

Abstract
The pathogen Brucella abortus resides inside macrophages within a unique, replication-permissive organelle that is derived from the endoplasmic reticulum (ER). Although dependent on the Brucella type IV secretion system VirB, the mechanisms governing the biogenesis of this compartment remain elusive. Here, we investigated a putative role of the early secretory pathway in ER membrane accretion by the Brucella-containing vacuoles (BCVs). We show that BCVs interact with ER exit sites (ERES), and blockade of Sar1 activity, which disrupts ERES, prevents intracellular replication of Brucella. In cells expressing the dominant interfering form Sar1[T39N], BCVs do not acquire ER membranes, suggesting that they are unable to mature into replicative organelles. By contrast, treatments that block subsequent secretory events do not affect bacterial replication. We propose that Sar1-dependent ERES functions, but not subsequent secretory events, are essential for the biogenesis of the Brucella replicative compartment and, thus, bacterial replication. These results assign an essential role for Sar1 in pathogenesis of an intracellular bacterium.
Keywords: macrophage, secretory pathway, endoplasmic reticulum, pathogenesis
Brucella abortus ensures its survival and replication within host macrophages by avoiding fusion of its membrane-bound compartment, the Brucella-containing vacuole (BCV), with late endocytic compartments and controlling the conversion of the BCV into an endoplasmic reticulum (ER)-derived organelle that is permissive for replication (1). The latter phenomenon occurs at late stages of the vacuolar maturation process by means of sustained interactions with the ER. Such interactions culminate in limited fusion of the BCV with the ER and depend on the Brucella type IV secretion system VirB (1, 2). Brucella replicative organelles are characterized by the presence of ER markers on the vacuolar membrane and the absence of the lysosomal-associated membrane glycoprotein 1 (LAMP-1), which is transiently acquired by BCVs during maturation (1, 3, 4). Immature BCVs harbor LAMP-1 but do not display ER markers on their membrane despite being in contact with ER (1). BCV–ER intimate contacts and fusion likely occur at sites where dynamic ER membrane fission and fusion events take place. Such events occur within compartments of the early secretory pathway, where protein export from the ER toward the Golgi apparatus is initiated. ER exit sites (ERES) are discrete domains of the ER membrane organized by means of the assembly of the coat complex COPII, where transport vesicles are formed through COPII-dependent vesiculation and tubulation of ER membranes (5–7). The organization and functionality of these specialized ER domains are regulated by the activity of the small GTPase Sar1, which controls the assembly of COPII complexes on ER membranes (7, 8). Subsequent protein transport through the ER–Golgi intermediate compartment (ERGIC), and retrograde transport of ER-resident proteins, is mediated by the action of the COPI coat complex, the assembly and functions of which are controlled by the ADP-ribosylation factor 1 (ARF1) (5, 9). The COPII and COPI complexes thus act sequentially in ER-to-Golgi transport (5, 6, 9). By using fluorescence microscopy, COPII-positive compartments were juxtaposed to COPI-positive compartments, because of the physical proximity of these two subdomains, but segregation occurred during transport of COPI-coated vesicular-tubular complexes to the Golgi apparatus (6). Our previous observations, using brefeldin A (BFA) to block ARF1/COPI-dependent vesicular transport in Brucella-infected macrophages, suggested that these secretory events are not required for ER membrane acquisition by B. abortus (1). We hypothesized that BCVs instead interact with BFA-insensitive compartments of the early secretory pathway, such as ERES (10). Here, we investigated whether such interactions occur and examined the role of Sar1 in the biogenesis of the Brucella replicative organelle.
