

Abstract
Background
The aim of this study was to elucidate whether genetic screening test results of pediatric steroid-resistant nephrotic syndrome (SRNS) patients vary with ethnicity.
Methods
Using high-throughput DNA sequencing, 28 nephrotic syndrome-related genes were analyzed in 110 children affected with SRNS and 10 children with isolated proteinuria enrolled by 5 centers in China (67 males, 53 females). Their age at disease onset was 1 day to 208 months (median, 48.8 months). Patients were excluded if their age of onset of disease was beyond 18 years or if they were diagnosed as Alport’s syndrome.
Results
A genetic etiology was identified in 28.3% of our cohort and the likelihood of establishing a genetic diagnosis decreased as the age of onset of nephrotic syndrome increased. The most common mutated genes were ADCK4 (6.67%), NPHS1 (5.83%), WT1 (5.83%), and NPHS2 (3.33%), and the difference in the frequencies of ADCK4 and NPHS2 mutations between this study and a study on monogenic causes of SRNS in the largest international cohort of 1,783 different families was significant. A case with congenital nephrotic syndrome was attributed to a homozygous missense mutation in ADCK4, and a de novo missense mutation in TRPC6 was detected in a case with infantile nephrotic syndrome.
Conclusions
Our results showed that, in the first and the largest multicenter cohort of Chinese pediatric SRNS reported to date, ADCK4 is the most common causative gene, whereas there is a low prevalence of NPHS2 mutations. Our data indicated that the genetic testing results for pediatric SRNS patients vary with different ethnicities, and this information will help to improve management of the disease in clinical practice.
Keywords: children, Chinese, gene, steroid-resistant nephrotic syndrome
Introduction
In childhood, the most common form of nephrotic syndrome (NS), characterized by massive proteinuria, hypoalbuminemia, edema and hyperlipidemia, is idiopathic NS. Most of children with idiopathic NS are steroid responsive and have a favorable outcome. However, approximately 10~20% of the children with idiopathic NS do not respond to steroid therapy, called steroid-resistant NS (SRNS), and 20~40% of these patients will progress to end-stage renal disease (ESRD) [1–5]. Focal segmental glomerulosclerosis (FSGS) is most frequently seen in SRNS. Advances in molecular genetics have revealed that a considerable proportion of cases with SRNS are caused by single-gene mutations leading to profound podocyte dysfunction, and these cases are usually also resistant to immunosuppressive agents [6–7]. Therefore, in clinical practice, it is important to identify patients with genetic causes of SRNS to provide personalized therapy.
To date, there are approximately 30 genes known to cause SRNS and/or FSGS with autosomal recessive, autosomal dominant, and X-linked recessive modes of inheritance, and genetic SRNS can be isolated or syndromic [8–11]. Because of the heterogeneous nature of SRNS, several mutated gene prioritization strategies have been developed based on age at onset, family history, or renal histology [8, 12–13]. However, genetic testing using the conventional Sanger method is quite laborious and expensive. To simultaneously screen multiple genes related to SRNS/FSGS, some recent studies have shown that targeted next generation sequencing (NGS) is a cost-effective strategy, allowing for a more complete characterization of correlations between genotype and phenotype as well as between genotype and treatment responses [7, 14–17]. However, it is not clear whether the frequencies of damaged alleles vary among different ethnicities in children with SRNS, which will influence the strategy for genetic testing. The aim of the present study was to elucidate whether the genetic testing results of pediatric SRNS vary with ethnicity.
Materials and Methods
Patients
The patients were recruited between 2012 and 2014 by pediatric nephrologists from 5 centers in China (two in eastern China, one in southern China, one in central China and one in northern China) based on fulfillment of one of the following two criteria: i) steroid-resistant idiopathic NS (including those that occurred in the first year of life), and ii) proteinuria with FSGS or a positive family history of proteinuria. Patients were excluded if their age of onset of disease was beyond 18 years or if they were diagnosed with Alport’s syndrome (based on meeting at least one of the following four criteria: i) abnormal staining of the type IV collagen α5 chain in the epidermal basement membrane; ii) abnormal staining of type IV collagen α chains in the glomerular basement membrane; iii) mutations of COL4A3, COL4A4, or COL4A5; and iv) ultrastructural changes in the glomerular basement membrane typical of Alport’s syndrome). The procedures were approved by the ethics committees of the 5 centers. After receiving informed consent from the patients or their family members, blood samples and comprehensive clinical data were collected and analyzed. When available, relatives’ DNAs samples were obtained.
NS was defined by urinary protein excretion ≥50 mg/kg per 24 hours or urine protein/creatinine > 3 mg/mg with hypoalbuminemia. Steroid resistance was defined by persistent proteinuria after 8 weeks of daily 2 mg/kg prednisone treatment (maximum dose 60 mg/day). Patients with a positive family history of proteinuria and/or renal failure were defined as familial cases, otherwise patient were defined as sporadic cases.
Targeted NGS and data analyses
Using a QIAamp DNA Blood Mini Kit (Qiagen), genomic DNA was isolated from peripheral blood leukocytes in all participants. Using the Nimblegen SeqCap EZ Choice system, targeted sequencing capture DNA probes were designed for exons and the flanking 30bp intronic sequences of 28 known NS genes (ADCK4, ARHGDIA, CD2AP, CFH, COQ2, COQ6, CUBN, DGKE, ITGA3, ITGB4, LAMB2, MEFV, MYO1E, NPHS1, NPHS2, PDSS2, PLCE1, PTPRO, SCARB2, SMARCAL1, TTC21B, ACTN4, ARHGAP24, INF2, LMX1B, PAX2, TRPC6, and WT1). Targeted sequencing was performed on the HiSeq2000 or Hiseq2500 platform (Illumina) as paired end 100 bp reads (PE100) by BGI-Tianjin, China.
Image analyses, error estimation and base calling were performed using the Illumina Pipeline (version 1.3.4) to generate the primary sequence data. Low-quality reads and potential adaptor contaminations were removed from the primary data using a local algorithm. The clean short-reads were mapped to the human genome (hg19) using BWA software (Burrows Wheeler Aligner http://sourceforge.net/projects/bio-bwa/). SOAPsnp software (http://soap.genomics.org.cn/) and SAM tools Pileup software (http://sourceforge.net/projects/samtools/) were used to detect single nucleotide variants and small insertion and deletions, respectively. Copy number variants (CNV) were identified by using a BGI in-house pipeline called batCNV1.0. All variants were annotated using a BGI in house developed annotation pipeline (available on demand).
Variants with support reads <10 and a ratio <10% were removed, and variants with a minor allele frequency ≥ 1% in the control database such as NCBI dbSNP build141 (http://www.ncbi.nlm.nih.gov/SNP/), 1000 Genomes Project (http://www.1000genomes.org/), Exome sequencing project (ESP6500. http://evs.gs.washington.edu/EVS/), Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/) and BGI in-house database, were filtered out as polymorphisms. The functional significance of unpublished variants was predicted using SIFT, PolyPhen 2 and Condel. When no prediction scores were available, Mutation Taster was used. Splice site effect prediction was performed using Human Splicing Finder (Version 3.0). The evolutionary conservation of the variant sites was evaluated using the PhyloP Primates tool. Variants were interpreted based on the American College of Medical Genetics and Genomics (ACMG) recommended standards [18–19].
