

Abstract
Rationale:
Immunoglobulin G4 (IgG4)-related hypophysitis is a type of IgG4-related disease (IgG4-RD), which is characterized by plasma cells infiltration in the pituitary causing functional changes and (or) space-occupying effect in the pituitary. IgG4-related hypophysitis is sensitive to hormone therapy in most patients, but recurrence is very likely.
Patient concerns:
Here, we report a 57-year-old male patient with bilateral eye redness as the initial presentation. He later presented with pituitary hypofunction that involved multiple organs, including eyes, lacrimal gland, pituitary, lung, gall bladder, and intestine. There was an elevation of C-reactive protein and blood sedimentation, but the IgG and IgG4 levels of the serum and the cerebrospinal fluid did not increase obviously following irregular glucocorticoid therapy. Magnetic resonance imaging revealed enlarged pituitary and obviously thickened pituitary stalk. IgG4-related hypophysitis was confirmed by biopsy of the pituitary.
Diagnoses:
The patient was diagnosis of IgG4-related hypophysitis with ophthalmopathy by pathological and molecular tests.
Interventions:
The patient responded to methylprednisolone pulse therapy but relapsed during the maintenance therapy using small-dose hormones combined with azathioprine. Methylprednisolone pulse therapy was given for 3 days followed by rituximab injection for 4 weeks.
Outcomes:
After use methylprednisolone pulse therapy with rituximab the patient achieved complete remission.
Lessons:
Rituximab achieved good effect for recurrent IgG4-related hypophysitis after glucocorticoid therapy combined with immunosuppressant in this case. Moreover, comparative analysis was carried out with other reported cases of IgG4-related hypophysitis in terms of clinical features, treatment, and follow-up.
Keywords: glucocorticoid, hypophysitis, IgG4-related disease, pituitary hypofunction, rituximab
1. Introduction
IgG4-related disease (IgG4-RD) is a newly discovered autoimmune disease manifested as chronic progression of systemic inflammation and fibrosis of unknown etiology. This term was first used by Hamano et al[1] in 2001 when describing 1 case of sclerosing pancreatitis. This case attracts great attention due to the elevation of the serum IgG4 level. As found by subsequent studies, such type of patients is combined with similar lesions in other organs, including bile duct, salivary gland, orbits, lymph nodes, retroperitoneum, mediastinum, soft tissue, and nerve system.[2,3] They are all characterized by extensive interstitial fibrosis, diffuse lymphoplasmacytic infiltration with obstructive phlebitis and significant elevation of the serum IgG4 level. If the pituitary is involved, it is known as IgG4-related hypophysitis. IgG4-related hypophysitis is featured by hypofunction of anterior pituitary and (or) diabetes insipidus, IgG4-related hypophysitis is featured by hypofunction of anterior pituitary and (or) diabetes insipidus, sometimes with space-occupying effect and multiorgan involvement. The elevated serum IgG4 level is considered an important diagnostic clue. Typical imaging findings include mass in the saddle area or thickening of pituitary stalk. The diagnosis can be confirmed by pathological examination. IgG4-related hypophysitis is usually sensitive to glucocorticoid therapy, but recurrence is very likely. This disease is rare and the diagnosis is difficult with high misdiagnosis rate. We present 1 recurrent case of IgG4-related hypophysitis with ophthalmopathy as the initial presentation and multiorgan involvement in the hope of benefiting the understanding on IgG4-related hypophysitis.
2. Materials and methods
The 57-year-old male, who was admitted to hospital in April, had bilateral eye redness for 1 year, with poor appetite and malaise. The patient reported the onset of bilateral eye swelling 1 year ago, particularly in the right eye, which had conjunctival and scleral congestion with progressive vision loss. He responded poorly to irregular hormone therapy. Four months before hospitalization, the patient showed the symptoms of malaise, poor appetite, nausea after eating greasy food without vomiting, fear of cold, and loss of weight. He then presented with compulsive polydipsia and polyuria with daily water uptake reaching 4.5 L. Fever of unknown reason occurred 1 month ago, with body temperature of 37.3 to 38.5°C and concurrent headache. The headache was aggravated when the body temperature rose. Antibiotic therapy achieved no effect. After admission, the patient suffered from chest tightness, suffocation, and intermittent diarrhea.