Methods
Bacterial Strains. The bacterial strains used in this study were the smooth virulent B. abortus strain 2308 and an isogenic, in-frame deletion mutant of the virB9 gene (2308 ΔvirB9). The virB9 in-frame deletion mutant of B. abortus 2308 was constructed by allelic exchange of the wild-type (WT) gene with an allele deleted of codons 7–279. We first constructed the counterselectable suicide vector pJC80 for allelic replacement in Brucella by ligating the NdeI-AatII sacBR fragment from pKNG101 (11) into the corresponding sites of pSP72 (Promega). To engineer a deletion of codons 7–279 in virB9, a 5′ fragment containing 788 bp upstream of and the first six codons of virB9 was generated by PCR amplification by using the primers JC42 (5′-CGCGGATCCCGTCAACAAAAGCGTTCCGGCTAAACC-3′) and JC43 (5′ CCCGGGCGATTTGATGACCGCAAGCAGGAATCTTTTCATTGC-3′) and a3′ fragment containing the 10 last codons of virB9 and the 1,300 bp downstream region by using primers JC44 (5′-AAAAGATTCCTGCTTGCGGTCATCA A ATCGCCCGGG-3′) and JC45 (5′-GGA AGATCTCGCTCGCAGA ACACT TCGCCCGGCCGG-3′). Both hemifragments were fused subsequently by overlap extension PCR amplification using primers JC42 and JC45. The resulting fragment was cloned into pJC80 by using the BamHI and BglII sites present on primers JC42 and JC45, respectively, to give pJC81. This plasmid then was introduced by electroporation into the WT strain 2308. Carbenicillin-resistant, sucrose-sensitive electroporants corresponding to clones in which pJC81 had integrated into the chromosome were subcultured further in tryptic soy (TS) broth and plated onto 5% sucrose TS agar plates to select sucrose-resistant, carbenicillin-sensitive recombinants that had lost the plasmid by cointegrate resolution. Recombinant clones were screened by PCR with primers flanking the virB9 gene to identify those harboring the ΔvirB9 in-frame deletion. Several independently isolated clones were assayed for intracellular growth and trafficking in both murine bone marrowderived macrophages (BMDMs) and HeLa cells, and all were unable to replicate and generate an ER-derived replicative organelle (data not shown), consistent with previous observations (1, 2, 4, 12). GFP-expressing bacteria were generated by electroporating into either 2308 or 2308 ΔvirB9 pJC43, a derivative of pBBR1MCS-2 (13) carrying the gfp-mut3 gene under the control of the constitutively active kanamycin-resistance gene aphA3 promoter. Bacteria were grown in TS broth (Sigma) or on TS agar plates (Sigma), supplemented with either kanamycin (50 μg/ml), or carbenicillin (100 μg/ml) or sucrose (5% wt/vol) when required. For infection of eukaryotic cells, 2 ml of TS broth was inoculated with a single bacterial colony from a freshly streaked TS agar plate and grown at 37°C for 15 h (early stationary phase) up to an OD600 of ≈2.0.
Plasmids. cDNAs encoding the untagged Sar1, Sar1[H79G], and Sar1[T39N] proteins were kindly provided by Jennifer Lippincott-Schwartz (National Institute of Child Health and Human Development, National Institutes of Health, Bethesda). They were used to generate N-terminal hemagglutinin (HA)-tagged alleles of the different Sar1 cDNAs by PCR amplification, which subsequently were cloned into pcDNA 3.1(–) (Invitrogen). A derivative of pCLXSN (Imgenex, San Diego) encoding GFP-p58 was kindly provided by Craig Roy (Yale University, New Haven, CT) and was used together with pCL-Eco (Imgenex) to produce retroviral particles in 293T cells as described in ref. 14. Plasmids encoding HA-tagged ARF1 and ARF1[T31N] proteins are described in ref. 15.
Cell Culture and Infection. To obtain BMDMs, bone marrow cells were isolated from femurs of 6- to 10-week-old C57BL/6 female mice and differentiated into macrophages as described in ref. 16. To express GFP-p58 in BMDMs, a 293T cell supernatant containing the packaged retroviral vector was added to the BMDM culture medium (2:5 ratio vol/vol) 5 days after isolation, and retroviral transduction was left to proceed for 2 days before infections were performed. HeLa cells (American Type Culture Collection clone CCL-2) were cultured at 37°C in a 7% CO2 atmosphere in DMEM supplemented with 10% FCS and 2 mM l-glutamine and seeded 24 h before infection on 12-mm glass coverslips (5 × 104 cells per coverslip).
