

Abstract
Histidine is a unique amino acid with an imidazole side chain in which both of the nitrogen atoms are capable of serving as a proton donor and proton acceptor in hydrogen bonding interactions. In order to probe the functional role of histidine involved in hydrogen bonding networks, fine-tuning the hydrogen bonding potential of the imidazole side chain is required but not feasible through traditional mutagenesis methods. Here, we show that two close mimetics of histidine, 3-methyl-histidine and thiazole alanine can be genetically encoded using an engineered pyrrolysine incorporation machinery. Replacement of the three histidine residues predicted to be involved in an extended charge-relay system in alanine racemase with 3-methyl-histidine or thiazole alanine shows dramatic loss in the enzyme's catalytic efficiency, implying the role of this extended charge-relay system in activating the active site residue Y265, a general acid/base catalyst in the enzyme.
Keywords: noncanonical amino acid, histidine analogs, thiazole alanine, 3-methyl histidine, amber suppression
Among the 20 canonical amino acids, histidine is unique in many ways. It is the only amino acid with an aromatic but hydrophilic side chain. As illustrated in Figure 1, the imidazole side chain can attain two possible neutral forms, in which either of the two nitrogen atoms can serve as a hydrogen bond donor or a hydrogen bond acceptor. When protonated, the positive charge can oscillate between the two nitrogen atoms for initiating favorable charge-charge/dipole interactions in proteins. Due to this unique feature histidine has been found in many catalytic charge relay systems in enzymes, coordinating charge or proton transfer during catalysis, most commonly observed in catalytic triads and catalytic diads in proteases and other hydrolytic enzymes.(1, 2) To probe the role of histidine in enzymatic reactions, it is usually mutated to another residue such as alanine, asparagine or phenylalanine. Although useful for the investigation of histidine functions, none of these amino acids can accurately imitate the unique characteristics of histidine. Genetic incorporation of structurally similar histidine analogs can prove to be a very effective tool in studying biological roles of histidine residues in enzymes by fine tuning hydrogen bonding potentials of the histidine side chain. As demonstrated by Hsieh et al. (1979), to ascertain the role of a histidine residue in a hydrogen-bonding network in an enzyme, substitution of this residue by histidine derivatives so as to deprive the availability of proton on pyrrole (-NH) nitrogen or to block the proton acceptor site of pyridine (-N) nitrogen of histidine residue can greatly help in determining its function as a proton donor/acceptor or a general acid-base in the enzyme.(3) Besides genetic incorporation of histidine mimetics, techniques such as native chemical ligation (NCL)(4-6) or the use of auxotrophic bacterial strains(7, 8) can also be utilized for studying a system involving histidine mimetics. However limitations such as the modifications possible(9) and the length of the peptide(10) that can be synthesized via NCL, toxicity of noncanonical amino acid (NCAA) being incorporated in cellular system and undesirable misincorporation via the use of auxotrophic strains(11) render these techniques inadequate for studying complex enzymatic systems, consequently directing the use of genetic incorporation of NCAAs as the preferred technique.
Figure 1.
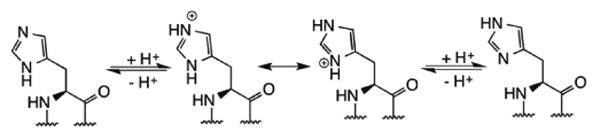
Neutral and charged states of histidine in proteins.
In order to create a mutagenic system that genetically incorporates NCAAs that closely mimic histidine for fine tuning of histidine-involved hydrogen bonding interactions, we referred to the engineering of the pyrrolysine incorporation machinery.(12) We set our sights on the incorporation of histidine analogs such as 3-methyl-histidine (Mh) and thiazole alanine (Ta), as both are close histidine mimetics. We have previously shown that a rationally designed pyrrolysyl-tRNA synthetase variant with the mutations N346A/C348A (PylRS(N346A/C348A)) can recognize a large number of phenylalanine derivatives and, together with its cognate , mediate their incorporation into proteins at amber (TAG) codons in Escherichia coli and mammalian cells.(13),(14) Given the similarity between some phenylalanine derivatives and histidine analogs we predicted that this same enzyme can also recognize these NCAAs and direct their incorporation into proteins at amber codons. To test this proposition, E. coli BL21 cells coding PylRS(N346A/C348A), , and superfolder green fluorescent protein (sfGFP) with an amber mutation at the S2 position were grown in minimal media supplemented with 5 mM Mh or Ta. For both NCAAs, their presence in the growth media promoted the suppression of the amber mutation in sfGFP and the subsequent full-length protein expression. Without providing a NCAA, no full-length sfGFP was expressed (Figure 2A). The electrospray ionization mass spectrometry (ESI-MS) analysis of the purified proteins detected molecular weights of 27,732 Da for the Mh-incorporated protein and 27,735 Da for the Ta-incorporated protein that agree well with the theoretical molecular weights (27,733 and 27,736 Da, respectively), confirming the selective incorporation of these two NCAAs and the high fidelity of the incorporation system (Figure 2B). Mh has also been genetically encoded using an alternative system.(15)
Figure 2.
