08 Ma. Because the divergence ages between R. roxellana and R. brelichi were 1.57 Ma in ML methods, which was coincide with the uplift of the Tibetan plateau—Yuanmu movement in approximately 1.6 million years ago; and the divergence time of the R. roxellana was from ca. 0.08–0.1 Ma in ML methods, which agreed with the penultimate glaciation (0.13–0.3 million years ago; Zhou et al., 2016). So we thought the divergence ages of R. roxellana acquired from ML methods were closer to reality.
4.3. Functional genes and environmental adaptability of mtDNA
Mitochondria play important roles in generating energy by oxidative phosphorylation and taking part in energy metabolism. The oxidative phosphorylation system consists of 13 essential proteins encoded by corresponding genes, including ATP 6, ATP 8, COI‐III, ND 1‐6 and 4L, and Cytb. These 13 genes are particularly used as effective genetic markers to investigate the molecular basis of organismal adaptation to environments (Fonseca, Johnson, O'Brien, Ramos, & Antunes, 2008; Zheng, Xu, & Shen, 2002). Previous studies showed that cytochrome c oxidase genes of Caprini antelope, camelids and Tibetan antelope (Di, Zambelli, & Rioja, 2009; Hassanin, Ropiquet, Couloux, & Cruaud, 2009; Ning, Xiao, Li, Hua, & Zhang, 2010), NADH dehydrogenase genes of Tibetan horses (Xu et al., 2005), cytochrome b geneand ATP synthase of alpacas (Xu et al., 2007) went through adaptive evolution. The ND 2 and ND 6 of R. roxellana presented significantly positive adaptation to high altitude and cold weather stress. In the study, 1 gene (ND 1) showed positive selection, three genes (ND 2, COXI, and COXII) displayed neutral selection, and the others exhibited purifying selection in the complete mitochondrial genome sequence of SNJ R. roxellana. These results obviously indicated more purifying selections on mitochondrial proteins in SNJ R. roxellana, consistent with strong purifying selections on mitochondrial proteins in primates (Felsenstein, 1985). Purifying selections was also known as negative selections, that is, more unfavorable environmental factors acted on SNJ R roxellana. The more unfavorable environmental factors resulted in more abundant genetic divergence and less similarity.
5. CONCLUSIONS
We obtained the complete mitochondrial genome sequence of SNJ R. roxellana with a total length of 16,552 bp by NGS and Sanger sequencing. The analyses on genetic composition, phylogenetic tree, and divergence time showed that SNJ R. roxellana mtDNA had the most genetic divergence, the least structure, and the more recent divergence times of R. roxellana, which indicated SNJ R. roxellana was a newest rapid evolutionary geographic population of R. roxellana and should be protected as a unit. Our findings are more comprehensive and promote the development of a more accurate protection policy for SNJ R. roxellana.
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGMENTS
This work was supported by the Hubei Provincial Key Laboratory of Conservation Biology for Shennongjia Golden Monkey (grant no. 2013SNJB01), the project from the Education Department of Hunan Province “the Reform of Theory Teaching and Teaching Methods of Bioinformatics” (grant no. 2013‐160) and the Hunan Provincial Collaborative Innovation Center for Control of Key Agricultural Pest (grant no. 2015‐2).