BMDM infections were performed as described in ref. 1. HeLa cell infections were performed at either a multiplicity of infection of 500:1 (untreated cells) or 100:1 [cytotoxic necrotizing factor 1 (CNF1)-treated cells] as described in refs. 3 and 17. The Escherichia coli CNF1 toxin was kindly provided by Emmanuel Lemichez (Faculté de Médecine, Nice, France) and was applied to HeLa cells for 1 h at a concentration of 0.5 nM to induce membrane ruffling before infection with Brucella. CNF1 treatment was performed to increase the percentage of cells infected with Brucella and allow statistically significant scoring in bacterial replication assays. CNF1-induced membrane ruffling affected neither Brucella intracellular trafficking nor replication (17), and identical results were obtained in untreated or CNF1-treated HeLa cells. The expression of the various Sar1 and ARF1 alleles was performed by transfecting HeLa cells using the FuGENE transfection reagent (Roche), according to the manufacturer's instructions. Transfections were performed 90 min after Brucella infection and were left to proceed until the time of analysis.
Fluorescence Microscopy. To investigate the interactions of BCVs with COPI-, COPII-, or p58-labeled intracellular compartments, BMDMs were infected with B. abortus strains 2308 or 2308 ΔvirB9 or their GFP-expressing derivatives for various times. When necessary, BFA (Sigma) was applied to BMDM at a final concentration of 10 μg/ml for 2 h. To examine the role of Sar1 and ARF1 GTPases on bacterial replication and BCV maturation, HeLa cells were infected with GFP-B. abortus strain 2308 and transfected as described above for 24 h before processing. Cells were fixed with 3% paraformaldehyde (pH 7.4) at 37°C for 10 min or with ice-cold 100% methanol for 30 s (anti-sec31 antibody staining), then processed for immunofluorescence staining as described in ref. 3. The primary antibodies used were as follows: rabbit polyclonal anti-sec31 (18); anti-β-COP (Affinity BioReagents, Golden, CO); anti-LAMP-1 (19); anti-human cathepsin D (DAKO); anti-calnexin (Stressgen Biotechnologies, Victoria, Canada) antibodies; mouse monoclonal anti-HA antibody, clone 16B12 (Covance, Princeton); and anti-giantin (20). The secondary antibodies used were as follows: Alexa Fluor 488-conjugated and Alexa Fluor 594-conjugated goat anti-rabbit (Molecular Probes); cyanin-5-conjugated donkey anti-mouse (Jackson ImmunoResearch). Propidium iodide (Molecular Probes) was used as described in ref. 21 to stain intracellular Brucella DNA. Specimens were observed either on a Leica DMRBE epifluorescence microscope for quantitative analysis of bacterial replication in HeLa cells or a Zeiss LSM 510 laser scanning confocal microscope for quantitative analysis and image acquisition. Images of 1024 × 1024 pixels were acquired and assembled by using Adobe photoshop 7.0 (Adobe Systems, San Jose, CA). To analyze and quantitate BCV docking to β-COP-, sec31- and/or p58-positive compartments, single confocal sections of random fields were acquired, and colocalization of markers were scored as positive when nonsaturated signals partially overlapped.
Single-Cell Bacterial Replication Assays. Brucella replication in HeLa cells was assessed by scoring the number of intracellular bacteria in individual infected cells at 24 h postinfection (p.i.). At this time point, cells containing less than five bacteria were scored as negative. Cells containing at least 10 bacteria were scored as positive for bacterial replication. For each condition, 100 infected cells were analyzed. Replication analysis in HeLa cells expressing Sar1 or ARF1 alleles was compared systematically to both untransfected cells on the same coverslip and independent untransfected controls, which paralleled in all cases. Results were expressed as the percentage of infected cells with replicating bacteria.
Results and Discussion
BCVs Interact with COPII-Positive, but Not COPI-Positive, Structures. We first focused on the potential interactions of BCVs with ERES. Because the COPII complex is localized to ERES (6), we first analyzed whether BCVs are found in the vicinity of, or colocalize with, COPII-coated structures. By confocal microscopy, vacuoles containing WT B. abortus were apposed to vesicular structures decorated with COPII, as judged by staining of the COPII subunit sec31 (Fig. 1A Insets). The percentage of BCVs in close apposition to COPII-positive compartments increased during the first 12 h p.i. and thereafter reached a plateau (50.9 ± 7.3% at 12 h p.i.; 51.7 ± 10.6% at 24 h p.i.), concomitant with bacterial replication after 12 h p.i. (Fig. 1 A). These appositions, often polar, were neither qualitatively nor quantitatively altered by BFA treatment (Fig. 1 A and B), which does not affect COPII assembly (10), suggesting the COPII-positive compartments in close contact to BCVs are ERES. Importantly, vacuoles containing B. abortus ΔvirB9 initially interacted with COPII-positive compartments to levels similar to that of WT BCVs by 4 h p.i. (33.5 ± 6.1% vs. 34.8 ± 3.0%, respectively; Fig. 1B). However, these interactions decreased rapidly thereafter with only 17.0 ± 4.5% of positive BCVs by 24 h p.i. (Fig. 1 A and B). This phenomenon is consistent with our previous results highlighting the inability of VirB-deficient brucellae to sustain interactions with the ER (1) and indicates that BCV interactions with COPII-positive compartments at times after 4 h p.i. are VirB-dependent.
