

Abstract
Hepatitis C virus (HCV) is a major cause of chronic hepatitis and liver carcinoma and new therapies based on novel targets are needed. The tight junction protein claudin 1 (CLDN-1) is essential for HCV cell entry and spread, and anti-CLDN-1 rat and mouse mAbs are safe and effective in preventing and treating HCV infection in a human liver chimeric mouse model. To accelerate translation of these observations into a novel approach to treat HCV infection and disease in humans, we screened a phage display library of human single-chain antibody fragments by using a panel of CLDN-1-positive and -negative cell lines and identified phage specifically binding to CLDN-1. The 12 clones showing the highest levels of binding were converted into human IgG4. Some of these mAbs displayed low-nanomolar affinity, and inhibited infection of human hepatoma Huh7.5 cells by different HCV isolates in a dose-dependent manner. Cross-competition experiments identified six inhibitory mAbs that recognized distinct epitopes. Combination of the human anti-SRB1 mAb C-1671 with these anti-CLDN-1 mAbs could either increase or reduce inhibition of cell culture-derived HCV infection in vitro. These novel human anti-CLDN-1 mAbs are potentially useful to develop a new strategy for anti-HCV therapy and lend support to the combined use of antibodies targeting the HCV receptors CLDN-1 and SRB1, but indicate that care must be taken in selecting the proper combination.
Introduction
Hepatitis C virus (HCV) infection represents a major public health concern, with approximately 170 million people chronically infected (Dustin & Rice, 2007), resulting in about 470 000 deaths each year. HCV chronic infection leads to fibrosis, cirrhosis and liver carcinoma (50–76 % of all liver cancers), which are the primary reasons for liver transplantation (LT), and this is set to increase (Davis et al., 2003).
The mechanisms by which HCV escapes the immune response and establishes a chronic infection are not completely defined. HCV replicates at a high rate (Neumann et al., 1998) and its RNA-dependent RNA polymerase lacks proofreading capacity. This results in high genetic variability and rapid evolution under selction either by the immune system or in the presence of antiviral drugs. Combination of pegylated-IFN-α and ribavirin (PegIFN/RBV) is effective in only about 50 % of patients and is often poorly tolerated (Manns et al., 2007). New antiviral drugs (direct-acting antivirals, DAAs) targeting the virus polymerase or protease, such as telaprevir or boceprevir (in clinical use since 2011), also show toxic side effects and their use has been associated with the emergence of drug-resistant variants (Welsch et al., 2008; Pawlotsky, 2011; Dabbouseh & Jensen, 2013; Liang & Ghany, 2013; Chung & Baumert, 2014). By early 2015, the US Food and Drug Administration and the European Medical Agency (EMA) had approved novel DAAs, including a second-generation protease inhibitor (simeprevir), the nucleotide polymerase inhibitor sofosbuvir, and the first HCV-NS5A inhibitor (daclatasvir, EMA approval only). Thus, new, more potent DAAs are being developed (Pawlotsky et al., 2015; Wedemeyer et al., 2015), but it will be essential to maintain a healthy pipeline of anti-HCV drugs, focused on a range of viral and host targets, to ensure that future interventions are not threatened by antiviral resistance. This is particularly true for patients that undergo LT for HCV-related liver disease, where finding appropriate regimens is still challenging (Kwo & Badshah, 2015).
mAbs are a validated class of therapeutics with proven safety and efficacy in a number of clinical applications, and with an established production process. Furthermore, with the advent of fully human mAbs, there are no risks of immune reaction against the antibody (Brekke & Sandlie, 2003; Adams & Weiner, 2005; Imai & Takaoka, 2006; Fuh, 2007). Overall, human mAbs may have superior safety and pharmacokinetic profiles to the current antivirals and emerging DAAs, particularly in the highly susceptible population of HCV-positive LT patients.
Antibodies that are capable of preventing HCV infection may represent novel therapy in two different settings: (i) treatment of chronically infected patients with a combination of mAbs and DAAs to prevent reinfection by drug-resistant mutants; (ii) treatment of patients undergoing LT, either as a monotherapy or in combination with DAAs during the anhepatic phase and during the first days or weeks following transplantation to prevent reinfection of the graft. One approach would be to use antibodies that target the virus directly. However, passive immunotherapy in LT patients with polyclonal antibodies or mAbs against the viral envelope has so far only achieved transient reduction in viral loads and has been unable to prevent reinfection, presumably owing to the high level of heterogeneity in the infecting viral population (Davis et al., 2005; Schiano et al., 2006). In addition, high-density lipoprotein can reduce the neutralizing effect of anti-HCV antibodies (Dreux et al., 2006; Voisset et al., 2006), raising additional concerns about their use being an effective antiviral therapy. For these reasons, the recent discovery of cell surface molecules required for HCV entry may present novel promising targets complementary to those of DAAs.
Four different cell surface receptors are necessary for HCV infection of liver cells: the tetraspanin CD81, the human scavenger receptor class B type I (SRB1), the tight junction claudin 1 (CLDN-1) and occludin (OCLN) (Scarselli et al., 2002; Bartosch et al., 2003; Catanese et al., 2007; Evans et al., 2007; Ploss et al., 2009). mAbs against CD81 and SRB1 have been shown to inhibit HCV infection in vitro and in vivo. In particular, we previously showed that the human anti-SRB1 mAb C-1671 completely prevents infection and intrahepatic spread of multiple HCV genotypes in vivo in human liver-chimeric mice (Meuleman et al., 2012). Human mAbs against CLDN-1 and OCLN have not been reported yet, but rat, mouse and mouse/human chimeric anti-CLDN-1 antibodies were found to efficiently inhibit infection by HCV of major genotypes in cell culture (Fofana et al., 2010; Yamashita et al., 2015) and some of them eliminate chronic HCV infection without detectable toxicity when administered to human liver-chimeric mice (Fukasawa et al., 2015; Mailly et al., 2015).
Two other members of the CLDN protein family (CLDN-6 and CLD-9) could be used alternatively for cell entry by those viruses with broad CLDN tropism (Haid et al., 2014) and thus could be used as additional targets for therapy. However, while some strains efficiently use CLDN-6 as well as CLDN-1, some other strains, such as JFH-1 and J6, solely use CLDN-1 to access cells and this can explain the efficacy of the above-mentioned anti-CLDN-1 antibodies.
To circumvent the potential problems associated with non-fully human antibodies (Getts et al., 2010), we have generated a panel of novel fully human mAbs endowed with binding specificity for CLDN-1. These mAbs recognize different CLDN-1 epitopes and inhibit in vitro infection of Huh7.5 by different viral isolates. Finally, one of them strongly increases antiviral potency when used in combination with the anti-SRB1 mAb C-1671, highlighting the potential synergistic effect of using antibodies targeting different HCV receptors.