Hong Y, Duo H, Hong J, et al. Resequencing and comparison of whole mitochondrial genome to gain insight into the evolutionary status of the Shennongjia golden snub‐nosed monkey (SNJ R. roxellana). Ecol Evol. 2017;7:4456–4464. https://doi.org/10.1002/ece3.3011
REFERENCES
- Chang, Z. F. , Liu, Z. J. , Yang, J. Y. , Li, M. , & Vigilant, L. (2012). Noninvasive genetic assessment of the population trend and sex ratio of the Shennongjia population of Sichuan snub‐nosed monkeys (Rhinopithecus roxellana). Chinese Science Bulletin, (10), 1135–1141. [Google Scholar]
- Chang, Z. F. , Luo, M. F. , Liu, Z. J. , Yang, J. Y. , Xiang, Z. F. , Li, M. , Vigilant, L. (2012). Human influence on the population decline and loss of genetic diversity in a small and isolated population of Sichuan snub‐nosed monkeys (Rhinopithecus roxellana). Genetica, 140, 105–114. [DOI] [PubMed] [Google Scholar]
- Di, R. F. , Zambelli, A. D. , & Rioja, L. B. V. (2009). Identification of camelid specific residues in mitochondrial ATP synthase subunits. Journal of Bioenergetics and Biomembranes, 41, 223–228. [DOI] [PubMed] [Google Scholar]
- Driscoll, C. A. , Yamaguchi, N. , & Bar, G. K. (2009). Mitochondrial phylogeography illuminates the origin of the extinct caspian tiger and its relationship to the Amur Tiger. PLoS One, 14(4), e4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, A. J. , Suchard, M. A. , Xie, D. , & Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, 29, 1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, C. J. , Magee, D. A. , Park, S. D. E. , Paul, A. M. , Amanda, J. L. , Alison, M. , et al. (2010). A complete mitochondrial genome sequence from a mesolithic wild aurochs (Bos primigenius). PLoS One, 17(5), e9255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekblom, R. , & Galindo, J. (2011). Applications of next generation sequencing in molecular ecology of non‐model organisms. Heredity, 107, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J. (1985). Confidence limits on phylogenies: An approach using the bootstrap. Evolution, 39, 783–791. [DOI] [PubMed] [Google Scholar]
- Finstermeier, K. , Zinner, D. , Brameier, M. , Meyer, M. , Kreuz, E. , & Hofreiter, M. (2013). A mitogenomic phylogeny of living primates. PLoS One, 16(8), e69504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca, D. A. , Johnson, R. R. , O'Brien, W. E. , Ramos, M. J. , & Antunes, A. (2008). The adaptive evolution of the mammalian mitochondrial genome. BMC Genomics, 9, 119–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann, T. , Lwin, N. , Aung, S. S. , Aung, T. N. , Aung, Z. M. , & Aung, T. H. (2011). A new species of snub‐nosed monkey, genus Rhinopithecus Milne‐Edwards, 1872 (Primates, Colobinae), from northern Kachin State, northeastern Myanmar. American Journal of Primatology, 73, 96–107. [DOI] [PubMed] [Google Scholar]
- Green, R. E. , Malaspinas, A. S. , & Krause, J. (2008). A complete Neandertal mitochondrial genome sequence determined by high‐throughput sequencing. Cell, 134, 416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa, M. , Cao, Y. , & Yang, Z. (1998). Preponderance of slightly deleterious polymorphism in mitochondrial DNA: nonsynonymous/synonymous rate ratio is much higher within species than between species. Molecular Biology and Evolution, 15, 1499–1505. [DOI] [PubMed] [Google Scholar]
- Hassanin, A. , Ropiquet, A. , Couloux, A. , & Cruaud, C. (2009). Evolution of the mitochondrial genome in mammals living at high altitude: new insights from a study of the tribe Caprini (Bovidae, Antilopinae). Journal of Molecular Evolution, 68, 293–310. [DOI] [PubMed] [Google Scholar]
- He, L. , Zhang, Y. G. , Peng, H. , Li, D. Q. , & Li, D. Q. (2010). Genetic diversity of Rhinopithecus roxellana in shennongjia national nature reserve as estimated by non‐invasive DNA technology. Acta Ecologica Sinica, 30(16), 4340–4350. [Google Scholar]
- Karanth, K. P. , Singh, L. , Collura, R. V. , & Stewart, C. B. (2008). Molecular phylogeny and biogeography of langurs and leaf monkeys of South Asia (Primates: Colobinae). Molecular Phylogenetics and Evolution, 46, 683–694. [DOI] [PubMed] [Google Scholar]