Fig. 1.

BCVs interact with COPII-positive compartments in macrophages. (A) Confocal microscopy micrographs of BMDMs infected for 4, 12, or 24 h with either WT or ΔvirB9 B. abortus strains expressing GFP. When required, BFA was applied to infected BMDMs for 2 h before analysis. Fixed cells were stained with anti-sec31 antibodies (red) to label COPII-positive compartments. (Insets) GFP-Brucella in close apposition to COPII-positive compartments. Arrows indicate examples of BCVs in close contact with COPII-labeled compartments. (B) Quantitation of BCV apposition to COPII-positive compartments over time. Single confocal microscopy 0.2-μm sections of random fields were acquired, and the appositions of BCVs to COPII-positive compartments were counted as positive when a coincident yellow fringe was observed (see A Insets). Data are means ± SD of three independent experiments. (C) Confocal microscopy micrographs of BMDMs infected with either WT or ΔvirB9 B. abortus strains for 4, 12, or 24 h. Fixed cells were stained with anti-β-COP antibodies to label COPI-positive compartments (green) and propidium iodide (red) to detect bacterial and host cell DNA. Arrows indicate BCVs positioned away from COPI-positive compartments. (D) Quantitation of BCV apposition to COPI-positive compartments over a 24-h infection period. Single confocal microscopy sections of random fields were acquired, and the appositions of BCVs to COPI-positive compartments were counted as positive when a coincident yellow fringe was observed. Data are means ± SD of three independent experiments. All micrographs shown are projections of three consecutive confocal sections. (Scale bars: 10 μm; Insets, 0.5 μm.)
In contrast, the majority of vacuoles containing either WT or VirB-deficient B. abortus neither interacted significantly with COPI-positive compartments at various times p.i. nor were labeled with COPI (Fig. 1C). A consistently small number of BCVs containing WT or ΔvirB9 Brucella were found in the vicinity of COPI-positive compartments (Fig. 1D), which likely represents BCVs located in the vicinity of COPI-positive compartments, because of their interactions with COPII-positive compartments. Thus, the observed BCV juxtapositions to COPI-positive structures do not depend on VirB and are unlikely to be related to BCV maturation events. The ARF1 GTPase, which controls COPI assembly on membranes, also was not detected on BCVs or in their vicinity at any time p.i. (data not shown). Taken together, these results demonstrate that BCVs interact with COPII-coated, but not COPI-coated, compartments in a VirB-dependent manner with kinetics comparable with those of BCV–ER interactions (1). Similar interactions were observed in HeLa epithelial cells (data not shown), consistent with the indistinguishable intracellular trafficking of Brucella in these two cell types (1, 3). Given the sequential mode of action of COPII and COPI complexes in ER to Golgi transport (5, 6, 9), our results strongly suggest that BCVs display a significant tropism toward ER export domains.
BCVs Interact with Functional ERES. To confirm that the COPII-positive compartments apposed to BCVs are actually ERES, we also examined the localization of the cargo receptor p58 (22), which accumulates in the ERGIC and also decorates functional ERES under steady-state conditions (22). In BMDMs expressing GFP-p58, a significant number of BCVs (up to 51.2 ± 2.4% at 8 h p.i.) were found in close contact to p58-positive vesicular structures (Fig. 2) with kinetics similar to those of COPII apposition (Fig. 1B). At 4 and 12h p.i., 97.2 ± 3.9% and 98.2 ± 2.5% of p58-positive compartments apposed to BCVs also were labeled with the COPII subunit sec31, indicating they are functional ERES and not ERGIC (Fig. 2 A Insets and data not shown). BFA treatment redistributes p58 to ERES only, due to the disruption of the ERGIC and Golgi (5, 10). In BFA-treated cells, BCVs remained in contact with p58 and sec31-labeled compartments (Fig. 2 A Insets). Furthermore, the percentage of BCVs apposed to p58-positive compartments in untreated and BFA-treated BMDM expressing GFP-p58 was not significantly different at any time point examined (Fig. 2B). Thus, all p58-positive compartments apposed to BCVs are functional ERES.