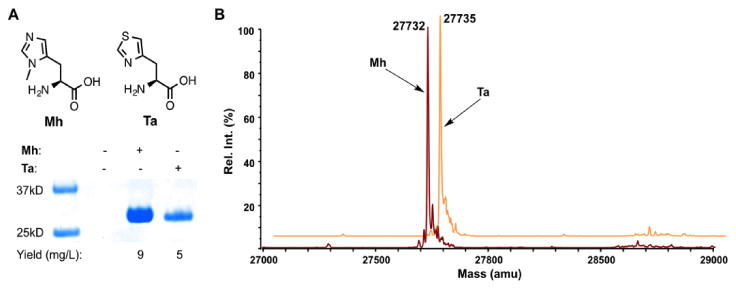
(A) The selective incorporation of Mh and Ta into sfGFP at its S2 position. (B) ESI-MS analysis of the two purified proteins incorporated with Mh and Ta.
We envisioned that the incorporation of both Mh and Ta can be applied to probe fundamental roles of histidine in charge-relay systems of enzymes. Instead of probing well studied catalytic triads and diads, we chose to investigate histidine functions in a potentially extended charge-relay system in alanine racemase (EC 5.1.1.1). Alanine racemase has become a promising drug target since the discovery of its key role in the formation of peptidoglycan in bacterial cell walls.(16-20) Mechanistic illustration of this enzyme may facilitate the identification of inhibitors(21-27) that can serve as novel antibiotics. This Pyridoxal 5′-Phosphate (PLP)-dependent enzyme catalyzes the interconversion of L-alanine to D-alanine.(28-30) Although it is one of the most well studied enzymes, information on several aspects of its mechanism remains elusive. It is known that alanine racemase employs a two-base catalytic mechanism in which K39 and Y265 residues act as general acid/base catalysts(31) (Figure 3A). In the racemization reaction from D-alanine to L-alanine, D-alanine first reacts with PLP to form an external aldimine. K39 then abstracts the α proton from this external aldimine to generate a quinonoid intermediate that receives a proton from Y265 to form a second external aldimine. L-alanine is then released from this external aldimine as the final product. The whole catalytic process is fully reversible. pH profile studies have indicated that the phenolic hydroxide of Y265 has a pKa value around 7.1-7.4, contributing to a very high catalytic rate of wild-type alanine racemase with a kcat/KM of 1.6 (0.2)×106 M-1s-1.(32) We predicted the existence of an extended charge-relay system and hydrogen bonding network involving residues E161, H127, H200, R219, H166, and Y265 in alanine racemase that are potentially involved in bringing down the pKa of the Y265 side chain hence affecting the catalytic activity of the enzyme (Figure 3B). E161 is solvent exposed as determined by the active site structure of alanine racemase from Bacillus stearothermophilus (PDB: 1SFT).(33) There are three histidine residues involved in this proposed charge-relay system. It has been shown that H166 coordinates the activation of Y265 by R219.(32) Although H127 and H200 are distal from Y265, they potentially serve critical roles in the activation of Y265 through this charge-relay system with the extended hydrogen bonding network. To test this proposition and directly observe how hydrogen bonding interactions contribute to the final activation of Y265, we decided to substitute these two residues in addition to H166 with either Mh or Ta using our established system for the incorporation of these two NCAAs. In total, six alanine racemase mutants containing these NCAAs were expressed and purified to homogeneity. Circular dichroism analysis of purified proteins indicates that they all fold like the wild-type enzyme (Figure 4). This is an expected observation given that both Mh and Ta are highly analogous to histidine with Ta being almost spatially identical to histidine. Two mutants H166Ta and H166Mh were also characterized by the mass spectrometry analysis to confirm the incorporation of Ta and Mh (Supplementary Figures S2 and S3). Catalytic activity of these mutant enzymes along with wild-type alanine racemase enzyme was characterized according to the methods of Sun et al.(32) and the determined values are presented in Table 1.