Results
Selection of CLDN-1-specific single-chain antibody fragments (scFvs)
The strategy used for the isolation of anti-CLDN-1 scFvs consisted of multiple selections from an scFv phage library on CLDN-1-bearing cells, for enrichment of binders, and CLDN-1-negative cell lines, to eliminate phage that bind to common cell surface antigens. This approach was devised to guarantee efficient selection and increase the number of different clones specifically binding to CLDN-1.
In the first selection scheme, we used human hepatoma Huh7.5 cells as antigen-positive cells, which naturally express high levels of CLDN-1 as well as the other HCV receptors CD81 and SRB1. These cells were chosen because they can be infected by HCVs representative of different genotypes (Gottwein et al., 2009) and, therefore, represent a bona fide source of CLDN-1 displayed in the right conformation for HCV recognition and entry. HEK 293 cells lacking CLDN-1 were used for the subtractive step.
In the other two selections, HEK 293 cells and Chinese hamster ovary (CHO) cells were either transduced or transfected, respectively, with human CLDN-1, and used for the positive selection steps, while mock-transduced or mock-transfected parental cells were used for the subtraction step. The choice of these two cell lines was based on the hypothesis that a possible mechanism for anti-CLDN-1 antiviral activity would be the disruption of the interaction between CLDN-1 and CD81 (Harris et al., 2010; Mailly et al., 2015) or other HCV receptors; therefore, we reasoned that using CLDN-1-transfected CHO cells (which do not express either CD81 or SRB1) or HEK 293 cells (not expressing SRB1) would render the surfaces of interaction of CLDN-1 with other HCV receptors more accessible to scFv binding.
Initially, two rounds of selection were performed either with Huh7.5 or with HEK 293-CLDN-1 cells in suspension. Phage exhibiting selective binding to CLDN-1-expressing cells were submitted to two additional rounds of selection, either by using the same combination of cells or by crossing the selection with the other cell line (see Fig. 1a), to further increase the chance of selecting phage with a high degree of specificity for CLDN-1.
Fig. 1.

Selection and screening of positive phage by cell ELISA on CLDN-1-positive and -negative cells. (a) Schematic representation of parallel and crossed selection on different cell lines. Positive phage, which selectively bound to each CLDN-1-positive cell line, were submitted to two additional rounds of selection, using either the same combination of cells or crossing the selection with the other one. (b) Binding to HEK 293-CLDN-1 of phage selected either by conventional panning on Huh7.5 or HEK 293-CLDN-1 cells in suspension (black bars), or by crossed selections (grey bars) on both Huh7.5 and HEK 293-CLDN-1 cells and indicated with (X), or by panning on adherent CHO cells (hatched bars) and indicated with (C). As a control, all the selected phage were tested on control mock-transduced HEK 293 cells (white bars). Error bars indicate sd.
The third strategy was performed by panning the library directly on adherent CHO-CLDN-1 cells after the subtraction of non-specific clones on adherent mock-transfected CHO cells, in order to select scFvs against CLDN-1 in its natural environmental within intercellular contacts.
After three or four rounds of selection, individual phage were tested by ELISA on CLDN-1-negative and -positive cells to identify CLDN-1-specific binders. Several phage specifically binding to CLDN-1-positive cells were identified from all the three strategies (Fig. 1b). All positive phage were sequenced and 20 different clones were identified. Specific binding was confirmed by expressing the corresponding soluble scFvs and by testing them in cell ELISA as described for phage (data not shown). Twelve clones derived from different selections were chosen for further studies.
Generation and characterization of human anti-CLDN-1 mAbs
The anti-CLDN-1 scFvs were converted into fully human IgG4 by subcloning the variable regions into two eukaryotic vectors for the expression of heavy and light chains, and by transfecting the resulting recombinant plasmids into EBNA 293 cells. The secreted antibodies were affinity purified and tested in cell ELISAs for their selective binding to cells expressing CLDN-1. All the resulting antibodies specifically recognized CLDN-1 displayed on Huh7.5 cells and on HEK 293-CLDN-1 cells (Fig. 2a) and immunoprecipitated CLDN-1 after binding to Huh7.5 cells (Fig. 2b).
Fig. 2.

Binding affinity and specificity of the novel anti-CLDN-1 mAbs. (a) Binding of the 12 novel anti-CLDN-1 mAbs (at a concentration of 100 nM) to Huh7.5 (black bars), mock-transduced HEK 293 (white bars) and HEK 293-CLDN-1-transduced cells (grey bars). Error bars indicate sd. (b) Western blotting analyses with the commercial murine anti-CLDN-1 mAb of lysates of CLDN-1-positive and -negative cells previously immunoprecipitated with each mAb. Lane 1, HEK 293 cell extract; lane 2, HEK 293-CLDN-1 cell extract; lane 3, HEK 293-CLDN-1 cell extract pre-cleared with protein A-Sepharose; lane 4, aliquot of protein A-Sepharose incubated with HEK 293-CLDN-1 cell extract; lanes 5–18, HEK 293-CLDN-1 cell extract immunoprecipitated with commercial anti-CLDN-1 (C-terminus), anti-CLDN-1 (loop I), or each selected mAb, A2, B9, C6, C8, B9X, D10X, A11X, D12C, H9C, F7C, E9C, C10C, respectively; lane 19, HEK 293 cell extract immunoprecipitated with an anti-CLDN-1 mAb; lane 20, HEK 293-CLDN-1 cell extract immunoprecipitated with an irrelevant IgG. (c) Kapp values obtained from the binding curves of the 12 anti-CLDN-1 mAbs to HEK 293-CLDN-1 cells.
We then determined the apparent affinity of the mAbs by dose titration in cell ELISA using CLDN-1-transduced HEK 293 cells. Ten mAbs displayed good apparent affinities in the low-nanomolar range (0.3–16 nM; Fig. 2c), while mAbs E9C and C10C showed a lower binding affinity (Kapp = 117 and 190 nM, respectively).
Anti-CLDN-1 mAbs inhibit Huh7.5 infection by HCV from different strains
The anti-CLDN-1 mAbs were tested for their ability to inhibit HCV infection of cultured Huh7.5 hepatocytes. With this aim, cell culture-derived HCV (HCVcc) from five different HCV strains representative of genotypes 1 and 2 were used: the reference cell culture-adapted Japanese fulminant hepatitis 1 clone JFH-1 (gt2a), two chimeric viruses [J6/JFH-1 (envelope proteins from gt2a) and H77/JFH-1 (envelope proteins from gt1a)], and two additional JFH-1 chimeras generated by using clinically relevant gt2b glycoproteins from patient-derived isolates – 2B1.1/JFH-1 and 2B2.8/JFH-1.