As well as being consistent with the reported inheritance pattern, the candidate variants were considered disease-causing based on meeting at least one of the following criteria: (1) truncating mutations (nonsense, obligatory splice site, and frameshift), (2) variants previously described as disease causing in a patient with a similar phenotype, or (3) amino acid substitutions that exhibited high evolutionary conservation (PhyloP Primates Score >1.5) and two of four prediction scores classified the allele as disease causing: SIFT, Mutation Taster, Polyphen 2 prediction Humvar Score 0.8–1, or Condel Score >0.5.
Validation of disease-causing mutations and segregation analysis
All disease-causing mutations identified with NGS were confirmed in probands by conventional PCR and Sanger sequencing using an ABI 3730XL automatic sequencer (SinoGenoMax, China), and segregation analyses was performed for first degree relatives whenever their DNA was available.
Since parental DNA samples were unavailable, segregation analyses were performed with molecular cloning followed by Sanger sequencing of relevant clones in one patient with compound heterozygous missense variants in SMARCAL1 (the protocol and primer information are available upon request).
Results
A total of 120 unrelated patients (67 males, 53 females) from 24 provinces, municipalities, and autonomous regions in China, were included. Almost all of them were of Han Chinese ethnicity, with one patient from Uighur. Their median age at disease onset was 48.8 (range 1 day to 208 months) months old. Of these patients, 110 cases were diagnosed with SRNS, and 10 cases had isolated proteinuria. The clinical characteristics are summarized in Table 1.
Table 1.
clinical feature of 120 patients in the present study
| Parameter | Age of onset | ||||
|---|---|---|---|---|---|
|
| |||||
| ≤3 mo | 4 to 12 mo | 13 mo to 5 yr | 6 to 12 yr | 13 to 17 yr | |
| Patients with steroid-resistant nephrotic syndrome | |||||
| Patients (n) | 12 | 16 | 50 | 31 | 1 |
| Renal biopsy (n) | (n=1) | (n=7) | (n=32) | (n=30) | IgM nephropathy |
| FSGS:1 | FSGS:5 | FSGS:17 | FSGS:14 | ||
| MCD:1 | MCD:10 | MCD:5 | |||
| IgM nephropathy:1 | MsPGN:3 | MN:5 | |||
| C3 nephropathy:1 | MsPGN:2 | ||||
| IgM nephropathy:1 | sclerosing glomerulonephritis:2 | ||||
| IgA nephropathy:1 | |||||
| MPGN:1 | |||||
| Follow up time, mo | (n=6) | (n=10 ) | (n=45) | (n=30) | |
| 1–204 (median: 39.3) | 1–60 (median: 18.6) | 1–159 (median:37.7) | 1.5–156 (median: 34.3) | 26 | |
| 2 progressed to ESRD and died at the age of 1 yr | 8 progressed to ESRD | 6 progressed to ESRD | increased serum creatine level | ||
| 3 died | 1 had increased serum creatine level | 4 had increased serum creatine level | 7 had increased serum creatine level | ||
| Familial patients, no. (%) | 2 (16.7%) | 0 | 5 (10%) | 5 (16.1%) | 0 |
| Sporadic patients, no. (%) | 10 (83.3%) | 13 (81.2%) | 38 (76%) | 25 (80.6%) | 1 (100%) |
| ND patients, no. (%) | 0 | 3 (18.8%) | 7 (14%) | 1 (3.2%) | 0 |
| Patients with isolated proteinuria | |||||
| Patients (n) | 0 | 2 | 4 | 3 | 1 |
| Renal biopsy (n) | 0 | (n=2 ) | (n=2 ) | (n=3 ) | MsPGN |
| FSGS:2 | sclerosing glomerulonephritis:1 MsPGN:1 |
FSGS:3 | |||
| Follow up time, mo | 0 | (n=2 ) | (n=4) | (n=3) | (n=1) |
| 4, 28 | 5, 23, 48, 48 | 1, 12, 72 | 18 | ||
| Normal renal function | 1 died | Normal renal function | Normal renal function | ||
| Familial patients, no. (%) | 0 | 1 (50%) | 4 (100%) | 1 (33.3%) | 1 (100%) |
| Sporadic patients, no. (%) | 0 | 0 | 0 | 2 (66.7%) | 0 |
| ND patients, no. (%) | 0 | 1 (50%) | 0 | 0 | 0 |
mo, month; yr, year; FSGS, focal segmental glomerulosclerosis; MCD, minimal change disease; MsPGN: mesangioproliferative glomerulonephritis; MN: membranous glomerulonephritis; MPGN, membranoproliferative glomerulonephritis; ESRD, end stage renal disease; Familial, a positive family history with proteinuria and/or renal failure; Sporadic, a negative family history with proteinuria and/or renal failure; ND, no data of a family history.
Gene analysis
Forty-two functionally significant mutations in 10 of the 28 targeted genes, including 20 missense, 11 nonsense, 8 splice site and 3 small insertions, were identified in 34 of the 120 patients, giving a mutation detection rate of 28.3%. Of the 42 mutations identified, 25 (59.5%) were novel, and the remaining 17 mutations had been reported elsewhere [12, 20–35]. None of the 34 patients were from consanguineous families, whereas 35.3% of them (12 patients) had a positive family history of proteinuria, and 2.9% (1 patient) had a positive family history of renal failure. Pathogenic mutations were detected in 68.4% of 19 familial patients (13 patients) and 23.6% of 89 sporadic patients (21 patients). Of 24 patients harboring disease-causing mutations in recessive NS genes, homozygous mutations were found in 6 patients and compound heterozygous mutations were found in 18 patients. A description of all of the detected pathogenic mutations and the clinical phenotypes of the patients are given in Tables 2–3 and Supplemental Table 1.
Table 2.