Figure 3.
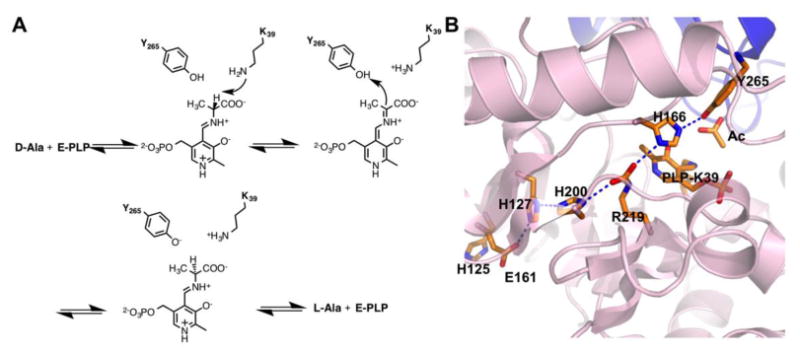
(A) The catalytic mechanism of alanine racemase. (B) The potentially extended charge-relay system for the activation of Y265 in the alanine racemase active site.
Figure 4.
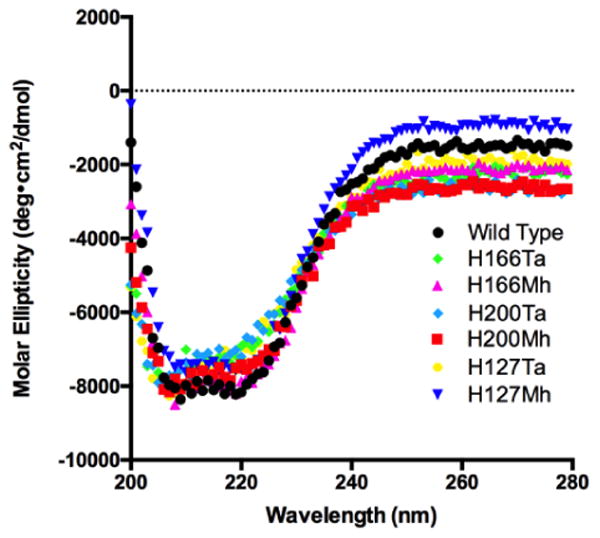
Circular dichroism spectra of wild-type alanine racemase and its six mutants.
Table 1. Catalytic parameters of wild-type alanine racemase and its mutants.
| Enzyme | kcat (s-1) | Rel. kcat | KM (mM) | kcat/KM (M-1s-1) | Rel. kcat/KM |
|---|---|---|---|---|---|
| wild type | 3.5 (0.1) × 103 | 1 | 2.2 | 1.6 (0.2) × 106 | 1 |
| H166Ta | 1.7 (0.1) × 101 | 0.005 | 0.5 | 3.3 (0.2) × 104 | 0.02 |
| H166Mh | 8.4 (0.3) × 100 | 0.002 | 0.5 | 1.8 (0.2) × 104 | 0.01 |
| H200Ta | 1.2 (0.5) × 101 | 0.003 | 1.6 | 7.3(0.8) × 103 | 0.005 |
| H200Mh | 5.2 (0.2) × 100 | 0.002 | 1.6 | 3.2 (0.4) × 103 | 0.002 |
| H127Ta | 1.2 (0.4) × 101 | 0.003 | 1.2 | 1.0 (0.1) × 104 | 0.006 |
| H127Mh | 2.2 (0.1) × 101 | 0.006 | 1.8 | 1.2 (0.2) × 104 | 0.007 |
| H125Ta | 4.1 (0.2) × 103 | 0.8 | 4.7 | 8.7 (0.4) × 105 | 0.5 |
| H125A | 4.4 (0.2) × 102 | 0.08 | 0.5 | 8.2 (0.4) × 105 | 0.5 |
| H166A | 3.5 (0.2) × 100 | 0.001 | 1.6 | 2.2 (0.3) × 103 | 0.001 |
| H166F | 3.9 (0.1) × 100 | 0.001 | 3.9 | 1.0 (0.1) × 103 | 0.0006 |
| H127A/H166A/H200A | 2.8 (0.4) × 10-1 | 0.00008 | 0.8 | 3.6(0.5) × 102 | 0.0002 |
Substituting any of the three histidines predicted to be involved in the charge-relay system with either Mh or Ta has a dramatic effect on the enzyme's catalytic activity. All of the generated mutants displayed diminished activities with more than 100-fold decrease in kcat in comparison to their wild-type counterpart. Given the established role of H166 in the direct activation of Y265, low activities of mutants H166Mh and H166Ta are expected. However, H127 and H200 are distal from Y265 with no direct interaction possible between the tyrosine and histidine residues. Both of these residues are more than 10Å away from the catalytic site and more than 15Å away from the phenolic hydroxide group on the side chain of Y265. Therefore, such a drastic decrease in activity observed for H127 and H200 mutants strongly suggests that the side chains on these histidine residues are involved in activation of Y265 residue. Although the methyl group of Mh potentially disrupts the orientation of vicinal amino acid side chains of H127 and H200 when these two sites are substituted with Mh, Ta is spatially similar to histidine and therefore its substitution at H127 and H200 is expected to have minimal impact on the local protein structure around H127 and H200 residues. Unlike histidine whose two side chain nitrogen atoms can both serve as donor and acceptor roles in hydrogen bonding interactions, the low pKa (∼2) of the thiazole side chain of Ta dictates that both nitrogen and sulfur in the Ta side chain can only serve as hydrogen