Five out of 12 mAbs inhibited all five isolates, although to a different degree (B9X, D10X, E9C, F7C and H9C), while mAb A2 was effective against four HCVcc isolates (Fig. 3). All other mAbs did not show significant inhibitory effects (data not shown). Three of the active mAbs – A2, B9X and D10X – showed an overall higher potency, with low and comparable IC50 values (lower than 20 μg ml− 1) against two isolates from different genotypes, 1a (H77/JFH-1) and 2a (JFH-1), with the latter mAb displaying high inhibition also against the J6/JFH-1 virus (Fig. 3 and Table 1). As controls, neutralization curves were performed with another anti-HCV receptor C-1671 mAb (anti-SRB1), with 1 : 7 mAb (anti-HCV envelope) and anti-tetanus toxin IgG (isotype-matched irrelevant control) (Fig. 4a). The 1 : 7 mAb showed IC50 values comparable with those of B9X and D10X on some strains, such as H77/JFH-1 and JFH-1, whereas for C-1671, owing to its different mode of action, the IC50 values are typically lower. As expected, the control anti-tetanus toxin IgG did not show significant effects.
Fig. 3.

Neutralization curves of the six novel anti-CLDN-1 antibodies. The mAbs were tested on Huh7.5 cells treated with 2B2.8 (▾), JFH-1 (•), J6/JFH1 (▪), 2B1.1 (▴) and H77/JFH-1 (♦). All the mAbs had neutralization capabilities against the isolates tested. Antibody preparations were pre-incubated at the indicated concentrations with Huh7.5 cells for 1 h. Cells were thoroughly washed with PBS and virus was added to the cells. Infection was determined by staining for NS5A after 72 h. Statistical analyses were performed at 100 μg ml− 1 via one-way ANOVA with Bonferroni correction. ****P < 0.0001. Error bars indicate sd.
Table 1. Comparison of binding affinity and inhibition activity of the anti-CLDN-1 mAbs.
Kapp values obtained by ELISAs on CLDN-1-positive cells and IC50 values calculated by the inhibition curves of the indicated HCV isolates for the six human anti-CLDN-1 mAbs.
| mAb | Kapp (nM) | IC50 (μg ml− 1) | ||||
|---|---|---|---|---|---|---|
| H77/JFH-1 | JFH-1 | 2B1.1/JFH-1 | 2B2.8/JFH-1 | J6/JFH-1 | ||
| A2 | 16.0 | 14 | 18 | >100 | >100 | 68 |
| B9X | 13.0 | 9 | 6 | 63 | 65 | 64 |
| D10X | 2.9 | 11 | 8 | 57 | 61 | 17 |
| H9C | 0.8 | 52 | 45 | 32 | 20 | 55 |
| E9C | 117.2 | 35 | 44 | 56 | 31 | 95 |
| F7C | 0.9 | 21 | 34 | 57 | 62 | 53 |
Fig. 4.

Neutralization curves of control antibodies and schematic representation for the competitive binding of the six anti-CLDN-1 antibodies. (a) As controls, neutralization assays were performed with anti-SRB1 receptor C-1671 mAb (Δ), anti-HCV envelope 1 : 7 mAb (▪) and anti-tetanus toxin IgG irrelevant control (♦). The mAbs were tested on Huh7.5 cells treated with 2B2.8, JFH-1, J6, 2B1.1 and H77/JFH-1 as described for Fig. 3. Statistical analyses were performed at 100 μg ml− 1 via one-way ANOVA with Bonferroni correction. ****P < 0.0001. Error bars indicate sd. (b) Percentage binding of each anti-CLDN-1 clone was calculated as the ratio of the absorbance (at 450 nm) of each bound scFv-phage detected in the absence or in the presence of the indicated mAbs.
All six inhibitory anti-CLDN-1 mAbs recognize different CLDN-1 epitopes
There was no correlation between the binding affinity to CLDN-1 and the IC50 of the six active mAbs (Table 1), suggesting that they recognize different epitopes on CLDN-1. To test this hypothesis we performed cross-competition ELISAs on Huh7.5 cells. In a first series of experiments anti-CLDN-1 mAbs were pre-incubated with Huh7.5 cells at saturating concentrations and then further incubated with phage displaying the different scFvs. For all six clones, binding of the phage was significantly inhibited only by the homologous mAb (Fig. 4b). These results were confirmed by labelling all six mAbs with biotin and by competitive ELISA performed by measuring cell binding of each biotinylated mAb in the absence or presence of all other six unlabelled mAbs (data not shown). These data strongly suggested that all six mAbs were able to bind to different epitopes on the extracellular region of CLDN-1.
Different combinations of anti-CLDN-1 mAbs and the human anti-SRB1 mAb C-1671 resulted in either strong synergistic or antagonistic inhibitory activity
Anti-HCV E2 and anti-CD81 antibodies were previously shown to improve their inhibitory activity when used in combination (Ashfaq et al., 2011). To investigate whether this would also be the case with antibodies targeting different HCV receptors, we tested B9X, D10X and the anti-SRB1 mAb C-1671 alone and also in combination to determine the combination index (CI). The results of this experiment were evaluated by the median effect analysis method using the CompuSyn program (Chou, 2006) to calculate the CI. CI values < 0.9, 0.9–1.1 and >1.1 indicate synergy, an additive effect and antagonism, respectively.
As shown in Fig. 5, the combination of D10X and C-1671 showed moderate or strong synergistic activity at IC50, IC75 and IC90 against the J6 (0.24 at IC90), 2B1.1 (0.66 at IC90), 2B2.8 (0.56 at IC90) and H77 (0.48 at IC90) chimeras, whereas the combination of B9X with C-1671 was antagonistic for all the viruses tested (>1.5 for all IC values).
Fig. 5.

Combinatorial treatment of D10X and B9X with the anti-SRB1 C-1671 mAb. (a) Inhibition curves (obtained on J6/JFH-1) of D10X (▪), B9X (○), anti-SRB1 C-1671 mAb (▵) or the combination of D10X with C-1671 (▴) and of B9X with C-1671 (•). (b) CI values obtained for J6/JFH-1 and the other isolates tested. CI values < 0.9, 0.9–1.1 and >1.1 indicate synergy, an additive effect and antagonism, respectively. Black bars, IC50; grey bars, IC75; white bars, IC90.