3. Results
Physical examination upon admission revealed: thin skin, scattered petechiae under the skin, muscular atrophy of the 4 limbs, no facial deformity, missing of several teeth, bilateral eyelid edema, conjunctival and scleral congestion of the right eye (Fig. 1), flexible movement of bilateral eyes on the thick side. Ophthalmologic examination revealed visual field defect and obvious visual impairment of the right eye. Auxiliary examinations revealed pituitary hypofunction, elevation of blood sedimentation and CRP, negative indicators related to tuberculosis and infections (Tables 1–3). The results were all negative for 5 antituberculosis antibodies, 6 anti-ENA antibodies and anti-cardiolipin antibodies; the tumor markers were negative; IgG4 83.9 mg/dL and IgG 1260.0 mg/dL in peripheral blood (Table 4); IgG 12.5 mg/dL, IgA 1.16 mg/dL, and IgM 0.21 mg/dL in the cerebrospinal fluid (Table 5). Smears showed no fungi or bacteria; negative for Hu Yo Ri and antigens. Only a few lymphocytes without cancer cells were observed in pathological examination. MRI indicated space-occupying lesions in the saddle area and the basilar clivus as well as cysts in the pituitary stalk (Fig. 2); MRI of the orbits indicated inflammatory lesion of the right eye (Fig. 3); chest CT scan revealed bilateral lung nodules suspected as granulomas (Fig. 4). PET-MRI indicated multiorgan involvement, that is, systemic inflammatory lesions intracranially, in the right salivary gland, sinus nasalis, chest and paravertebrally, gall bladder, colon, and rectum. Under the navigation of MRI, biopsy of the saddle area was performed via the transsphenoidal approach and IgG4-related hypophysitis (Fig. 5) was confirmed. The patient was sensitive to methylprednisolone pulse therapy at 36 mg/d for 4 weeks. The symptoms of headache, ophthalmopathy (Fig. 1), chest tightness, and diarrhea were alleviated obviously. But the pituitary function was not improved. Therefore, Euthyrox and Minirin replacement therapy was given. The dose of methylprednisolone was decreased at the rate of 4 mg per 2 weeks until 8 mg/d, and maintenance therapy was given along with azathioprine at 100 mg/d. The patient relapsed 4 months later, showing such symptoms again as headache, bilateral eye redness, and swelling (particularly in the right eye), and vision impairment (Fig. 2). Methylprednisolone pulse therapy was given at 600 mg/d for 3 days, followed by rituximab injection at 100 mg/wk for 4 weeks. The patient achieved complete remission (Figs. 1 and 2).
Figure 1.
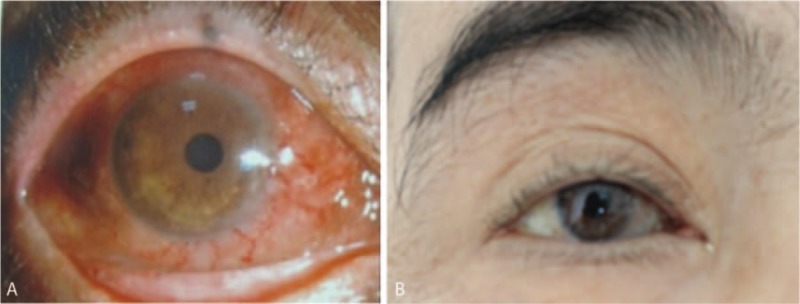
Examination of the eye. A, Upon admission and before glucocorticoid pulse therapy: Redness of the right eye with conjunctival and scleral congestion. B, After glucocorticoid pulse therapy combined with rituximab: obvious improvement of symptoms in the right eye.
Table 1.
Gonadal hormone concentrations.
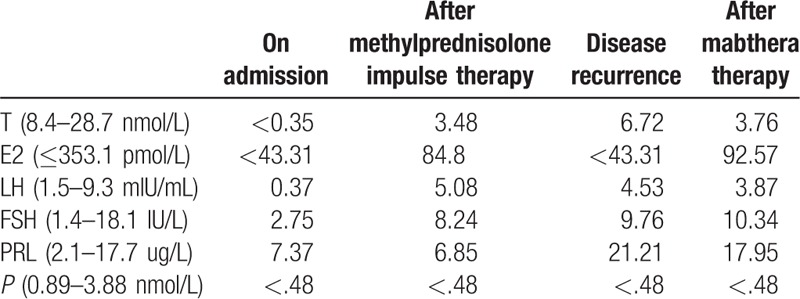
Table 3.
Thyroid function.
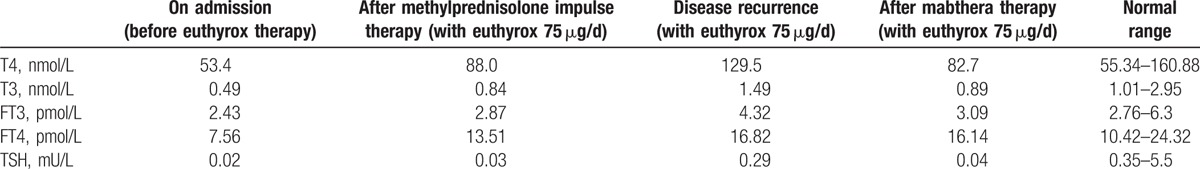
Table 4.
Other laboratory results.