Genotype and phenotype of mutations in recessive steroid-resistant nephrotic syndrome -causing genes detected in Chinese children with steroid-resistant nephrotic syndrome#
| patient | Nucleotide alteration | Deduced protein change | Location (Zygosity, segregation) | Gender | Age at onset | kidney disease | Biopsy (at age) | Extrarenal presentations | Follow up (at age) | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| NPHS1 (accession no: NM_004646.3) | ||||||||||
| 22 | c.313G>A | p.Asp105Asn | EX3 (het) | F | 5 mo | NS | MCD(6 mo) | No | Normal renal function (10 mo) | 20 |
| c.385C>T | p.Leu129Phe | EX3 (het) | 21 | |||||||
| 1 | c.802C>T | p.R268* | EX7 (het) | F | 1d | NS | ND | Atrial septal defect | Died | 22 |
| c.3325C>T | p.Arg1109* | EX26 (het) | 23 | |||||||
| 6 | c.928G>A | p.Asp310Asn | EX8 (het, m) | M | 13d | SRNS | ND | Umbilical hernia | ND | 24 |
| c.1931-1_1931insT | p.Tyr644Leufs*30 | EX15 (het, f) | Novel | |||||||
| 10 | c.967G>T | p.Val323Leu | EX8 (het, m) | F | 34d | SRNS | FSGS(8 yrs, 15 yrs) | No | Normal renal function (17 yrs) | Novel |
| c.1570G>A | p.Ala524Thr | EX12 (het, f) | Novel | |||||||
| 2 | c.2663G>A | p.Arg888Lys | EX19 (het, m) | M | 2d | NS | ND | Small for gestational age | ND | Novel |
| c.3286+5G>A | Splice | IVS24 (het, f) | Novel | |||||||
| 8 | c.3027C>G | p.Tyr1009* | EX22 (het) | M | 19d | NS | ND | Premature infant, enlarged placenta | ND | Novel |
| c.3478C>T | p.Arg1160* | EX27 (het) | 25 | |||||||
| 9 | c.3118C>T | p.Gln1040* | EX23 (het, m) | M | 24d | NS | ND | Bilateral inguinal hernia, enlarged placenta | Died (1 yr) | Novel |
| c.3325C>T | p.Arg1109* | EX26 (het, f) | 23 | |||||||
| NPHS2 (accession no: NM_014625.2) | ||||||||||
| 34 | c.211C>T | p.Arg71* | EX1 (hom) | M | 3 yrs 5 mo | SRNS | FSGS(3 yrs 9 mo) | Right inguinal hernia | ESRD (10 yrs 4 mo) | 26 |
| 29 | c.211C>T | p.Arg71* | EX1 (het, f) | F | 2 yrs 5 mo | SRNS | ND | ND | ESRD (7 yrs 8 mo) | 26 |
| c.467_468insT | p.Leu156Phefs*11 | EX4 (het, m) | 27 | |||||||
| 32 | c.412C>T | p.Arg138* | EX3 (het) | F | 2 yrs 10 mo | SRNS | ND | ND | ND | 28 |
| c.535-1G>A | splice | IVS4 (het) | Novel | |||||||
| 41 | c.467_468insT | p.Leu156Phefs*11 | EX4 (het, f) | M | 2 yrs | SRNS | ND | Left testiculus dysplasia | ESRD (6 yrs 1 mo) | 27 |
| c.738+2T>C | splice | IVS5 (het, m) | Novel | |||||||
| ADCK4 (accession no: NM_024876.3) | ||||||||||
| 119 | c.241G>T | p.Glu81* | EX4 (het, m) | M | 11 yrs | nephrotic-level proteinuria, steroid resistant | FSGS (11 yrs) | Low level of of serum C3 concentration | Normal renal function (12 yrs) | Novel |
| c.1468C>T | p.Arg490Cys | EX15 (het, f) | Novel | |||||||
| 83 | c.448C>T | p.Arg150* | EX6 (het, m) | F | 8 yrs | SRNS | FSGS(10 yrs 9 mo) | ND | ESRD (11 yrs 8 mo) | Novel |
| c.748G>C | p.Asp250His | EX9 (het, f) | 29 | |||||||
| 117 | c.532C>T | p.Arg178Trp | EX7 (het, m) | F | 9 yrs 2 mo | SRNS | Sclerosing glomerulonephritis (9 yrs 2 mo) | ND | ESRD (11 yrs) | 30 |
| c.748G>C | p.Asp250His | EX9 (het, f) | 29 | |||||||
| 82 | c.737G>A | p.Ser246Asn | EX9 (hom, f, m) | F | 8 yrs 2 mo | SRNS | FSGS(8 yrs 10 mo) | ND | Normal renal function (9 yrs 2 mo) | Novel |
| 120 | c.737G>A | p.Ser246Asn | EX9 (hom, f, b) | F | 17 yrs 4 mo | proteinuria | MsPGN (18 yrs 10mo) | ND | Normal renal function (18 yrs 10 mo) | Novel |
| 4 | c.748G>C | p.Asp250His | EX9 (hom, f, m) | F | 10d | NS | ND | ND | ND | 29 |
| 30 | c.748G>C | p.Asp250His | EX9 (hom, f, m) | F | 1 yr 7 mo | SRNS | FSGS(1 yr 7 mo) | ND | ESRD (6 yrs) | 29 |
| 16 | c.748G>C | p.Asp250His | EX9 (het, m) | F | 6 yrs | proteinuria | FSGS(10 yrs) | ND | Normal renal function (12 yrs) | 29 |
| c.1093C>G | p.Gln365Glu | EX12 (het, f) | Novel | |||||||
| CUBN (accession no: NM_001081.3) | ||||||||||
| 53 | c.4837C>T | p.Arg1613* | EX32 (het, m) | F | 1 yr 6 mo | SRNS | MsPGN(1 yr 6 mo) | ND | Normal renal function (11 yrs 6 mo) | Novel |
| c.8938G>A | p.Asp2980Asn | EX57 (het, f) | Novel | |||||||
| 36 | c.7561C>G | p.Pro2521Ala | EX49 (het, m) | F | 2 yrs 8 mo | SRNS | ND | ND | ESRD (2 yrs 10 mo) | Novel |
| c.9335T>C | p.Phe3112Ser | EX59 (het, f) | Novel | |||||||
| LAMB2 (accession no: NM_002292.3) | ||||||||||
| 7 | c.2044_2045insTT | p.Cys682Phefs*13 | EX16 (hom, m, f) | F | 19d | NS | ND | Premature infant, bilateral myosis | ND | Novel |
| PLCE1 (accession no: NM_016341.3) | ||||||||||
| 13 | c.4301G>A | p.Arg1434Gln | EX17 (het, m) | M | 7 mo | SRNS | FSGS(1 yr 3 mo) | Movement development retardation | ESRD (11 mo) | Novel |
| c.4852G>T | p.Glu1618* | EX21 (het, f) | Novel | |||||||
| SMARCAL1 (accession no: NM_014140.3) | ||||||||||
| 66 | c.2443G>A | p.Glu815Lys | EX16 (het, not located in the same allele with p.Arg817His) | F | 5 yrs 11 mo | SRNS | FSGS(6 yrs) | Scattered cafe-au-lait-spots in right armpit and abdomen, growth retardation | Normal renal function (6 yrs 2 mo) | Novel |
| c.2450G>A | p.Arg817His | EX16 (het, not located in the same allele with p.Glu815Lys ) | Novel | |||||||
1. Patient 7 was Uighur, and the remainders were Han Chinese ethnicity.
2. Patients 6, 29, 32. 83, 117, 82, 120, 4, 30 and 16 had a positive family history with proteinuria and/or renal failure, and the remainders had a negative family history with proteinuria and/or renal failure.
3. Except that disease did not reoccur after patient 117 underwent renal transplantation, no data of disease recurrence if transplanted in other patients were available.