To test whether the antagonism between B9X and C-1671 was due to an unexpected capacity of B9X to hinder the interactions of C-1671 with the hepatocytes, a competitive ELISA was performed by measuring the binding to Huh7.5 cells of biotinylated anti-SRB1 mAb in the absence or presence of the unlabelled D10X or B9X mAbs. Simultaneous binding of anti-CLDN-1 and anti-SRB1 mAbs revealed that the two mAbs did not interfere in their interactions with their respective receptors (Fig. 6a).
Fig. 6.
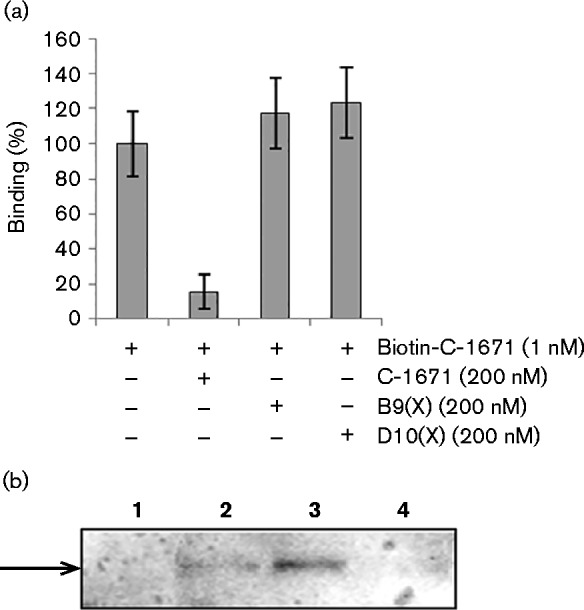
Mechanism of action of D10X and B9X. (a) Competitive ELISA of D10X or B9X with anti-SRB1 C-1671 mAb. The percentage binding to Huh7.5 cells of biotinylated anti-SRB1 mAb in the absence or presence of unlabelled anti-CLDN-1 D10X or B9X mAbs was detected with HRP-streptavidin. (b) Internalization of anti-CLDN-1 mAbs. The intracellular levels of the antibodies in target cells were measured by Western blotting with an HRP-conjugated anti-human IgG. Lanes 1–4: immunoreactive proteins in the extracts of cells untreated (lane 1) or treated with B9X (lane 2), with D10X (lane 3) or with unrelated control antibody (lane 4). Error bars indicate sd.
To test whether the different mechanism of action of D10X and B9X was due to a different ability of these anti-CLDN-1 mAbs to induce CLDN-1 endocytosis and thus be internalized by hepatocytes, we analysed their presence in the lysate of treated cells. Briefly, Huh7.5 cells were incubated with D10X or B9X mAbs (10 μg ml− 1) for 2 h at 37 °C, stripped of surface-bound protein with a low pH glycine/NaCl buffer, and lysed.
Aliquots of cell extracts containing equal amounts of protein were analysed by immunoblotting using goat HRP-conjugated anti-human IgG. As shown in Fig. 6(b), an immunoreactive band with the molecular mass expected for the IgG was clearly observed in the intracellular fraction of cells treated with D10X. The signal intensity was much stronger than that observed for cells treated with B9X, whereas no signal was detected in the extracts of untreated control cells or control cells treated with an irrelevant isotype-matched control antibody.
These results indicate that D10X can induce CLDN-1 endocytosis more efficiently than B9X and that this could be at least in part responsible for their different mechanism of action.
All six inhibitory anti-CLDN-1 mAbs do not show toxicity in cell culture
To address potential toxic effects of anti-CLDN-1 mAbs in vitro, we performed cell viability and lysis analyses on Huh7.5 cells treated with the six mAbs with anti-HCV inhibitory activity. Following incubation of hepatoma cells with each of the anti-CLDN-1 antibodies for 72 h at 37 °C, tests based on 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and lactate dehydrogenase (LDH) release were carried out. As shown in Fig. 7, no toxic effects were observed up to high mAb concentrations (100 μg ml− 1).
Fig. 7.

Cytotoxic effects on Huh7.5 cells of the anti-CLDN-1 mAbs and their competitive binding with anti-SRB1 mAb. (a) Percentage cell lysis after incubation with each indicated anti-CLDN-1 mAb; as positive control, cells were treated with 1 % Triton X-100. (b) Percentage of cell viability after 72 h incubation with each indicated anti-CLDN-1 mAb measured by MTT. Error bars indicate sd.
Discussion
The tight junction protein CLDN-1 is an essential receptor for HCV infection of human hepatocytes in vitro and in vivo (Fofana et al., 2010; Mailly et al., 2015). Recently, it was shown that rat, mouse and human/mouse hybrid mAbs are effective against HCV infection in vitro and in vivo without detectable toxicity when administered to human liver-chimeric mice (Fukasawa et al., 2015; Mailly et al., 2015). Thus, antibodies against CLDN-1 could represent a novel class of anti-HCV drugs to be used alone or in combination with DAAs for treatment of chronic hepatitis C and to prevent reinfection of liver allografts in LT patients.
A critical step to be taken toward this end would be to generate human anti-CLDN-1 mAbs with similar characteristics to those described, but amenable to clinical use in humans in light of potential unwanted anti-rat or anti-mouse IgG immune responses, which are likely to reduce efficacy of the treatment. We report here the successful generation by phage display technology of six novel fully human anti-CLDN-1 mAbs capable of selectively binding to CLDN-1 displayed on cells and of inhibiting infection of cultured Huh7.5 cells by five different HCVccs representative of genotypes 1 and 2.
To generate human mAbs against human CLDN-1, we selected human scFvs from a phage display library with alternate cycles of positive selection on CLDN-1-positive cells and of subtraction with CLDN-1-negative cells to eliminate phage-binding to other cell surface proteins. While being more laborious, this strategy of library panning on live cells offers the advantage of the target protein being in its native conformation (Sasso et al., 2015). Using this approach, we previously succeeded in generating human mAbs with in vitro and in vivo biological activity against the transferrin receptor, the ErbB2 and EphA2 tyrosine kinase receptors and the HCV receptor SRB1 (De Lorenzo et al., 2002; Catanese et al., 2007; Ansuini et al., 2009; Khalaj-Kondori et al., 2011).
The selected scFvs were converted into fully human IgG4 because this isotype was shown to have reduced complement and cell-mediated cytotoxicity, while displaying long half-life (Vidarsson et al., 2014). All resulting human mAbs conserved their specificity of binding to cell-displayed CLDN-1, with the majority of them showing apparent affinities in the low-nanomolar range, and did not show cytotoxic activity when incubated with human hepatoma cells in vitro.