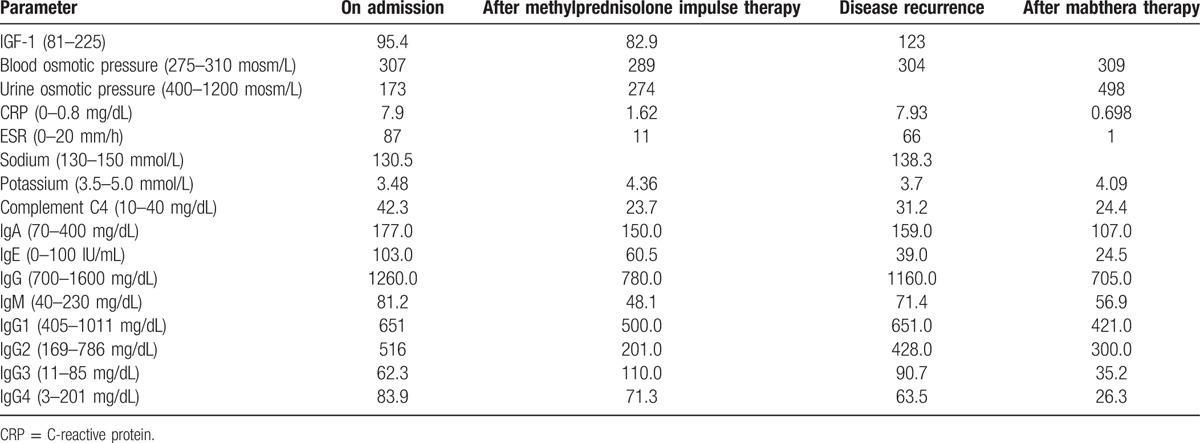
Table 5.
Results of cerebrospinal fluid test.
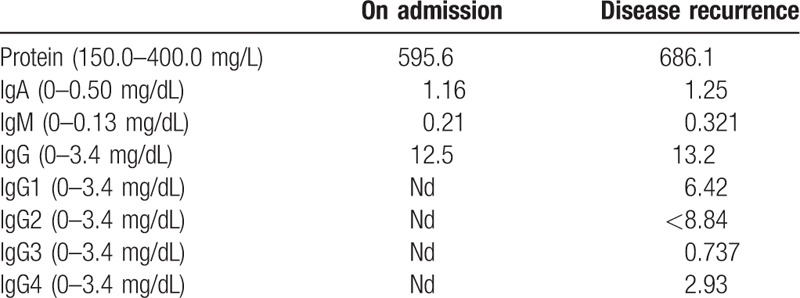
Figure 2.
MRI of the pituitary. A (a–d), Upon admission: the pituitary was enlarged with height of 10 mm and obvious thickening of pituitary stalk, showing equal enhancement on T1- and T2-weighted MRI images; the hypersignals in the posterior pituitary disappeared without depression of the sellar floor; the chiasma opticum was elevated due to compression, primarily on the left, showing as obvious nonuniform enhancement in contrast-enhanced scan; the boundary with bilateral cavernous sinus was unclear, with lesions extending posteroinferiorly; the posterior clinoid process disappeared, and the top of the clivus was involved. Nonenhanced area was seen in the pituitary stalk, and no abnormal signals or abnormal enhancement was seen in the pineal body. B (e–h), After hormone pulse therapy: the inflammatory lesion in the saddle was reduced obviously. C (i–l), Relapse after maintenance therapy using small-dose hormone combined with azathioprine several months later: Sella turcica was reduced and morphologically abnormal; the anterior pituitary was reduced with slender pituitary stalk. D (m–p), After methylprednisolone pulse therapy combined with rituximab, the symptoms were effectively controlled: Sella turcica was reduced and morphologically abnormal; the anterior pituitary was reduced with slender pituitary stalk, showing no obvious changes as before. MRI = magnetic resonance imaging.