Ref, Reference; EX, Exon; IVS, Intron; het, heterozygous; hom, homozygous; F, female; M, male; f: father; m: mother; b: brother; d, day; mo, month; yrs, years; NS, nephrotic syndrome; SRNS, steroid nephrotic syndrome; MCD, minimal change disease; FSGS, focal segmental glomerulosclerosis; MsPGN, mesangial proliferative glomerulonephritis; ND, no data or not done; ESRD, end-stage renal disease.
Table 3.
Genotype and phenotype of mutations in dominant steroid-resistant nephrotic syndrome -causing genes detected in Chinese children with steroid-resistant nephrotic syndrome#
| patient | Nucleotide alteration | Deduced protein change | Location (Zygosity, segregation) | Gender | Age at onset | kidney disease | Biopsy (at age) | Extrarenal presentations | Follow up (at age) | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| WT1 (accession no: NM_024426.4) | ||||||||||
| 3 | c.1339+1G>C(de novo) | Splice | IVS8 (het) | F | 6 d | NS | ND | ND | Died (1 mo) | Novel |
| 25 | c.1339+5G>A(de novo) | Splice | IVS8 (het) | M | 7 mo | SRNS | ND | Male pseudo-hermaphroditis, cryptorchidism, karyotype was 46, XY | ND | 12 |
| 27 | c.1384C>T(de novo) | p.Arg462Trp | EX9 (het) | M | 9 mo | SRNS | ND | Hypospadias,cryptorchidism, right preauricular fistula | ND | 31 |
| 75 | c.1432+1G>A | splice | IVS9 (het) | F | 1yr 1 mo | SRNS | FSGS (1 yr 7 mo) | ND | ND | 32 |
| 35 | c.1432+4C>T | splice | IVS9 (het) | F | 4 yrs 8 mo | SRNS, right kidney dysplasia, left renal cyst | ND | ND | CKD stage 2 (6 yrs 2 mo) | 33 |
| 33 | c.1432+5G>A | splice | IVS9 (het) | F | 2 yrs 4 mo | SRNS | FSGS (4 yrs) | ND | Normal renal function (3 yrs 4 mo) | 34 |
| 78 | c.1432+5G>A | splice | IVS9 (het) | F | 1 yr 11 mo | SRNS | ND | ND | Normal renal function (3 yrs 2 mo) | 34 |
| LMX1B (accession no: NM_001174146.1) | ||||||||||
| 113 | c.737G>A | p.Arg246Gln | EX4 (het, m ) | F | 1yr 1mo | proteinuria | ND | ND | Normal renal function (5 yrs 1 mo) | 35 |
| 116 | c.737G>A | p.Arg246Gln | EX4 (het, f ) | M | 5 yrs 2 mo | proteinuria | MsPGN (5 yrs 3 mo) | ND | Normal renal function (7 yrs 1 mo) | 35 |
| TRPC6 (accession no: NM_004621.5) | ||||||||||
| 15 | c.523C>T(de novo) | p.Arg175Trp | EX2 (het) | M | 4 mo | SRNS | FSGS (1 yr 4 mo) | ND | Normal renal function (1 yr 4 mo) | Novel |
1. Patients listed in this table were Han Chinese ethnicity, and no data of disease recurrence if transplanted were available. Patients 33, 113 and 116 had a positive family history with proteinuria and/or renal failure, and the remainders had a negative family history with proteinuria and/or renal failure.
2. PCR amplification of homo sapiens sex determining region Y (SRY, accession no: NM_003140) was performed in DNA samples for patients 3, 27, 33, 35, 75 and 78, and agarose gel electrophoresis showed positive for patients 27, 33 and 35, whereas negative for patients 3, 75 and 78.
Ref, Reference; EX, Exon; IVS, Intron; het, heterozygous; F, female; M, male; m: mother; f: father; d, day; mo, month; yrs, years; NS: nephrotic syndrome, SRNS: steroid nephrotic syndrome; ND, no data or not done; FSGS, focal segmental glomerulosclerosis; MsPGN, mesangial proliferative glomerulonephritis; CKD, chronic kidney disease.
In patients with disease onset before age 13 years, the likelihood of establish genetic diagnosis decreased as the age of onset for NS increased (Figure 1). We detected the disease-causing mutation in 75% of children in the first 3 months of life, 27.8% at age 4–12 months, 25.9% at age 13 months–5 years, and 14.7% at age 6–12 years. Meanwhile, the distribution of the detected causative genes according to age at onset of NS is given in Table 4 and Figure 1. Mutations in NPHS1, WT1, LAMB2 and PLCE1 were responsible for 12 of the 30 patients (40%) with NS onset within the first year of life, and NPHS1 was the most frequent causative gene. Mutations in ADCK4 and TRPC6, but not NPHS2, were also responsible for this form of SRNS. WT1 mutations were detected in 7 of the 84 patients (8.3%) of NS with onset within 5 years, which could be used to help diagnose Denys-Drash syndrome in Patients 25 and 27, Frasier syndrome in Patients 33 and 35, and isolated NS in Patients 3, 77 and 78. ADCK4 was the most frequent genetic basis for pediatric SRNS, especially for SRNS with onset at age 6–12 years (14.7%).
Fig. 1.
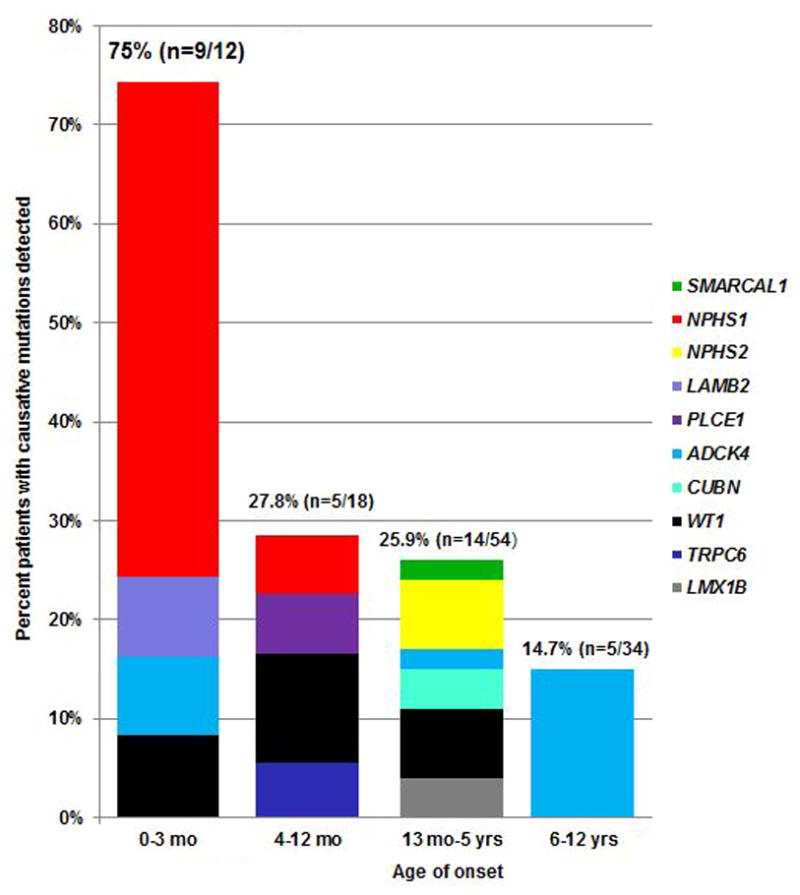
Percentage of patients with genetic diagnose per gene per age group. Histograms indicate fraction (in percentage) of patients with causative gene detected per age group.