Fig. 2.

BCVs interact with functional ERES. (A) Confocal microscopy micrographs of GFP-p58 expressing BMDMs infected with WT B. abortus for either 4 or 12 h. Fixed cells were stained with anti-sec31 antibodies (red) to label COPII-positive compartments and propidium iodide (pseudocolored in blue) to detect bacterial and host cell DNA. GFP-p58 is shown in green. Arrows indicate BCVs apposed to p58- and sec31-positive ERES. (Insets) Magnified views of typical BCV–ERES appositions. The micrographs shown are projections of three consecutive confocal sections. (Scale bars: 10 μm; Insets, 1 μm.) (B) Quantitation of BCVs apposed to p58-positive compartments in untreated or BFA-treated BMDMs expressing GFP-p58. Data are means ± SD of three independent experiments. Where indicated, BFA was applied for 2 h before processing to ensure that GFP-p58 was relocated to ERES after disruption of the secretory pathway.
Disruption of ERES Prevents Brucella Intracellular Replication. Given that BCV-ERES interactions occur at times where BCVs undergo maturation (during the first 12 h p.i.), we sought to determine whether ERES are functionally important to the biogenesis of the Brucella replicative compartment. To examine this hypothesis, we took advantage of the differential effects that expression of the constitutively active, GTP-bound form Sar1[H79G] or dominant interfering, GDP-bound form Sar1[T39N] have on the secretory pathway. Whereas the former disrupts the ERGIC and the Golgi compartment yet leaves ERES functional, the latter disrupts functional ERES by preventing COPII assembly (10). We analyzed bacterial replication in cells expressing either of the Sar1 dominant alleles as a measure of the biogenesis of the replicative organelle. Because of their inhibition of the secretory pathway, Sar1 or ARF1 dominant allele constructs cannot be used in conjunction with retroviral systems. Therefore, we expressed these constructs in HeLa cells by using classical transfection methods. We have shown previously that the nature and mechanisms of biogenesis of the Brucella replicative compartment are directly comparable in macrophages and epithelial cells (1, 3). Although overexpression of the WT Sar1 clustered ERES around the Golgi apparatus (Fig. 3A and data not shown), it did not significantly affect bacterial replication (Fig. 3B). In Sar1-overexpressing cells, BCVs localized to clustered ERES during maturation (at 8 h p.i.; data not shown), yet replicative vacuoles were positioned away from clustered ERES at 24 h p.i. (Fig. 3A). These results suggest that physical interactions with functional ERES occur during BCV maturation, but not once bacterial replication occurs. Interestingly, expression of Sar1[H79G] did not affect bacterial replication (Fig. 3), despite disrupting the secretory pathway (Fig. 5, which is published as supporting information on the PNAS web site) and clustering ERES at the microtubule organizing center (10). This finding suggests that disrupting the secretory pathway does not affect Brucella replication, provided that Sar1 activity is maintained. In agreement, blocking Sar1 activity via the expression of Sar1[T39N] disrupted ERES (Fig. 3A) and significantly impaired Brucella replication (Fig. 3), demonstrating that functional ERES are required for Brucella replication. In addition, blocking downstream secretory events by overexpressing the dominant interfering, GDP-bound form ARF1[T31N], which prevents protein transport without altering Sar1 activity, did not affect bacterial replication in HeLa cells (Fig. 3). This result is consistent with results obtained with BFA in macrophages (1). Taken together, our data demonstrate that Sar1 activities involved in ERES functions, but not ARF1 activities, are essential to intracellular replication of Brucella.
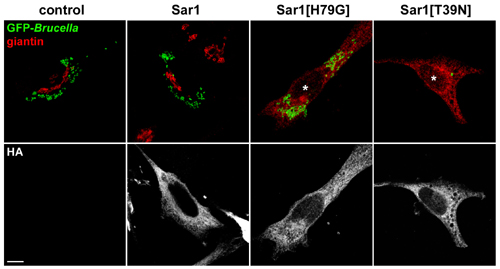
Fig. 3.