bond acceptors. This implies that the proton transfer chain as suggested by us, where the phenolic group of Y265 accepts a proton from the side chain of H166 which in turn accepts a proton from R219 and then extending this charge relay system, the side chain of H200 donates a proton to R219 while accepting a proton from the side chain of H127, would be interrupted on changing the proton donating ability of all the histidine residues involved. This is illustrated by the diminished activity of enzyme observed by replacing H127, H200, and H166 with Ta, suggesting that the hydrogen bonding network in the predicted extended charge-relay system is disrupted and subsequently prohibits the direct activation of Y265 by this charge-relay system. All the mutant enzymes retain similar basal level activities possibly due to the access of Y265 to water molecule which acts as a substitute to the charge-relay system in activating the Y265 residue.
On the contrary to their dramatic decreased kcat values in comparison to the wild-type enzyme, H200Ta, H200Mh, H127Ta, and H127Mh have Km values almost identical to that of the wild-type enzyme. This is expected since both H200 and H127 are distal to the active site and their replacement with Ta and Mh minimally affects the active site structure. However, both H166Ta and H166Mh have Km values 4-fold lower than that of the wild-type enzyme. In comparison to histidine, both Ta and Mh are more hydrophobic. Their replacement of H166 may create a more hydrophobic active site environment that favors the substrate reaction with the bound PLP. For comparison purpose, we also expressed and determined catalytic activities of H166A and H166F mutants. Although both mutants show kcat values not much lower than H166Ta and H166Mh, they have much higher Km values, leading to overall kcat/Km values more than 10 fold lower than those of H166Ta and H166Mh. This observation may be because Ta and Mh mimic histidine better than ala and phe and therefore minimally disrupt the substrate binding when replacing H166. When H166, H200, and H127 are all replaced with Ala, the afforded mutant has a very low activity as expected. We also mutated H125, a histidine not part of the potential charge relay system, to Ta and determined the activity of the afforded H125Ta mutant to serve as a control. As expected we observe no significant change in kcat by introducing this mutation, the result indicates that H125Ta is solvent exposed and does not play any significant role in the activity of the enzyme as this surfaced exposed residue is far away from the active site of the enzyme. A similar mutation at H125 to alanine also caused insignificant change to the catalytic activity of the enzyme. CD spectra of the wild-type enzyme and two H125 mutants are very similar, indicating no significant structural differences between them (Figure S4).