Table 4.
Distribution of causative genes among different age of onset groups
| Age of onset | |||||
|---|---|---|---|---|---|
|
|
|||||
| ≤3 mo | 4 to 12 mo | 13 mo to 5 yr | 6 to 12 yr | 13 to 17 yr | |
| Patients (n) | 12 | 18 | 54 | 34 | 2 |
| Causative genes, no (%) | WT1:1 (8.3%) | TRPC6:1 (de novel, 5.6%) | WT1:4 (7.4%) | ADCK4:5 (14.7%) | ADCK4:1 (50%) |
| NPHS1:6 (50%) | WT1:2 (11.1%) | LMX1B: 2 (3.7%) | |||
| ADCK4:1 (8.3%) | NPHS1:1 (5.6%) | NPHS2: 4 (7.4%) | |||
| LAMB2:1 (8.3%) | PLCE1:1 (5.6%) | CUBN: 2 (3.7%) | |||
| ADCK4:1 (1.9%) | |||||
| SMARCAL1:1 (1.9%) | |||||
mo, month; yr, year.
Sadowski et al. [16] published the results of monogenic causes of SRNS in the largest international cohort of 1,783 different families (2,016 individuals). We therefore compared the mutation detection rate and most common causative genes between Sadowski’s study and our study. Except for the NEIL1 gene, the targeted NGS panel used by the present study included genes analyzed in Sadowski’s study and two additional genes (TTC21B and PAX2). The top 3 frequently mutated genes in Sadowski’s study were NPHS2 (9.93%. 95% CI =8.58~11.41%), NPHS1 (7.34%. 95% CI =6.18~8.66%), and WT1 (4.77%. 95% CI =3.83~5.86%), whereas ADCK4 was the least common mutated gene (0.17%. 95% CI =0.03~0.5%); in our study, the most common mutated genes were ADCK4 (6.67%. 95% CI =2.92~12.71%), NPHS1 (5.83%. 95% CI =2.38~11.65%), WT1 (5.83%. 95% CI =2.38~11.65%), and NPHS2 (3.33%, 95% CI =0.92~8.31%). The difference in the frequencies of ADCK4 and NPHS2 mutations between these two studies was significant (no overlap of the 95% CI between different studies was considered significant, P < 0.05).
Renal biopsy
Seventy-nine of the 120 patients (65.8%) underwent renal biopsy (Tables 1 and 5), and 42 patients had FSGS. Genetic alterations were found in 12 of the 42 patients with FSGS (28.6%) and 3 of the 7 patients with mesangioproliferative glomerulonephritis. Of 34 patients with a single-gene cause, 17 patients underwent renal biopsy and diffuse mesangial sclerosis was not observed (Table 5).
Table 5.
Causative mutation distribution among different renal biopsy findings
| Biopsy pattern | No biopsy performed or no data | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| FSGS | MCD | MsPGN | Sclerosing glomerulonephritis | Others# | ||
| Patients, no. | 42 | 16 | 7 | 3 | 11 | 41 |
| Patients with identified mutation, no. (%) | 12 (28.6%) | 1 (6.3%) | 3 (42.9%) | 1 (33.3%) | 0 | 17 (41.5%) |
FSGS, focal segmental glomerulosclerosis; MCD, minimal change disease; MsPGN: mesangioproliferative glomerulonephritis; #: included 5 patients with membranous glomerulonephritis, 3 patients with IgM nephropathy, 1 patient with C3 nephropathy, 1 patient with IgA nephropathy and 1 patient with membranoproleferative glomerulonephritis.
Genotypic and phenotypic features of ADCK4-related nephropathy (Table 2)
ADCK4 was the most common mutated gene in our study, so we analyzed it’s genotypic and phenotypic characteristics. Five missense mutations (including two previously reported mutations) and two nonsense mutations were identified in 8 patients.
The missense mutation p. Asp250His was found in five patients, either in the homozygous state (Patients 4 and 30) or in compound heterozygosity with another mutation on the second allele (Patients 83, 117 and 16). The age at first feature identified in these patients ranged from 10 days to 9.2 years, with an average age of 5 years. Patients 83, 117, 4 and 30 presented with SRNS, whereas Patient 16 presented only with proteinuria. Patients 83, 117, and 30 developed ESRD at age 11.7 years, 11years and 6 years, respectively, whereas Patient 16 had normal renal function at age 12 years. FSGS was diagnosed in Patients 83, 30 and 16, and sclerosing glomerulonephritis was diagnosed in Patient 117.
Patients 82 and 120 were homozygotes for the novel missense mutation p.Ser246Asn. Patient 82 manifested SRNS at age 8.2years, with renal histology showing FSGS, and Patient 120 had proteinuria at age 17.3 years, with the renal biopsy revealing mild mesangial proliferative glomerulonephritis. Both of these patients had normal renal function after a one-year follow up. The older brother of Patient 82 had ESRD and proteinuria (3+) at age 11years, and renal ultrasound examination revealed renal atrophy. He died after 4 months of dialysis. Her parents and a younger brother who carried heterozygous p.Ser246Asn had no renal symptoms. The older brother of Patient 120 had ESRD and proteinuria (3+) at age 21 years, and a renal ultrasound examination showed renal atrophy. He underwent kidney transplantation with their mother as the donor. His kidney function was stable on immunosuppressive drugs at 1 year after transplantation. Segregation analyses showed he was also a homozygote for p.Ser246Asn. The healthy father carried heterozygous p.Ser246Asn, but the mother’s blood sample was not available.
Patient 119 was a compound heterozygote for p.Glu81* from his mother and p.Arg490Cys from his father. He presented with steroid resistant nephrotic-level proteinuria at the age of 11 years, with the renal biopsy showing FSGS. His renal function was in the normal range at the age of 12 years. The serum C3 levels of him and his father were 0.446 g/L and 0.371 g/L (normal range 0.6~1.5 g/L), respectively. Their serum C4 levels were normal. His parents had no renal manifestations.
Discussion
The application of genetic testing in pediatric SRNS has significant implications for the comprehensive management of this disease. Therefore, the present study, by simultaneous screening 28 NS-related genes, characterized the molecular spectrum in the first and largest multicenter cohort of Chinese pediatric SRNS patients reported to date. The most important finding is that Chinese pediatric SRNS patients are genetically different, with the ADCK4 gene being shown to be important.