- Kimura, M. (1980). A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution, 16, 111–120. [DOI] [PubMed] [Google Scholar]
- Kolleck, J. , Yang, M. , Zinner, D. , & Roos, C. (2013). Genetic diversity in endangered Guizhou snub‐nosed monkeys (Rhinopithecus brelichi): Contrasting results from microsatellite and mitochondrial DNA data. PLoS One, (8):e73647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause, J. , Fu, Q. , Good, J. M. , Bence Viola, B. , Shunkov, M. V. , Derevianko, A. P. , et al. (2010). The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature, 464, 894–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, K. Q. (2015). Tonkin snub‐nosed monkey Rhinopithecus avunculus (Dollman 1912) In Schwitzer Khau Ca, et al. (Eds.), Primates in Peril: The World's 25 Most Endangered Primates 2014–2016 (pp. 81–83). [Google Scholar]
- Le, K. Q. , & Covert, H. H. (2010). Another population of the Tonkin snub‐nosed monkey (Rhinopithecus avunculus) discovered in Ha Giang Province, Vietnam. Vietnamese Journal of Primatology, 4, 19–25. [Google Scholar]
- Li, R. Q. , Fan, W. , Tian, G. , Zhu, H. M. , He, L. , Cai, J. , et al. (2010). The sequence and de novo assembly of the giant panda genome. Nature, 463, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M. , Liu, Z. J. , Gou, J. X. , Ren, B. P. , Pan, R. L. , Su, Y. J. , et al. (2007). Phylogeography and population structure of the golden monkeys (Rhinopithecus roxellana): inferred from mitochondrial DNA sequences. American Journal of Primatology, 69, 1195–1209. [DOI] [PubMed] [Google Scholar]
- Liedigk, R. , Yang, M. Y. , Jablonsk, N. G. , Momberg, F. , Geissmann, T. , & Lwin, N. (2012). Evolutionary history of the odd‐nosed monkeys and the phylogenetic position of the newly described myanmar snub ‐nosed monkey Rhinopithecus strykeri . PLoS One, 16(7), e37418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist, C. , Schuster, S. C. , Sun, Y. , Talbot, S. L. , Qi, J. , Ratan, A. , et al. (2010). Complete mitochondrial genome of a Pleistocene jawbone unveils the origin of polar bear. Proceedings of the National Academy of Sciences of the United States of America, 107, 5053–5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, M. F. , Liu, Z. J. , Pan, H. J. , Zhao, L. , & Li, M. (2012). Historical geographic dispersal of the golden snub‐nosed monkey (Rhinopithecus roxellana) and the influence of climatic oscillations. American Journal of Primatology, 74, 91–101. [DOI] [PubMed] [Google Scholar]
- Miller, W. , Drautz, D. I. , Janecka, J. E. , Lesk, A. M. , Ratan, A. , Tomsho, L. P. , et al. (2009). The mitochondrial genome sequence of the Tasmanian tiger (Thylacinus cynocephalus). Genome Research, 19, 213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, W. , Schuster, S. C. , Welch, A. J. , Ratan, A. , Bedoya‐Reina, O. C. , Zhao, F. Q. , et al. (2012). Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proceedings of the National Academy of Sciences of the United States of America, 109, 2382–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin, P. A. , Archer, F. I. , Foote, A. D. , Vilstrup, J. , Allen, E. E. , Wade, P. , et al. (2010). Complete mitochondrial genome phylogeographic analysis of killer whales (Orcinus orca) indicates multiple species. Genome Research, 20, 908–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning, T. , Xiao, H. , Li, J. , Hua, S. , & Zhang, Y. P. (2010). Adaptive evolution of the mitochondrial NADH6 gene in the domestic horse. Genetics and Molecular Research, 9, 144–150. [DOI] [PubMed] [Google Scholar]
- Osterholz, M. , Walter, L. , & Roos, C. (2008). Phylogenetic position of the langur genera Semnopithecus and Trachypithecus among Asian colobines, and genus affiliations of their species groups. BMC Evolutionary Biology, 8, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, D. , Hu, H. X. , Men, S. J. , Men, Z. M. , Fu, Y. X. , & Zhang, Y. P. (2009). Population analysis of golden monkey using mitochondrial control region: high level of polymorphism and its implications. International Journal of Primatology, 30, 337–351. [Google Scholar]