Six out of 12 mAbs (A2, B9X, D10X, E9C, F7C and H9C) inhibited infection of human hepatoma Huh7.5 by five different HCV isolates representing different genotypes and subtypes in a dose-dependent manner. All but one of these mAbs, mAb A2, were active against all five viruses tested, though with different potency, ranging from 6 to over 50 μg ml− 1, and with H77/JFH-1 and JFH-1 being on average the most sensitive isolates.
The ability of anti-CLDN-1 human mAbs to inhibit infection by different HCV isolates was not unexpected and was in line with previous observations made with rat and mouse anti-CLDN-1 antibodies (Fofana et al., 2010; Fukasawa et al., 2015; Mailly et al., 2015; Yamashita et al., 2015). Most importantly, mAbs B9X and D10X displayed binding affinity for cell-displayed CLDN-1 and IC50 values comparable with the rat anti-CLDN-1 mAb OM-7D3, which was recently shown to prevent HCV infection and to clear persistent infection in a human liver-chimeric mouse model without detectable toxicity (Mailly et al., 2015).
Interestingly, there was no correlation between the binding affinity and the inhibition activity since the mAb showing the highest binding affinity (i.e. mAb F7C) was less effective at inhibiting infection than mAbs A2, B9X and D10X. These data, together with the observation that only half of the tested mAbs showed inhibition activity against HCV infection, suggested that the selected scFvs recognize different CLDN-1 epitopes. Cross-competition experiments between the six active mAbs confirmed this hypothesis and further validated our selection strategy on different cell lines aimed at identifying a large number of different binders specific for different epitopes of CLDN-1.
We also investigated whether the CLDN-1 antibodies B9X and D10X could synergize with the anti-SRB1 antibody C-1671, which potently inhibits infection and spread in vivo in the human liver-chimeric mouse model (Meuleman et al., 2012). Combination of C-1671 with D10X resulted in measurable synergy when tested against four virus strains, including two clinically derived isolates. In contrast, combination of the anti-SRB1 mAb with B9X resulted in reduced efficacy despite the absence of binding interference of the two mAbs to their respective targets.
To explain the different mechanisms of action of B9X and D10X, we tested their ability to induce CLDN-1 endocytosis by measuring their internalization in treated hepatocytes. D10X induces CLDN-1 endocytosis more efficiently than B9X, and thus the hypothesis can be made that it could neutralize the infection not only by blocking virus binding and cell entry (like C-1671) but also in the post-entry events within the endosomes, thus potentiating the effects of anti-SRB1 antibody more efficiently than B9X.
Another promising antiviral approach is represented by the combination of DAAs and viral cell entry inhibitors. A recent report (Xiao et al., 2015) showed that combining DAAs and anti-CLDN-1 or anti-SRB1 mAb results in synergy in treatment of HCV infection either in vitro or in vivo. Thus, even if the efficacy of our anti-CLDN-1 antibodies is lower [half-maximal effective concentration (EC50) ranging from 6 to over 50 μg ml− 1] than that showed for DAAs (EC50 ranging from 0.01 μg ml− 1 for simeprevir to 0.5 μg ml− 1 for boceprevir in in vitro assays of Huh7.5.1 cells infected with HCVcc), they could be useful for combinatorial treatment, which might be promising for prevention of liver graft infection.
The human anti-CLDN-1 mAbs described here represent a first step toward development of potent HCV entry inhibitors for clinical use. To this end, we are currently generating a second generation of anti-CLDN-1 antibodies by affinity maturation. In any case, the data presented in this work provide for the first time, to the best of our knowledge, clear-cut evidence for synergistic activity of anti-CLDN-1 and anti-SRB1 antibodies, useful for developing more effective anti-HCV therapy, whilst at the same time highlighting the need for careful screening of the right combination prior to further development.
Methods
Cell cultures
The human embryonic kidney HEK 293T and HEK 293-EBNA, and the human hepatoma Huh7.5 cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) with the addition of non-essential amino acid solution (Gibco). HEK 293T cells transduced with the gene encoding CLDN-1 were grown in DMEM containing blasticidin (2 μg ml− 1; Gibco). CHO cells (American Type Culture Collection) were cultured in F12 medium (Gibco). Media were supplemented with 10 % FBS, 50 U penicillin ml− 1, and 50 μg streptomycin ml− 1 (all from Gibco).
Antibodies
The following antibodies were used: mouse HRP-conjugated anti-M13 mAb (GE Healthcare Bio-Sciences), mouse HRP-conjugated anti-c-myc-tag mAb (Miltenyi Biotec), mouse anti-CLDN-1 (C-terminal end) mAb (Life Technology), rabbit anti-loop 1 of CLDN-1 polyclonal antibody (Abcam), goat HRP-conjugated anti-human IgG (Promega), goat HRP-conjugated anti-human Fc mAb (Immuno Reagents).
Preparation of phage particles from the phagemid library
Phage particles were recovered from the library by using the helper phage M13-K07, as described previously (De Lorenzo et al., 2002). Phage were purified, concentrated by two steps of polyethylene glycol (PEG) precipitation (Sambrook et al., 1989) and washed with 20 ml sterile water. After an additional PEG-precipitation step, phage were resuspended in PBS and centrifuged at 17 500 g for 15 min at 4 °C.
Selection of scFv-phage on live cells
The human Huh7.5 cell line, naturally expressing high levels of CLDN-1, the HEK 293T cells mock-transduced or transduced with CLDN-1, and the CHO cells mock-transfected or transfected with the vector encoding CLDN-1 were detached by using cell dissociation solution (Sigma-Aldrich) and washed twice with PBS. For each round of panning, phage (1013 c.f.u.) were blocked with 5 % milk powder (Sigma-Aldrich) in PBS for 15 min. The blocked phage were submitted to two more rounds of negative selection by two successive incubations with CLDN-1-negative cells (5 × 106), carried out by gently rotating the suspension for 2 h at 4 °C. The unbound phage, collected by centrifugation at 350 g min− 1 for 10 min, were then used for the positive selection performed by using CLDN-1-transduced HEK 293T (1 × 106) or Huh7.5 cells (1 × 106), incubated for 16 h by gently rotating at 4 °C. Cells were then recovered by centrifugation at 350 g for 10 min and washed twice with PBS. The positive selection on CLDN-1-transfected CHO cells was carried out by incubating the phage with 2 × 106 adherent cells. After extensive washes, bound phage from each selection were eluted from positive cells with a solution of 1 μg trypsin (Sigma-Aldrich) ml− 1, which was then stopped by the addition of complete EDTA-free protease inhibitor (Roche Diagnostic). The recovered phage were amplified by infecting E. coli TG1 cells to prepare phage for the next round of selection.