We identified pathogenic mutations in 28.3% of our cohort, which was very close to the highest previous mutation detection rate in the largest international cohort of SRNS (mutations were detected in 31.2% of families with SRNS manifesting before the age of 18 years)[16]. As in children of other ethnicities with SRNS, the percentage of detecting disease-causing mutations decreased as the age of at onset for NS increased in the present study [12, 16–17]. However, three genotypic features in our series were found: (1) no NPHS2 mutations were detected in congenital or infantile NS, which was consistent with the results of Japanese and Korean pediatric SRNS populations [20, 36], and this was contrary to the results in other reports [12, 16–17, 37]. A homozygous missense mutation in ADCK4 was identified in a case with congenital NS, and a de novo missense mutation in TRPC6 was detected in a case with infantile NS. To our knowledge, these patients are the youngest patients with the disease to be caused by ADCK4 and TRPC6 mutations. Both findings emphasized the necessity to include ADCK4 and TRPC6, but not NPHS2, in the mutation analysis of NS in the first year of life, in addition to NPHS1, WT1, LAMB2 and PLCE1. (2) Mutations in NPHS2 were detected less frequently in the present study than in the largest international cohort of SRNS (3.33% versus 9.93%) and in smaller series of pediatric SRNS (3.33% versus 4.5% to 40%) [12, 16, 27, 38–39]. Furthermore, no NPHS2 mutations were detected in our previous study [40] and in 18 African American children with SRNS [41], whereas a rate of 9.1% NPHS2 mutations was found in 22 Chinese children with SRNS [42]. In contrast to the low frequency of NPHS2 mutations, a surprisingly higher mutation rate of ADCK4 (6.67%) was observed in our series [16, 29, 43]. Therefore, for genetic diagnosis of SRNS with an age of onset beyond 1year, we suggest, when NGS technology is not available, the first step should be to screen for ADCK4. (3) We only detected NPHS1 and WT1 mutations in patients with an age of onset within the first year and 5 years, respectively, whereas a considerable fraction of the largest international cohort of SRNS presenting before age 18 years was found to have mutations in these two genes [16], and in Lipska’s study, WT1 mutations accounted for 3.9% of 227 nonsyndromic SRNS patients with onset of age between 10 to 20 years [44]. All of the above differences in genetics results between this study and previous studies especially the study of the largest international cohort of SRNS, indicated that a small study size may reduce the chance of detecting causative gene mutations, and ethnic differences exists in the frequencies of damaging alleles.
Previously, ADCK4 related glomerulopathy was identified in 0.17% 3.7% of SRNS patients, the disease first presented mostly in 10–20 years old patients, and FSGS/global glomerulosclerosis was observed in all biopsied patients [16, 29–30, 43]. However, in this study, the ADCK4 mutation detection rate was 6.67%, and the age at first feature identified was mostly 6–11 years old. In addition to FSGS and global glomerulosclerosis, mesangial proliferative glomerulonephritis was also observed. Similar to previously reported cases with ADCK4 mutations [29–30], onset with different levels of proteinuria was observed in our cohort, and progression to ESRD could occur before 10 years old. These findings demonstrated the heterogeneous renal phenotype of ADCK4 disease.
This study also had some limitations. First, our cohort was not large. However, study participants in the present study were from 24 of 34 provinces, municipalities, autonomous regions and special administrative regions in China and were enrolled by 5 centers located in eastern China, southern China, central China and northern China. It was the largest multicenter cohort of Chinese pediatric SRNS reported to date, which could balance the sampling bias. Second, patients with ADCK4 or CUBN mutations may be amenable to supplementation with coenzyme Q10 or vitamin B12. However, in our cohort, 4 patients progressed to ESRD before genetic diagnosis, and 6 patients were lost to follow-up at the time of genetic diagnosis. Thus, there was no chance to observe the effect of treatment with coenzyme Q10 or vitamin B12. Third, there were no data on extrarenal manifestations in 19 of the 34 patients (55.9%) with causative mutations, and renal biopsy was less frequently performed in our subjects with an onset age before 5 years, therefore it was difficult to delineate the causative gene associated phenotype and the association of genotype with histopathologic diagnoses completely.
In conclusion, we found that in Chinese pediatric SRNS patients, ADCK4 is the most frequent causative gene, whereas there is a low prevalence of NPHS2 mutations, and infantile-onset nephrotic syndrome can be caused by the TRPC6 mutation. Our data indicated that the genetic screening strategy of pediatric SRNS should vary with ethnicities, which will help to improve management of the disease during clinical practice and provide a valuable reference for genetic testing among different ethnic or racial populations.
Supplementary Material
Acknowledgments
We thank all patients and their families for participating in this study. This work was supported by National “Twelfth Five-Year” Science and Technology Support Project (No. 2012BAI03B02).
References
- 1.Tarshish P, Tobin JN, Bernstein J, Edelmann CM., Jr Prognostic significance of the early course of minimal change nephrotic syndrome: report of the International study of kidney disease in children. J Am Soc Nephrol. 1997;8:769–776. doi: 10.1681/ASN.V85769. [DOI] [PubMed] [Google Scholar]
- 2.Tune BM, Mendoza SA. Treatment of the idiopathic nephrotic syndrome: regimens and outcomes in children and adults. J Am Soc Nephrol. 1997;8:824–832. doi: 10.1681/ASN.V85824. [DOI] [PubMed] [Google Scholar]
- 3.Arbus GS, Poucell S, Bacheyie GS, Baumal R. Focal segmental glomerulosclerosis with idiopathic nephrotic syndrome: three types of clinical response. J Pediatr. 1982;101:40–45. doi: 10.1016/s0022-3476(82)80177-7. [DOI] [PubMed] [Google Scholar]
- 4.Kin JS, Bellew CA, Silverstein DM, Aviles DH, Boineau FG, Vehaskari VM. High incidence of initial and late steroid resistance in childhood nephrotic syndrome. Kidney Int. 2005;68:1275–1281. doi: 10.1111/j.1523-1755.2005.00524.x. [DOI] [PubMed] [Google Scholar]