Disruption of Sar1 activity and ERES prevents Brucella replication. (A) Confocal microscopy micrographs of B. abortus-infected HeLa cells transfected with various Sar1 or ARF1 alleles after 24 h of infection. HeLa cells were infected with WT GFP-expressing Brucella then transfected with plasmids encoding either HA-tagged Sar1, Sar1[T39N], Sar1[H79G], ARF1, or ARF1[T31N]. After 24 h of infection, cells were fixed and stained with rabbit anti-sec31 (red) and mouse anti-HA (Upper) antibodies to label ERES and detect transfected cells, respectively. The micrographs shown are projections of three consecutive confocal sections and depict typical bacterial replication patterns with the various GTPase alleles. Asterisks indicate clustered ERES either around the Golgi apparatus (Sar1) or in a juxtanuclear area (Sar1[H79G]). Only the expression of Sar1[T39N] prevents bacterial replication (arrow). (B) Quantitation of the effect of Sar1 and ARF1 alleles expression upon Brucella replication, expressed as the percentage of infected cells displaying bacterial replication. Scoring of bacterial replication was performed after 24 h of infection by epifluorescence microscopy analysis of untransfected (control) or transfected, infected cells. Data are means ± SD of five independent experiments.
Inhibition of Sar1 Activity Prevents ER Membrane Acquisition by BCVs. The inhibition of Brucella replication observed in cells expressing Sar1[T39N] could be due either to a blockade of BCV maturation into an ER-derived organelle, which consequently prevents bacterial replication, or to an inhibition of bacterial replication within mature replicative organelles. To discriminate between these two hypotheses, we examined for the presence of calnexin and LAMP-1 on BCVs in cells expressing various alleles of Sar1. Consistent with bacterial replication (Fig. 3B), ≈85% of BCVs in cells expressing either Sar1 or Sar1[H79G] were decorated with calnexin, but only ≈11% were LAMP-1-positive at 24 h p.i. (Fig. 4), indicating that these vacuoles displayed features of replicative organelles. By contrast, only 15.0 ± 2.2% of BCVs in cells expressing Sar1[T39N] were labeled with calnexin, but 80.7 ± 4.6% retained LAMP-1 (Fig. 4). These LAMP-1-positive vacuoles did not originate from fusion with lysosomes, because few of them (13.7 ± 2.7%) were labeled with the lysosomal cathepsin D (data not shown). Thus, BCVs in cells expressing Sar1[T39N] display features of immature vacuoles that have not acquired ER membrane. This finding implies that blocking Sar1 activity prevents ER membrane acquisition and the subsequent conversion of BCVs into ER-derived replicative organelles.
Fig. 4.

Inhibition of Sar1 activity prevents ER membrane acquisition by BCVs. (A) Quantitation of the presence of the ER marker calnexin or the late endosomal/lysosomal marker LAMP-1 on BCVs in HeLa cells expressing various Sar1 alleles. HeLa cells were infected with WT GFP-expressing B. abortus, then transfected with plasmids encoding either HA-tagged Sar1, Sar1[T39N], or Sar1[H79G]. After 24 h of infection, cells were fixed and stained with either rabbit anticalnexin or rabbit anti-LAMP-1 (red) and mouse anti-HA (blue) antibodies to detect transfected cells. Calnexin- or LAMP-1-positive BCVs were scored by confocal microscopy on single sections of untransfected cells (control) or cells transfected with either of the Sar1 alleles. Data are means ± SD of three independent experiments. (B) Confocal microscopy micrographs illustrating calnexin (Left) and LAMP-1 (Right) staining of BCVs in HeLa cells expressing either Sar1[T39N] (Upper) or Sar1[H79G] (Lower). GFP-Brucella are shown in green, calnexin or LAMP-1 in red, and HA-tagged Sar1 alleles in blue. The micrographs shown are projections of three consecutive confocal sections. (Insets) Magnifications of areas indicated by arrows, in which the HA staining (blue) has been removed for clarity. (Scale bars: 10 μm; Insets, 1 μm.)