Characterization of selected scFv-phage
A TG1 culture was infected with the eluted phage (after three or four rounds of panning) and plated on 2 × TY/agar containing glucose (1 %) and ampicillin (100 μg ml− 1). Individual clones were picked, transferred into 96-well plates and grown for the production of scFv-phage by superinfection with M13-K07 helper phage encoding trypsin-cleavable pIII. The plates were centrifuged at 350 g for 30 min at 4 °C to pellet the bacteria, and aliquots of 50 μl scFv-phage-containing supernatants were used for cell ELISA. The cDNA encoding the scFv of ELISA-positive clones was PCR-amplified and sequenced (Bio-Fab Research), then aligned by using gentle software (Magnus Manske, University of Cologne, Germany). Cultures of E. coli SF110 infected with anti-CLDN-1 scFv-phage were used for the expression of soluble scFvs, which were purified on immobilized-metal affinity chromatography, as described previously (De Lorenzo et al., 2002).
Production and purification of mAbs
For the conversion of the selected scFvs into whole IgG4, an In-Fusion HD cloning kit (Clontech Laboratories) was used. PCR products were purified and inserted in the appropriate vectors expressing the constant antibody heavy and light chains by using the In-Fusion HD Enzyme Premix (Clontech Laboratories), according to the manufacturer's recommendations. The variable heavy chain (VH) sequences were cloned in the linearized PEU 8.2 vector and the variable light chain (VL) sequences were cloned in linearized PEU 4.2 vector. A sequence analysis was then performed to confirm the successful cloning of the inserted sequence with the correct reading frame. Aliquots of 10 μg of the recombinant vectors encoding each VH and VL pair were co-transfected in HEK 293-EBNA by using Lipofectamine transfection reagent (Life Technologies) and grown for 10 days at 37 °C. The antibodies secreted into the medium were purified by using HiTrap Protein-A HP (GE Healthcare Life Sciences).
Cell ELISAs and competition analysis
To confirm the binding specificity for CLDN-1 of the selected scFv-phage or purified mAbs, cell ELISA was carried out by using CLDN-1-positive and -negative cells as previously described (De Lorenzo et al., 2002). To determine whether the novel mAbs recognize different CLDN-1 epitopes, CLDN-1-positive cells, detached with non-enzymic cell dissociation solution (Sigma-Aldrich), were resuspended in PBS/BSA (6 %), plated in 96-well plates (1 × 105 cells in each well) and incubated for 90 min with 200 nM each mAb in PBS/BSA (3 %), with agitation, at room temperature. Then the cells were resuspended in 100 μl each scFv-phage (1 × 1011 c.f.u.) prepared in a PBS/milk (2.5 %) solution and incubated for further 2 h, with agitation, at room temperature, before the bound phage was detected (De Lorenzo et al., 2002) with a mouse HRP-conjugated anti-M13 mAb (GE Healthcare). To determine whether the novel mAbs B9X and D10X interfere in the binding of C-1671 to the cells, Huh7.5 cells were incubated with each unlabelled mAb (200 nM) as described above and then further treated for 1 h with biotinylated anti-SRB1 mAb (1 nM), which was then detected with HRP-streptavidin (Bio-Rad).
Cell lysis, immunoprecipitation and Western blot analyses
CLDN-1-positive cells (2.5 × 107) were lysed as described previously (De Lorenzo et al., 2002). Each cell lysate was pre-cleared with protein A-Sepharose, then incubated at 4 °C for 16 h by gently rotating with 5 μg mouse anti-C-terminal region of CLDN-1 mAb (Life Technology), 5 μg rabbit anti-loop 1 of CLDN-1 polyclonal antibody (Abcam) or 10 μg each mAb selected by phage display. Each immune-complex was collected by adsorption to protein A-Sepharose and analysed by Western blotting as previously described (Laemmli, 1970; Palmer et al., 1999) by using a mouse anti-C-terminal region of CLDN-1 mAb (Life Technology).
Intracellular anti-CLDN-1 antibodies were detected by Western blotting of treated Huh7.5 cell extracts with goat HRP-conjugated anti-human IgG (Promega), The signal was visualized by enhanced chemiluminescence detection (ECL Western blotting detection kit; Amersham Biosciences). The signal intensity of reactive bands was measured with a phosphorimager (GS-710; Bio-Rad).
HCVcc generation, neutralization and combination assays
All chimeras were produced as previously described (Lindenbach et al., 2005). Neutralization assays were performed by seeding Huh7.5 cells (1.5 × 105) in a 96-well plate (BD Biosciences). After 24 h, each mAb was added to cells, incubated for 1 h at 37 °C with 5 % CO2, and washed twice with PBS. Virus was then added and incubated at 37 °C for 72 h. Infected cells were visualized by NS5A staining (Tellinghuisen et al., 2008) and the infectivity was defined as the percentage of stained foci compared with an untreated control culture. Synergistic, additive or antagonistic interaction by the antibodies for virus neutralization was evaluated by the median effect analysis method, as previously described (Chou, 2006; Lacek et al., 2014), using the CompuSyn software (ComboSyn). CI values of < 0.9, 0.9–1.1 and >1.1 indicate synergy, an additive effect and antagonism, respectively.
Cell viability and cytotoxicity assays
Huh7.5 cells were seeded in 96-well plates at a density of 1 × 104 per well and incubated at 37 °C with 5 % CO2 for 16 h. After the addition of 20 or 100 μg ml− 1 mAbs to the culture medium, cells were incubated for a further 72 h. Viable cells were counted by the trypan blue exclusion test and cell survival was expressed as a percentage of viable cells in the presence of the mAb under test with respect to negative control cultures grown in the absence of the protein. Cell lysis was determined by measuring the release of LDH using an LDH detection kit (Roche Diagnostic). Lysis was calculated as the percentage of cytolysis measured in the presence of each mAb, taking as 100 % the maximal LDH release, determined by lysis of target cells treated with 1 % Triton X-100.
Statistical analyses and reproducibility of results
Typically, the reported values were obtained from at least three independent experiments in which three determinations were performed for each sample. Standard deviations were calculated on the basis of the results obtained from all the experiments. Results are expressed as the mean+sd. Statistical analyses were performed via one-way ANOVA with Bonferroni correction.
Acknowledgements
This work was supported by the EU FP7 grant ‘HepaMAb’ (305600), the Medical Research Council UK (G0801169) and by POR ‘Rete delle Biotecnologie in Campania’ (DMMBM). The authors thank Dr Maria Teresa Catanese for providing HEK 293-CLDN-1 cells and for useful support, and Mats Persson, Takaji Wakita and Charles Rice for the generous provision of reagents.