- Perelman, P. , Johnson, W. E. , Roos, C. , Seuanez, H. N. , Horvath, J. E. , Moreira, M. A. M. , et al. (2011). A molecular phylogeny of living primates. PLoS Genetics, 7, e1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi, L. , Hodgson, J. A. , Burrell, A. S. , Sterner, K. N. , Raaum, R. L. , & Disotell, T. R. (2014). Primate phylogenetic relationships and divergence dates inferred from complete mitochondrial genomes. Molecular Phylogenetics and Evolution, 75, 165–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou, N. , & Nei, M. (1987). The neighbor‐joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4, 406–425. [DOI] [PubMed] [Google Scholar]
- Sterner, K. N. , Raaum, R. L. , Zhang, Y. P. , Stewart, C. B. , & Disotell, T. R. (2006). Mitochondrial data support an odd‐nosed colobine clade. Molecular Phylogenetics and Evolution, 40, 1–7. [DOI] [PubMed] [Google Scholar]
- Tamura, K. , Battistuzzi, F. U. , Billing‐Ross, P. , Murillo, O. , Filipski, A. , & Kumar, S. (2012). Estimating divergence times in large molecular phylogenies. Proceedings of the National Academy of Sciences, 109, 19333–19338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Nei, M. , & Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor‐joining method. Proceedings of the National Academy of Sciences of the United States of America, 101, 11030–11035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. , & Kumar, S. S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 2013(30), 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. X. , Jiang, X. L. , & Li, D. W. (1998). Classification and distribution of the extant subspecies of golden snub‐nosed monkey (Rhinopithecus roxellana) In Jablonski N. G. (Ed.), The natural history of the doucs and snub‐nosed monkeys (pp. 53–64). Singapore, Singapore: World Scientific Publishing. [Google Scholar]
- Xu, S. Q. , Luosang, J. B. , Hua, S. , He, J. , Asan, C. R. , Wang, W. , et al. (2007). High altitude adaptation and phylogenetic analysis of Tibetan horse based on the mitochondrial genome. Journal of Genetics and Genomics, 34, 720–729. [DOI] [PubMed] [Google Scholar]
- Xu, S. Q. , Yang, Y. Z. , Zhou, J. , Jing, J. E. , Chen, Y. T. , Wang, J. , et al. (2005). A mitochondrial genome sequence of the Tibetan antelope (Pantholops hodgsonii). Genomics, Proteomics & Bioinformatics, 3, 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, M. Y. , Yang, Y. Q. , Cui, D. Y. , Fickenscher, G. , Zinner, D. , Roos, C. , et al. (2012). Population genetic structure of Guizhou snub‐nosed monkeys (Rhinopithecus brelichi) as inferred from mitochondrial control region sequences, and comparison with R. roxellana and R. bieti . American Journal of Physical Anthropology, 147, 1–10. [DOI] [PubMed] [Google Scholar]
- Yann, S. G. , & Juan, I. M. B. (2010). Optimization of de novo transcriptome assembly from next‐generation sequencing data. Genome Research, 20, 1432–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, L. , Wang, X. P. , Ting, N. , & Zhang, Y. P. (2011). Mitogenomic analysis of Chinese snub‐nosed monkeys: Evidence of positive selection in NADH dehydrogenase genes in high‐altitude adaptation. Mitochondrion, 11, 497–503. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. P. (1998). Mitochondrial cytochrome b gene sequences of old world monkeys: With special reference on evolution of Asian colobines. Primates, 39, 39–49. [Google Scholar]
- Zheng, B. , Xu, Q. , & Shen, Y. (2002). The relationship between climate change and Quaternary glacial cycles on the Qinghai‐Tibetan Plateau: review and speculation. Quaternary International, 97–98, 93–101. [Google Scholar]
- Zhou, X. , Meng, X. , Liu, Z. , Chang, J. , Wang, B. , Li, M. , et al. (2016). Population genomics reveals low genetic diversity and adaptation to hypoxia in Snub‐Nosed Monkeys. Molecular Biology and Evolution, 33(10), 2670 https://doi.org/10.1093/molbev/msw150 [DOI] [PubMed] [Google Scholar]
- Zhou, X. M. , Wang, B. S. , Pan, Q. , Zhang, J. B. , Kumar, S. , Sun, X. Q. , et al. (2014). Whole‐genome sequencing of the snub‐nosed monkey provides insights into folivory and evolutionary history. Nature Genetics, 46, 1303–1312. [DOI] [PubMed] [Google Scholar]