We have demonstrated previously that BCVs interact with the ER to generate an ER-derived intracellular organelle permissive for replication and that the acquisition of ER membranes depends on a functional type IV secretion system VirB (1). Here, we show that interactions of BCVs with the ER actually occur at ER export sites, which are stable, functionally distinct subdomains of this organelle. We further identified the GTPase Sar1 as the first host molecule involved in the intracellular replication of Brucella. By intercepting ARF1-dependent vesicular traffic from ERES, another intracellular pathogen, Legionella pneumophila, also generates an ER-derived replicative organelle (23). However, unlike L. pneumophila, the formation of the Brucella replicative organelle does not require the BFA-sensitive ARF1/COPI-dependent vesicular transport, highlighting mechanistic differences between these two intracellular pathogens and their subversion of the secretory pathway. The dependence on Sar1 activity of the biogenesis of the Brucella replicative organelle is consistent with the specific interactions of BCVs with COPII-coated, but not with COPI-coated, compartments. Sar1 controls the assembly of the COPII complex onto the ER membrane, which mediates cargo selection and membrane deformation into budding vesicular and tubular carriers (8). It is likely that Brucella targets ERES and takes advantage of the membrane dynamics of these domains to fuse with, and acquire, ER membranes. ER membrane acquisition by BCVs has been typically analyzed by the presence on the vacuolar membrane of either ER chaperones, such as calnexin, calreticulin, the translocator sec61β, an ER resident enzyme PDI, or the activity of the glucose-6-phosphatase (1, 3, 4), most of which are absent from ERES (24, 25). Hence, the acquisition of ER resident proteins by BCVs is unlikely to result from interactions with ERES only. During BCV maturation, ER structures surround the bacterial vacuoles, but no obvious fusion is observed (1). ER resident proteins are only detected on the vacuolar membrane after 12 h p.i., when BCV maturation is completed and bacterial replication occurs (1). This finding suggests that mature BCVs also are able to fuse with ER domains other than ERES. Our results indicate that BCV–ERES interactions occur before ER resident proteins actually are displayed on BCVs and that these interactions are a prerequisite for the acquisition of ER membranes. It is, thus, possible that BCVs acquire fusion capabilities by means of their interactions with ERES, which subsequently allows them to fuse with other ER compartments. Further work is required to examine the possibility of BCV sequential interactions with different functional domains of the ER and to characterize the involvement of host and bacterial machineries.
Supplementary Material
Acknowledgments
We thank Leigh Knodler for critical reading of the manuscript. We are grateful to Jennifer Lippincott-Schwartz and Craig Roy for providing plasmids encoding Sar1 and ARF1 alleles and to Emmanuel Lemichez for providing purified CNF1. This work was supported by institutional grants from Institut National de la Santé et de la Recherche Médicale and Centre National de la Recherche Scientifique and by European Community Research and Technological Development Project NOVELTARGETVACCINES Research Contract ICA4-CT-1999-10001. S.P.S. was supported by the Fondation pour la Recherche Médicale.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: BCV, Brucella-containing vacuole; BFA, brefeldin A; BMDM, bone marrowderived macrophages; CNF1, cytotoxic necrotizing factor 1; ER, endoplasmic reticulum; ERES, ER exit sites; ERGIC, ER-Golgi intermediate compartment; HA, hemagglutinin; LAMP-1, lysosomal-associated membrane glycoprotein 1; p.i., postinfection; TS, tryptic soy.
See Commentary on page 1271.