References
- Adams G. P., Weiner L. M. (2005). Monoclonal antibody therapy of cancer Nat Biotechnol 231147–1157 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- Ansuini H., Meola A., Gunes Z., Paradisi V., Pezzanera M., Acali S., Santini C., Luzzago A., Mori F., other authors (2009). Anti-EphA2 antibodies with distinct in vitro properties have equal in vivo efficacy in pancreatic cancer J Oncol 2009951917. 10.1155/2009/951917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashfaq U. A., Qasim M., Yousaf M. Z., Awan M. T., Jahan S. (2011). Inhibition of HCV 3a genotype entry through host CD81 and HCV E2 antibodies J Transl Med 9194. 10.1186/1479-5876-9-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosch B., Vitelli A., Granier C., Goujon C., Dubuisson J., Pascale S., Scarselli E., Cortese R., Nicosia A., Cosset F. L. (2003). Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor J Biol Chem 27841624–41630 10.1074/jbc.M305289200. [DOI] [PubMed] [Google Scholar]
- Brekke O. H., Sandlie I. (2003). Therapeutic antibodies for human diseases at the dawn of the twenty-first century Nat Rev Drug Discov 252–62 10.1038/nrd984. [DOI] [PubMed] [Google Scholar]
- Catanese M. T., Graziani R., von Hahn T., Moreau M., Huby T., Paonessa G., Santini C., Luzzago A., Rice C. M., other authors (2007). High-avidity monoclonal antibodies against the human scavenger class B type I receptor efficiently block hepatitis C virus infection in the presence of high-density lipoprotein J Virol 818063–8071 10.1128/JVI.00193-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T. C. (2006). Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies Pharmacol Rev 58621–681 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- Chung R. T., Baumert T. F. (2014). Curing chronic hepatitis C — the arc of a medical triumph N Engl J Med 3701576–1578 10.1056/NEJMp1400986. [DOI] [PubMed] [Google Scholar]
- Dabbouseh N. M., Jensen D. M. (2013). Future therapies for chronic hepatitis C Nat Rev Gastroenterol Hepatol 10268–276 10.1038/nrgastro.2013.17. [DOI] [PubMed] [Google Scholar]
- Davis G. L., Albright J. E., Cook S. F., Rosenberg D. M. (2003). Projecting future complications of chronic hepatitis C in the United States Liver Transpl 9331–338 10.1053/jlts.2003.50073. [DOI] [PubMed] [Google Scholar]
- Davis G. L., Nelson D. R., Terrault N., Pruett T. L., Schiano T. D., Fletcher C. V., Sapan C. V., Riser L. N., Li Y., other authors (2005). A randomized, open-label study to evaluate the safety and pharmacokinetics of human hepatitis C immune globulin (Civacir) in liver transplant recipients Liver Transpl 11941–949 10.1002/lt.20405. [DOI] [PubMed] [Google Scholar]
- De Lorenzo C., Palmer D. B., Piccoli R., Ritter M. A., D'Alessio G. (2002). A new human antitumor immunoreagent specific for ErbB2 Clin Cancer Res 81710–1719. [PubMed] [Google Scholar]
- Dreux M., Pietschmann T., Granier C., Voisset C., Ricard-Blum S., Mangeot P. E., Keck Z., Foung S., Vu-Dac N., other authors (2006). High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI J Biol Chem 28118285–18295 10.1074/jbc.M602706200. [DOI] [PubMed] [Google Scholar]
- Dustin L. B., Rice C. M. (2007). Flying under the radar: the immunobiology of hepatitis C Annu Rev Immunol 2571–99 10.1146/annurev.immunol.25.022106.141602. [DOI] [PubMed] [Google Scholar]
- Evans M. J., von Hahn T., Tscherne D. M., Syder A. J., Panis M., Wölk B., Hatziioannou T., McKeating J. A., Bieniasz P. D., Rice C. M. (2007). Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry Nature 446801–805 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- Fofana I., Krieger S. E., Grunert F., Glauben S., Xiao F., Fafi-Kremer S., Soulier E., Royer C., Thumann C., other authors (2010). Monoclonal anti-claudin 1 antibodies prevent hepatitis C virus infection of primary human hepatocytes Gastroenterology 139953–964, e1–e4 10.1053/j.gastro.2010.05.073. [DOI] [PubMed] [Google Scholar]
- Fuh G. (2007). Synthetic antibodies as therapeutics Expert Opin Biol Ther 773–87 10.1517/14712598.7.1.73. [DOI] [PubMed] [Google Scholar]
- Fukasawa M., Nagase S., Shirasago Y., Iida M., Yamashita M., Endo K., Yagi K., Suzuki T., Wakita T., other authors (2015). Monoclonal antibodies against extracellular domains of claudin-1 block hepatitis C virus infection in a mouse model J Virol 894866–4879 10.1128/JVI.03676-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getts D. R., Getts M. T., McCarthy D. P., Chastain E. M., Miller S. D. (2010). Have we overestimated the benefit of human(ized) antibodies? MAbs 2682–694 10.4161/mabs.2.6.13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottwein J. M., Scheel T. K., Jensen T. B., Lademann J. B., Prentoe J. C., Knudsen M. L., Hoegh A. M., Bukh J. (2009). Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs Hepatology 49364–377 10.1002/hep.22673. [DOI] [PubMed] [Google Scholar]
- Haid S., Grethe C., Dill M. T., Heim M., Kaderali L., Pietschmann T. (2014). Isolate-dependent use of claudins for cell entry by hepatitis C virus Hepatology 5924–34 10.1002/hep.26567. [DOI] [PubMed] [Google Scholar]
- Harris H. J., Davis C., Mullins J. G., Hu K., Goodall M., Farquhar M. J., Mee C. J., McCaffrey K., Young S., other authors (2010). Claudin association with CD81 defines hepatitis C virus entry J Biol Chem 28521092–21102 10.1074/jbc.M110.104836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K., Takaoka A. (2006). Comparing antibody and small-molecule therapies for cancer Nat Rev Cancer 6714–727 10.1038/nrc1913. [DOI] [PubMed] [Google Scholar]
- Khalaj-Kondori M., Sadeghizadeh M., Behmanesh M., Saggio I., Monaci P. (2011). Chemical coupling as a potent strategy for preparation of targeted bacteriophage-derived gene nanocarriers into eukaryotic cells J Gene Med 13622–631 10.1002/jgm.1617. [DOI] [PubMed] [Google Scholar]
- Kwo P. Y., Badshah M. B. (2015). New hepatitis C virus therapies: drug classes and metabolism, drug interactions relevant in the transplant settings, drug options in decompensated cirrhosis, and drug options in end-stage renal disease Curr Opin Organ Transplant 20235–241 10.1097/MOT.0000000000000198. [DOI] [PubMed] [Google Scholar]