- 5.Mekahli D, Liutkus a, Ranchin B, Yu A, Bessenay L, Girardin E, Van Damme-Lombaerts R, Palcoux JB, Cachat F, Lavocat MP, Bourdat-Michel G, Nobili F, Cochat P. Long-term outcome of idiopathic steroid resistant nephrotic syndrome: a multicenter study. Pediatr Nephrol. 2009;24:525–532. doi: 10.1007/s00467-009-1138-5. [DOI] [PubMed] [Google Scholar]
- 6.Buscher AK, Kranz B, Buscher R, Hildebrandt F, Dworniczak B, Pennekamp P, Kuwertz-Broking E, Wingen AM, John U, Markus Kemper, Monnens L, Hoyer PF, Weber S, Konrad M. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2010;5:2075–2084. doi: 10.2215/CJN.01190210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giglio S, Provenzano A, Mazzinghi B, Becherucci F, Giunti L, Sansavini G, Ravaglia F, Roperto RM, Farsetti S, Benetti E, Rotondi M, Murer L, Lazzeri E, Lasagni L, Materassi M, Romagnani P. Heterogeneous genetic alterations in sporadic nephrotic syndrome associate with resistance to immunosuppression. J Am Soc Nephrol. 2015;26:230–236. doi: 10.1681/ASN.2013111155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joshi S, Andersen R, Jespersen B, Rittig S. Genetics of steroid-resistant nephrotic syndrome: a review of mutation spectrum and suggested approach for genetic testing. Acta Pædiatrica. 2013;102:844–856. doi: 10.1111/apa.12317. [DOI] [PubMed] [Google Scholar]
- 9.Brown EJ, Pollak MR, Barua M. Genetic testing for nephrotic syndrome and FSGS in the era of next-generation sequencing. Kidney Int. 2014;85:1030–1038. doi: 10.1038/ki.2014.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piscione TD, Licht C. Genetics of proteinuria: an overview of gene mutations associated with nonsyndromic proteinuric glomerulopathies. Adv Chronic Kidney Dis. 2011;18:273–289. doi: 10.1053/j.ackd.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Bierzynska A, Soderquest K, Koziell A. Genes and podocytes-new insights into mechanisms of podocytopathy. Front Endocrinol (Lausanne) 2015;5:226. doi: 10.3389/fendo.2014.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santín S, Bullich G, Tazon-Vega B, García-Maset R, Gimenez I, Silva I, Ruíz P, Ballarín J, Torra R, Ars E. Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2011;6:1139–1148. doi: 10.2215/CJN.05260610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benoit G, Machuca E, Antignac C. Hereditary nephrotic syndrome: a systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr Nephrol. 2010;25:1621–1632. doi: 10.1007/s00467-010-1495-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCarthy HJ, Bierzynska A, Wherlock M, Ognjanovic M, Kerecuk L, Hegde S, Feather S, Gilbert RD, Krischock L, Jones C, Sinha MD, Webb NJA, Christian M, Williams MM, Marks S, Koziell A, Welsh GI, Saleem MA. Simultaneous sequencing of 24 genes associated with steroid-Resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2013;8:637–648. doi: 10.2215/CJN.07200712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lovric S, Fang H, Vega-Warner V, Sadowski CE, Gee HY, Halbritter J, Shazia Ashraf, Saisawat P, Soliman NA, Kari JA, Otto EA, Hildebrandt F. Rapid detection of monogenic causes of childhood onset steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2014;9:1109–1116. doi: 10.2215/CJN.09010813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, Engelmann S, Vega-Warner V, Fang H, Halbritter J, Somers MJ, Tan W, Shril S, Fessi I, Lifton RP, Bockenhauer D, El-Desoky S, Kari JA, Zenker M, Kemper MJ, Mueller D, Fathy HM, Soliman NA, Hildebrandt F SRNS Study Group. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2015;26:1279–1289. doi: 10.1681/ASN.2014050489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bullich G, Trujillano D, Santín S, Ossowski S, Mendizábal S, Fraga G, Madrid Á, Ariceta G, Ballarín J, Torra R, Estivill X, Ars E. Targeted next-generation sequencing in steroidresistant nephrotic syndrome: mutations in multiple glomerular genes may influence disease severity. Eur J Hum Genet. 2015;23:192–199. doi: 10.1038/ejhg.2014.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Heqde MR, Lyon E, Ward BE Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 19.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. Standards and Guidelines for the Interpretation of Sequence Variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the association for molecular pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sako M, Nakanishi K, Obana M, Yata N, Hoshii S, Takahashi S, Wada N, Takahashi Y, Kaku Y, Satomura K, Ikeda M, Honda M, Iijima K, Yoshikawa N. Analysis of NPHS1, NPHS2, ACTN4, and WT1 in Japanese patients with congenital nephrotic syndrome. Kidney Int. 2005;67:1248–1255. doi: 10.1111/j.1523-1755.2005.00202.x. [DOI] [PubMed] [Google Scholar]
- 21.Schultheiss M, Ruf RG, Mucha BE, Wiggins R, Fuchshuber A, Lichtenberger A, Hildebrandt F. No evidence for genotype/phenotype correlation in NPHS1 and NPHS2 mutations. Pediatr Nephrol. 2004;19:1340–1348. doi: 10.1007/s00467-004-1629-3. [DOI] [PubMed] [Google Scholar]
- 22.Ulinski T, Aoun B, Toubiana J, Vitkevic R, Bensman A, Donadieu J. Neutropenia in congenital nephrotic syndrome of the Finnish type: role of urinary ceruloplasmin loss. Blood. 2009;113:4820–4821. doi: 10.1182/blood-2009-02-204099. [DOI] [PubMed] [Google Scholar]
- 23.Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A. Positionally cloned gene for a novel glomerular protein-nephrin-is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- 24.Shi Y, Ding J, Liu JC, Wang H, Bu DF. NPHS1 mutations in a Chinese family with congenital nephrotic syndrome. Zhonghua Er Ke Za Zhi. 2005;43:805–809. [PubMed] [Google Scholar]
- 25.Lenkkeri U, Mannikko M, McCready P, Lamerdin J, Gribouval O, Niaudet P, Antignac C, Kashtan CE, Holmberg C, Kestila M, Tryggvason K. Structure of the gene for congenital nephrotic syndrome of the finnish type (NPHS1) and characterization of mutations. Am J Hum Genet. 1999;64:51–61. doi: 10.1086/302182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun H, Zhou W, Wang J, Yin L, Lu Y, Fu Q. A novel mutation in NPHS2 gene identified in a Chinese pedigree with autosomal recessive steroid-resistant nephrotic syndrome. Pathology. 2009;41:661–665. doi: 10.3109/00313020903273118. [DOI] [PubMed] [Google Scholar]