In summary, we have shown that the two close mimetics of histidine, Mh and Ta, can be genetically incorporated into proteins at amber codons using an engineered pyrrolysine incorporation machinery. Given their spatial resemblance and contrasting hydrogen bonding characteristics, replacing a histidine in a protein with Mh or Ta is expected to introduce minimal impact on the protein secondary structure, allowing for the fine tuning of the hydrogen bonding interactions critical in their enzymatic activity. By tweaking the hydrogen bonding interactions in a potentially extended charge-relay system by replacement of histidine residues involved in this system with Mh and Ta, we have demonstrated the possibility that all the histidine residues in this charge-relay system, H166, H200 and H127, coordinate the activation of the Y265 residue at the active site for catalysis, therefore suggesting the existence of this extended charge-relay system. The reported method can be easily adapted to study functional roles of histidine in many proteins and enzymes and is expected to have profound impact in the biochemistry research area. One drawback of the reported method is the disruption of the hydrogen bonding system. An alternative solution is to the genetically incorporate fluorohistidines that allows fine tune histidine pKa while maintaining all the hydrogen bonding potential. The engineering of the pyrrolysine incorporation system for the genetic incorporation of fluorohistidines is currently underway.
Methods
E. coli Top10 were co-transformed with a pEVOL-pylT-PylRS(N346A/C348A) plasmid containing and two copies of PylRS N346A/C348A mutant (previously reported)(34) and a pBAD plasmid containing the gene encoding alanine racemase (AlaR). QuickChange® mutagenesis was performed to obtain three different alanine racemase mutants with the following mutations: H166TAG, H200TAG and H127TAG (See Supporting Info). The transformed cells were grown overnight at 37°C, 250 rpm, in auto-induction media(35) and supplemented with 2 mM of the NCAA to obtain six corresponding mutant enzymes (Yield and mass spectra data provided in Supporting Info).
To study the activity of the alanine racemase enzyme and its derivatives, the enzyme kinetic activity assay was employed. The assay consisted of coupled reactions between conversion of D-alanine to L-alanine and subsequent reaction of L-alanine with L-alanine dehydrogenase to form NADH which is detectable by its absorbance at 340 nm. All the reactions were carried out at room temperature in a buffer containing 50 μM PLP (Pyridoxal Phosphate), 0.1 M KCl, 0.1 M CHES (N-Cyclohexyl-2-aminoethanesulfonic acid) Buffer (pH 9.0), 10 mM NAD+, and 0.1 – 10 mM D-alanine and 2 units/ml of L-alanine dehydrogenase.(32) The extinction coefficient of NADH used for calculations was 6220 M-1 cm-1. The enzyme concentrations were 0.11 nM for WT, 0.09 μM for H166Ta, 0.16 μM for H166Mh, 0.17 μM for H200Ta, 0.12 μM for H200Mh, 0.17 μM for H127Ta and 0.37 μM for H127Mh. The calculated kinetic parameters kcat and kcat/KM values are shown in Table 1.
Supplementary Material
Acknowledgments
We thank M. Toney at University of California-Davis for providing us with the alanine racemase DNA, Y. Rezenom from the Laboratory for Biological Mass Spectrometry at Texas A&M University for characterizing our proteins with electrospray ionization mass spectrometry, Z. Ren for MS/MS analysis and C. Hilty and T. Begley for the access to the circular dichroism instrument in their group.
Funding Sources: This work was supported partially by the National Institute of Health (Grants R01CA161158 and R01GM121584) and the Welch Foundation (Grant A-1715).
Footnotes
Notes: The authors declare no competing financial interest.
Supporting Information. Amino acid sequence, plasmid construction, protein expression and purification, kinetic assay and circular dichroism details. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Rebek J. On the structure of histidine and its role in enzyme active sites. Struct Chem. 1990;1:129–131. [Google Scholar]