- Lacek K., Urbanowicz R. A., Troise F., De Lorenzo C., Severino V., Di Maro A., Tarr A. W., Ferrara F., Ploss A., other authors (2014). Dramatic potentiation of the antiviral activity of HIV antibodies by cholesterol conjugation J Biol Chem 28935015–35028 10.1074/jbc.M114.591826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4 Nature 227680–685 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Liang T. J., Ghany M. G. (2013). Current and future therapies for hepatitis C virus infection N Engl J Med 3681907–1917 10.1056/NEJMra1213651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach B. D., Evans M. J., Syder A. J., Wölk B., Tellinghuisen T. L., Liu C. C., Maruyama T., Hynes R. O., Burton D. R., other authors (2005). Complete replication of hepatitis C virus in cell culture Science 309623–626 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- Mailly L., Xiao F., Lupberger J., Wilson G. K., Aubert P., Duong F. H., Calabrese D., Leboeuf C., Fofana I., other authors (2015). Clearance of persistent hepatitis C virus infection in humanized mice using a claudin-1-targeting monoclonal antibody Nat Biotechnol 33549–554 10.1038/nbt.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manns M. P., Foster G. R., Rockstroh J. K., Zeuzem S., Zoulim F., Houghton M. (2007). The way forward in HCV treatment – finding the right path Nat Rev Drug Discov 6991–1000 10.1038/nrd2411. [DOI] [PubMed] [Google Scholar]
- Meuleman P., Catanese M. T., Verhoye L., Desombere I., Farhoudi A., Jones C. T., Sheahan T., Grzyb K., Cortese R., other authors (2012). A human monoclonal antibody targeting scavenger receptor class B type I precludes hepatitis C virus infection and viral spread in vitro and in vivo Hepatology 55364–372 10.1002/hep.24692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann A. U., Lam N. P., Dahari H., Gretch D. R., Wiley T. E., Layden T. J., Perelson A. S. (1998). Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-α therapy Science 282103–107 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- Palmer D. B., Crompton T., Marandi M. B., George A. J., Ritter M. A. (1999). Intrathymic function of the human cortical epithelial cell surface antigen gp200-MR6: single-chain antibodies to evolutionarily conserved determinants disrupt mouse thymus development Immunology 96236–245 10.1046/j.1365-2567.1999.00691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlotsky J. M. (2011). Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus Hepatology 531742–1751 10.1002/hep.24262. [DOI] [PubMed] [Google Scholar]
- Pawlotsky J. M., Feld J. J., Zeuzem S., Hoofnagle J. H. (2015). From non-A, non-B hepatitis to hepatitis C virus cure J Hepatol 62S87–S99 10.1016/j.jhep.2015.02.006. [DOI] [PubMed] [Google Scholar]
- Ploss A., Evans M. J., Gaysinskaya V. A., Panis M., You H., de Jong Y. P., Rice C. M. (2009). Human occludin is a hepatitis C virus entry factor required for infection of mouse cells Nature 457882–886 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E. F., Maniatis T. (1989). Molecular Cloning: a Laboratory Manual 2nd ednCold Spring Harbor; NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Sasso E., Paciello R., D'Auria F., Riccio G., Froechlich G., Cortese R., Nicosia A., De Lorenzo C., Zambrano N. (2015). One-Step Recovery of scFv Clones from High-Throughput Sequencing-Based Screening of Phage Display Libraries Challenged to Cells Expressing Native Claudin-1 Biomed Res Int 2015, 70321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarselli E., Ansuini H., Cerino R., Roccasecca R. M., Acali S., Filocamo G., Traboni C., Nicosia A., Cortese R., Vitelli A. (2002). The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus EMBO J 215017–5025 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiano T. D., Charlton M., Younossi Z., Galun E., Pruett T., Tur-Kaspa R., Eren R., Dagan S., Graham N., other authors (2006). Monoclonal antibody HCV-AbXTL68 in patients undergoing liver transplantation for HCV: results of a phase 2 randomized study Liver Transpl 121381–1389 10.1002/lt.20876. [DOI] [PubMed] [Google Scholar]
- Tellinghuisen T. L., Foss K. L., Treadaway J. (2008). Regulation of hepatitis C virion production via phosphorylation of the NS5A protein PLoS Pathog 4e1000032. 10.1371/journal.ppat.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidarsson G., Dekkers G., Rispens T. (2014). IgG subclasses and allotypes: from structure to effector functions Front Immunol 5520. 10.3389/fimmu.2014.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisset C., Op de Beeck A., Horellou P., Dreux M., Gustot T., Duverlie G., Cosset F. L., Vu-Dac N., Dubuisson J. (2006). High-density lipoproteins reduce the neutralizing effect of hepatitis C virus (HCV)-infected patient antibodies by promoting HCV entry J Gen Virol 872577–2581 10.1099/vir.0.81932-0. [DOI] [PubMed] [Google Scholar]
- Wedemeyer H., Dore G. J., Ward J. W. (2015). Estimates on HCV disease burden worldwide – filling the gaps J Viral Hepat 22 (Suppl. 1), 1–5 10.1111/jvh.12371. [DOI] [PubMed] [Google Scholar]
- Welsch C., Domingues F. S., Susser S., Antes I., Hartmann C., Mayr G., Schlicker A., Sarrazin C., Albrecht M., other authors (2008). Molecular basis of telaprevir resistance due to V36 and T54 mutations in the NS3-4A protease of the hepatitis C virus Genome Biol 9R16. 10.1186/gb-2008-9-1-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao F., Fofana I., Thumann C., Mailly L., Alles R., Robinet E., Meyer N., Schaeffer M., Habersetzer F., other authors (2015). Synergy of entry inhibitors with direct-acting antivirals uncovers novel combinations for prevention and treatment of hepatitis C Gut 64483–494 10.1136/gutjnl-2013-306155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M., Iida M., Tada M., Shirasago Y., Fukasawa M., Nagase S., Watari A., Ishii-Watabe A., Yagi K., Kondoh M. (2015). Discovery of anti-claudin-1 antibodies as candidate therapeutics against hepatitis C virus J Pharmacol Exp Ther 353112–118 10.1124/jpet.114.217653. [DOI] [PubMed] [Google Scholar]