- 27.Caridi G, Bertelli R, Carrea A, Di Duca M, Catarsi P, Artero M, Carraro M, Zennaro C, Candiano G, Musante L, Seri M, Ginevri F, Perfumo F, Ghiggeri GM. Prevalence, genetics, and clinical features of patients carrying podocin mutations in steroid-resistant nonfamilial focal segmental glomerulosclerosis. J Am Soc Nephrol. 2001;12:2742–2746. doi: 10.1681/ASN.V12122742. [DOI] [PubMed] [Google Scholar]
- 28.Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, Dahan K, Gubler MC, Niaudet P, Antignac C. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24:349–354. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 29.Korkmaz E, Lipska-Zietkiewicz BS, Boyer O, Gribouval O, Fourrage C, Tabatabaei M, Schnaidt, Gucer S, Kaymaz F, Mustafa Arici, Dinckan A, Mir S, Bayazit AK, Emre S, Ayse Balat, Rees L, Rukshana Shroff, Bergmann C, Mourani C, Antignac C, Ozaltin F, Schaefer F the PodoNet Consortium. ADCK4-associated glomerulopathy causes adolescence-onset FSGS. J Am Soc Nephrol. 2016;27:63–68. doi: 10.1681/ASN.2014121240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, Fang H, Song X, Cattran DC, Avila-Casado C, Paterson AD, Nitschké P, Bole-Feysot C, Cochat P, Esteve-Rudd J, Haberberger B, Allen SJ, Zhou W, Airik R, Otto EA, Barua M, Al-Hamed MH, Kari JA, Evans J, Bierzynska A, Saleem MA, Böckenhauer D, Kleta R, El Desoky S, Hacihamdioglu DO, Gok F, Washburn J, Wiggins RC, Choi M, Lifton RP, Levy S, Han Z, Salviati L, Prokisch H, Williams DS, Pollak M, Clarke CF, Pei Y, Antignac C, Hildebrandt F. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest. 2013;123:5179–5189. doi: 10.1172/JCI69000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pelletier J, Bruening W, Kashtan CE, Mauer M, Manivel JC, Striegel JE, Houghton DC, Junien C, Habib R, Fouser L, Fine RN, Silveman BL, Haber DA, Housman D. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell. 1991;67:437–447. doi: 10.1016/0092-8674(91)90194-4. [DOI] [PubMed] [Google Scholar]
- 32.Miyoshi Y, Santo Y, Tachikawa K, Namba N, Hirai H, Mushiake S, Nakajima S, Michigami T, Ozono K. Lack of puberty despite elevated estradiol in a 46,XY phenotypic female with Frasier syndrome. Endocr J. 2006;53:371–376. doi: 10.1507/endocrj.k05-180. [DOI] [PubMed] [Google Scholar]
- 33.Barbaux S, Niaudet P, Gubler MC, Grünfeld JP, Jaubert F, Kuttenn F, Fekete CN, Souleyreau-Therville N, Thibaud E, Fellous M, McElreavey K. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet. 1997;17:467–470. doi: 10.1038/ng1297-467. [DOI] [PubMed] [Google Scholar]
- 34.Bruening W, Bardeesy N, Silverman BL, Cohn RA, Machin GA, Aronson AJ, Housman D, Pelletier J. Germline intronic and exonic mutations in the Wilms’ tumour gene (WT1) affecting urogenital development. Nat Genet. 1992;1:144–148. doi: 10.1038/ng0592-144. [DOI] [PubMed] [Google Scholar]
- 35.Boyer O, Woerner S, Yang F, Oakeley EJ, Linghu B, Gribouval O, Tête MJ, Duca JS, Klickstein L, Damask AJ, Szustakowski JD, Heibel F, Matignon M, Baudouin V, Chantrel F, Champigneulle J, Martin L, Nitschké P, Gubler MC, Johnson KJ, Chibout SD, Antignac C. LMX1B mutations cause hereditary FSGS without extrarenal involvement. J Am Soc Nephrol. 2013;24:1216–1222. doi: 10.1681/ASN.2013020171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho HY, Lee JH, Choi HJ, Lee BH, Ha IS, Choi Y, Cheong HI. WT1 and NPHS2 mutations in Korean children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2008;23:63–70. doi: 10.1007/s00467-007-0620-1. [DOI] [PubMed] [Google Scholar]
- 37.Hinkes BG, Mucha B, Vlangos CN, Gbadegesin R, Liu JH, Hasselbacher K, Hangan D, Ozaltin F, Zenker M, Hildebrandt F. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2) Pediatrics. 2007;119:e907–919. doi: 10.1542/peds.2006-2164. [DOI] [PubMed] [Google Scholar]
- 38.Frishberg Y, Rinat C, Megged O, Shapira E, Feinstein S, Raas-Rothschild A. Mutations in NPHS2 encoding podocin are a prevalent cause of steroid-resistant nephrotic syndrome among Israeli-Arab children. J Am Soc Nephrol. 2002;13:400–405. doi: 10.1681/ASN.V132400. [DOI] [PubMed] [Google Scholar]
- 39.Ogino D, Hashimoto T, Hattori M, Sugawara N, Akioka Y, Tamiya G, Makino S, Toyota K, Mitsui T, Hayasaka K. Analysis of the genes responsible for steroid-resistant nephrotic syndrome and/or focal segmental glomerulosclerosis in Japanese patients by whole exome sequencing analysis. J Hum Genet. 2016;61:137–141. doi: 10.1038/jhg.2015.122. [DOI] [PubMed] [Google Scholar]
- 40.Yu ZH, Ding J, Huang JP, Yao Y, Xiao HJ, Zhang JJ, Liu JC, Yang JY. Mutations in NPHS2 in sporadic steroid-resistant nephrotic syndrome in Chinese children. Nephrol Dial Transplant. 2005;20:902–908. doi: 10.1093/ndt/gfh769. [DOI] [PubMed] [Google Scholar]
- 41.Chernin G, Heeringa SF, Gbadegesin R, Liu JH, Hinkes BG, Vlangos CN, Vega-Warner V, Hildebrandt F. Low prevalence of NPHS2 mutations in African American children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2008;23:1455–1460. doi: 10.1007/s00467-008-0861-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mao JH, Zhang Y, Du LZ, Dai YW, Gu WZH, Liu AM, Shang SHQ, Liang L. NPHS1 and NPHS2 gene mutations in Chinese children with sporadic nephrotic syndrome. Pediatr Res. 2007;61:117–122. doi: 10.1203/01.pdr.0000250041.19306.3d. [DOI] [PubMed] [Google Scholar]
- 43.Trautmann A, Bodria M, Ozaltin F, Gheisari A, Melk A, Azocar M, Anarat A, Caliskan S, Emma F, Gellermann J, Oh J, Baskin E, Ksiazek J, Remuzzi G, Erdogan O, Akman S, Dusek J, Davitaia T, Ozkaya O, Papachristou F, Firszt-Adamczyk A, Urasinski T, Testa S, Krmar RT, Hyla-Klekot L, Pasini A, Ozcakar ZB, Sallay P, Cakar N, Galanti M, Terzic J, Aoun B, Afonso AC, Szymanik-Grzelak H, Lipska BS, Schnaidt S, Schaefer F. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol. 2015;10:592–600. doi: 10.2215/CJN.06260614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lipska BS, Iatropoulos P, Maranta R, Carid G, Ozaltin F, Anarat A, Balat A, Gellermann J, Trautmann A, Erdogan O, Saeed B, Emre S, Bogdanovic R, Azocar M, Balasz-Chmielewska I, Benetti E, Caliskan S, Mir S, Melk A, Ertan P, Baskin E, Jardim H, Davitaia T, Wasilewska A, Drozdz D, Szczepanska M, Jankauskiene A, Higuita LMS, Ardissino G, Ozkaya O, Kuzma-Mroczkowska E, Soylemezoglu O, Ranchin B, Medynska A, Tkaczyk M, Peco-Antic A, Akil I, Jarmolinski T, Firszt-Adamczyk A, Dusek J, Simonetti GD, Gok F, Gheissari A, Emma F, Krmar RT, Fischbach M, Printza N, Simkova E, Mele C, Ghiggeri GM, Schaefer F the PodoNet Consortium. Genetic screening in adolescents with steroid resistant nephrotic syndrome. Kidney Int. 2013;84:206–213. doi: 10.1038/ki.2013.93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.