- 2.Schneider F. Histidine in enzyme active centers. Angew Chem Int Ed Engl. 1978;17:583–592. doi: 10.1002/anie.197805831. [DOI] [PubMed] [Google Scholar]
- 3.Hsieh K, Jorgensen EC. Angiotensin II Analogues. 14. Roles of the Imidazole Nitrogens of Position-6 Histidine in Pressor Activity. J Med Chem. 1979;22:1199–1206. doi: 10.1021/jm00196a010. [DOI] [PubMed] [Google Scholar]
- 4.Hackeng TM, Griffin JH, Dawson PE. Protein synthesis by native chemical ligation: Expanded scope by using straightforward methodology. Proc Natl Acad Sci. 1999;96:10068–10073. doi: 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang WH, Otting G, Jackson CJ. Protein engineering with unnatural amino acids. Curr Opin Struc Biol. 2013;23:581–587. doi: 10.1016/j.sbi.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 6.Dawson P, Muir T, Clark-Lewis I, Kent S. Synthesis of proteins by native chemical ligation. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 7.Ikeda Y, Kawahara S, Taki M, Kuno A, Hasegawa T, Taira K. Synthesis of a novel histidine analogue and its efficient incorporation into a protein in vivo. Protein Eng. 2003;16:699–706. doi: 10.1093/protein/gzg084. [DOI] [PubMed] [Google Scholar]
- 8.Klein DC, Weller JL, Kirk KL, Hartley RW. Incorporation of 2-Fluoro-L-Histidine into Cellular Protein. Mol Pharmacol. 1977;13:1105–1110. [PubMed] [Google Scholar]
- 9.Schwarzer D, Cole PA. Protein semisynthesis and expressed protein ligation: chasing a protein's tail. Curr Opin Chem Biol. 2005;9:561–569. doi: 10.1016/j.cbpa.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 10.Hemantha HP, Narendra N, Sureshbabu VV. Total chemical synthesis of polypeptides and proteins: chemistry of ligation techniques and beyond. Tetrahedron. 2012;68:9491–9537. [Google Scholar]
- 11.Beiboer SHW, Vandenberg B, Dekker N, Cox RC, Verheij HM. Incorporation of an unnatural amino acid in the active site of porcine pancreatic phospholipase A2. Substitution of histidine by 1,2,4-triazole-3-alanine yields an enzyme with high activity at acidic pH. Protein Eng. 1996;9:345–352. doi: 10.1093/protein/9.4.345. [DOI] [PubMed] [Google Scholar]
- 12.Nozawa K, O'Donoghue P, Gundllapalli S, Araiso Y, Ishitani R, Umehara T, Soll D, Nureki O. Pyrrolysyl-tRNA synthetase:tRNA(Pyl) structure reveals the molecular basis of orthogonality. Nature. 2009;457:1163–1167. doi: 10.1038/nature07611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuley A, Wang YS, Fang X, Kurra Y, Rezenom YH, Liu WR. The genetic incorporation of thirteen novel non-canonical amino acids. Chem Commun. 2014;50:2673–2675. doi: 10.1039/c3cc49068h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tharp JM, Wang YS, Lee YJ, Yang Y, Liu WR. Genetic incorporation of seven ortho-substituted phenylalanine derivatives. ACS Chem Biol. 2014;9:884–890. doi: 10.1021/cb400917a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao H, Peters FB, Yang P, Reed S, Chittuluru JR, Schultz PG. Genetic incorporation of histidine derivatives using an engineered pyrrolysyl-tRNA synthetase. ACS Chem Biol. 2014;9:1092–1096. doi: 10.1021/cb500032c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshimura T, Soda K. Alanine racemase: Structure and Function. Wiley-VCH Verlag GmbH; Weinheim, Germany: 2007. [Google Scholar]
- 17.Yokoigawa K, Okubo Y, Kawai H, Esaki N, Soda K. Structure and function of psychrophilic alanine racemase. J Mol Catal B-Enzyme. 2001;12:27–35. [Google Scholar]
- 18.Toney MD. Reaction specificity in pyridoxal phosphate enzymes. Arch Biochem Biophys. 2005;433:279–287. doi: 10.1016/j.abb.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 19.Lin Y, Gao J, Rubinstein A, Major DT. Molecular dynamics simulations of the intramolecular proton transfer and carbanion stabilization in the pyridoxal 5′-phosphate dependent enzymes L-dopa decarboxylase and alanine racemase. Biochim Biophys Acta. 2011;1814:1438–1446. doi: 10.1016/j.bbapap.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnston RB, Schreiber EC, Davis MP, Jillson L, Sorrell WT, Kirker ME. Catalytic properties of the active site of alanine racemase from B. subtilis. Prog Clin Biol Res. 1984;144A:339–350. [PubMed] [Google Scholar]