References
- 1.Celli, J., de Chastellier, C., Franchini, D. M., Pizarro-Cerda, J., Moreno, E. & Gorvel, J. P. (2003) J. Exp. Med. 198, 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Callaghan, D., Cazevieille, C., Allardet-Servent, A., Boschiroli, M. L., Bourg, G., Foulongne, V., Frutos, P., Kulakov, Y. & Ramuz, M. (1999) Mol. Microbiol. 33, 1210–1220. [DOI] [PubMed] [Google Scholar]
- 3.Pizarro-Cerda, J., Meresse, S., Parton, R. G., van der Goot, G., Sola-Landa, A., Lopez-Goni, I., Moreno, E. & Gorvel, J. P. (1998) Infect. Immun. 66, 5711–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Comerci, D. J., Martinez-Lorenzo, M. J., Sieira, R., Gorvel, J. P. & Ugalde, R. A. (2001) Cell Microbiol. 3, 159–168. [DOI] [PubMed] [Google Scholar]
- 5.Scales, S. J., Pepperkok, R. & Kreis, T. E. (1997) Cell 90, 1137–1148. [DOI] [PubMed] [Google Scholar]
- 6.Stephens, D. J., Lin-Marq, N., Pagano, A., Pepperkok, R. & Paccaud, J. P. (2000) J. Cell Sci. 113, 2177–2185. [DOI] [PubMed] [Google Scholar]
- 7.Barlowe, C., Orci, L., Yeung, T., Hosobuchi, M., Hamamoto, S., Salama, N., Rexach, M. F., Ravazzola, M., Amherdt, M. & Schekman, R. (1994) Cell 77, 895–907. [DOI] [PubMed] [Google Scholar]
- 8.Barlowe, C. (2002) Curr. Opin. Cell Biol. 14, 417–422. [DOI] [PubMed] [Google Scholar]
- 9.Aridor, M., Bannykh, S. I., Rowe, T. & Balch, W. E. (1995) J. Cell Biol. 131, 875–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ward, T. H., Polishchuk, R. S., Caplan, S., Hirschberg, K. & Lippincott-Schwartz, J. (2001) J. Cell Biol. 155, 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaniga, K., Delor, I. & Cornelis, G. R. (1991) Gene 109, 137–141. [DOI] [PubMed] [Google Scholar]
- 12.Delrue, R. M., Martinez-Lorenzo, M., Lestrate, P., Danese, I., Bielarz, V., Mertens, P., De Bolle, X., Tibor, A., Gorvel, J. P. & Letesson, J. J. (2001) Cell Microbiol. 3, 487–497. [DOI] [PubMed] [Google Scholar]
- 13.Kovach, M. E., Phillips, R. W., Elzer, P. H., Roop, R. M., II, & Peterson, K. M. (1994) BioTechniques 16, 800–802. [PubMed] [Google Scholar]
- 14.Naviaux, R. K., Costanzi, E., Haas, M. & Verma, I. M. (1996) J. Virol. 70, 5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peters, P. J., Hsu, V. W., Ooi, C. E., Finazzi, D., Teal, S. B., Oorschot, V., Donaldson, J. G. & Klausner, R. D. (1995) J. Cell Biol. 128, 1003–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Chastellier, C., Frehel, C., Offredo, C. & Skamene, E. (1993) Infect. Immun. 61, 3775–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chaves-Olarte, E., Guzman-Verri, C., Meresse, S., Desjardins, M., Pizarro-Cerda, J., Badilla, J., Gorvel, J. P. & Moreno, E. (2002) Cell Microbiol. 4, 663–676. [DOI] [PubMed] [Google Scholar]
- 18.Shugrue, C. A., Kolen, E. R., Peters, H., Czernik, A., Kaiser, C., Matovcik, L., Hubbard, A. L. & Gorelick, F. (1999) J. Cell Sci. 112, 4547–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steele-Mortimer, O., Meresse, S., Gorvel, J. P., Toh, B. H. & Finlay, B. B. (1999) Cell Microbiol. 1, 33–49. [DOI] [PubMed] [Google Scholar]
- 20.Schweizer, A., Fransen, J. A., Bachi, T., Ginsel, L. & Hauri, H. P. (1988) J. Cell Biol. 107, 1643–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coers, J., Kagan, J. C., Matthews, M., Nagai, H., Zuckman, D. M. & Roy, C. R. (2000) Mol. Microbiol. 38, 719–736. [DOI] [PubMed] [Google Scholar]
- 22.Lahtinen, U., Hellman, U., Wernstedt, C., Saraste, J. & Pettersson, R. F. (1996) J. Biol. Chem. 271, 4031–4037. [DOI] [PubMed] [Google Scholar]
- 23.Kagan, J. C. & Roy, C. R. (2002) Nat. Cell Biol. 4, 945–954. [DOI] [PubMed] [Google Scholar]
- 24.Kuehn, M. J., Herrmann, J. M. & Schekman, R. (1998) Nature 391, 187–190. [DOI] [PubMed] [Google Scholar]
- 25.Mezzacasa, A. & Helenius, A. (2002) Traffic 3, 833–849. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}