- 21.Lynch JL, Neuhaus FC. On the Mechanism of Action of the Antibiotic O-Carbamyl-d-Serine in Streptococcus faecalis. J Bacteriol. 1966;91:449–460. doi: 10.1128/jb.91.1.449-460.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Atherton FR, Hali MJ, Hassall CH, Lambert RW, Ringrose PS. Phosphonopeptides as Antibacterial Agents: Rationale, Chemistry, and Structure-Activity Relationships. Antimicrob Agents Ch. 1979;15:677–683. doi: 10.1128/aac.15.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erion MD, Walsh CT. 1-Aminocyclopropanephosphonate: time-dependent inactivation of 1-aminocyclopropanecarboxylate deaminase and Bacillus stearothermophilus alanine racemase by slow dissociation behavior. Biochemistry. 1987;26:3417–3425. doi: 10.1021/bi00386a025. [DOI] [PubMed] [Google Scholar]
- 24.Mobashery S, Johnston M. Inactivation of alanine racemase by beta-chloro-L-alanine released enzymatically from amino acid and peptide C10-esters of deacetylcephalothin. Bichemistry. 1987;26:5878–5884. doi: 10.1021/bi00392a045. [DOI] [PubMed] [Google Scholar]
- 25.Anthony KG, Strych U, Yeung KR, Shoen CS, Perez O, Krause KL, Cynamon MH, Aristoff PA, Koski RA. New Classes of Alanine Racemase Inhibitors Identified by High-Throughput Screening Show Antimicrobial Activity against Mycobacterium tuberculosis. PLoS ONE. 2011;6:e20374. doi: 10.1371/journal.pone.0020374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ciustea M, Mootien S, Rosato AE, Perez O, Cirillo P, Yeung KR, Ledizet M, Cynamon MH, Aristoff PA, Koski RA, Kaplan PA, Anthony KG. Thiadiazolidinones: A New Class of Alanine Racemase Inhibitors with Antimicrobial Activity against Methicillin- Resistant S. aureus. Biochem pharmacol. 2012;83:368–377. doi: 10.1016/j.bcp.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee Y, Mootien S, Shoen C, Destefano M, Cirillo P, Asojo OA, Yeung KR, Ledizet M, Cynamon MH, Aristoff PA, Koski RA, Kaplan PA, Anthony KG. Inhibition of Mycobacterial Alanine Racemase Activity and Growth by Thiadiazolidinones. Biochem pharmacol. 2013;86:222–230. doi: 10.1016/j.bcp.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Major DT, Gao J. A Combined Quantum Mechanical and Molecular Mechanical Study of the Reaction Mechanism and r-Amino Acidity in Alanine Racemase. J Am Chem Soc. 2006;128:16345–16357. doi: 10.1021/ja066334r. [DOI] [PubMed] [Google Scholar]
- 29.Amadasi A, Bertoldi M, Contestabile R, Bettati S, Cellini B, Salvo ML, Borri-Voltattorni C, Bossa F, Mozzarelli A. Pyridoxal 5′-Phosphate Enzymes as Targets for Therapeutic Agents. Curr Med Chem. 2007;14:1291–1324. doi: 10.2174/092986707780597899. [DOI] [PubMed] [Google Scholar]
- 30.Yoshimura T, Goto M. D-Amino acids in the brain: structure and function of pyridoxal phosphate-dependent amino acid racemases. FEBS J. 2008;275:3527–3537. doi: 10.1111/j.1742-4658.2008.06516.x. [DOI] [PubMed] [Google Scholar]
- 31.Watanabe A, Yoshimura T, Mikami B, Hayashi H, Kagamiyama H, Esaki N. Reaction Mechanism of Alanine Racemase from Bacillus stearothermophilus : X-Ray Crystillographic Studies of the Enzyme Bound With N-(5′-Phosphopyridoxyl) Alanine. J Biol Chem. 2002;277:19166–19172. doi: 10.1074/jbc.M201615200. [DOI] [PubMed] [Google Scholar]
- 32.Sun S, Toney MD. Evidence for a Two-Base Mechanism Involving Tyrosine-265 from Arginine-219 Mutants of Alanine Racemase. Biochemistry. 1999;38:4058–4065. doi: 10.1021/bi982924t. [DOI] [PubMed] [Google Scholar]
- 33.Shaw JP, Petsko GA, Ringe D. Determination of the structure of alanine racemase from Bacillus stearothermophilus at 1.9-A resolution. Biochemistry. 1997;36:1329–1342. doi: 10.1021/bi961856c. [DOI] [PubMed] [Google Scholar]
- 34.Wang YS, Fang X, Wallace AL, Wu B, Liu WR. A Rationally Designed Pyrrolysyl-tRNA Synthetase Mutant with a Broad Substrate Spectrum. J Am Chem Soc. 2012;134:2950–2953. doi: 10.1021/ja211972x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hammill JT, Miyake-Stoner S, Hazen JL, Jackson JC, Mehl RA. Preparation of site-specifically labeled fluorinated proteins for 19F-NMR structural characterization. Nat Protoc. 2007;2:2601–2607. doi: 10.1038/nprot.2007.379. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.