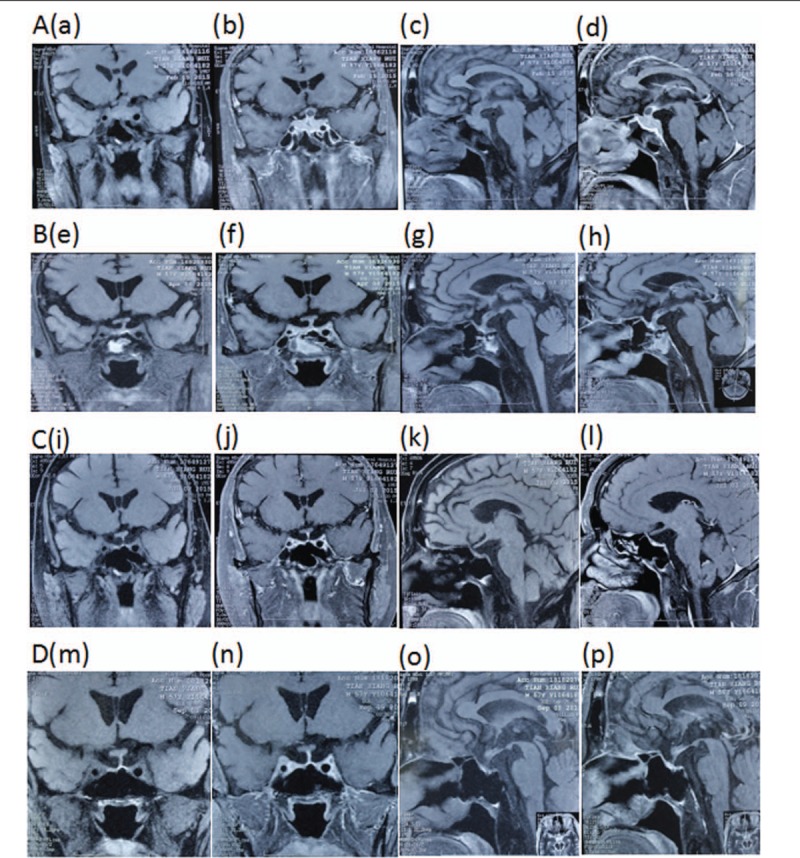
Figure 3.
Findings on MRI of the orbits. A, B, Abnormal signals of interior and exterior wall of right eye with enhancement, indicating inflammatory lesions. C, The symptoms of the right eye were greatly improved after hormone pulse therapy.
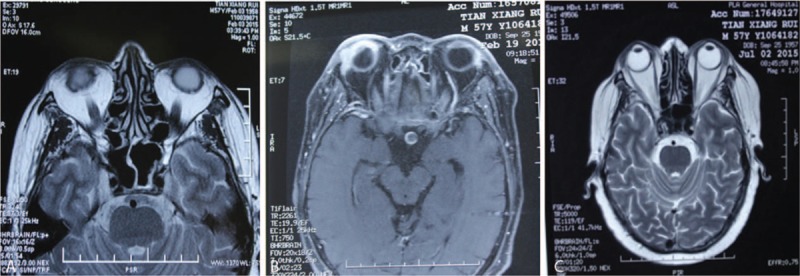
Figure 4.
Chest CT. A, Nodules in the apicoposterior segment of the left upper lobe and anterior basilar segment of the right lower lobe. B, Inflammation in bilateral lower lobes, pleural effusion in the right chest, atelectasis of the right lower lobe, and enlarged nodules in the apicoposterior segment of the left upper lobe and anterior basilar segment of the right lower lobe, as compared with previous results. C, Improved inflammation in bilateral lungs and improved peural effusion in the right chest; reduced nodules in the apicoposterior segment of the left upper lobe and anterior basilar segment of the right lower lobe. CT = computed tomography.
Figure 5.
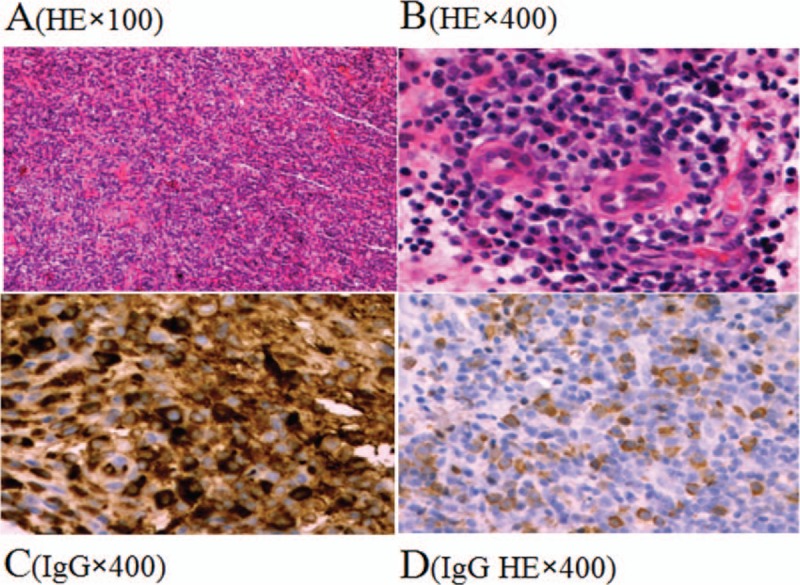
The results of immunohistochemistry. Infiltration of the pituitary by the plasma cells below the mucosa of the saddle area, with multinucleated giant cells seen locally and small patch-like inflammatory exudate; immunohistochemistry: IgG (+), IgG4 (partial +, accounting for 40% of IgG-positive cells, >10 cells in high power field), CD68 (scattered +), Syn (small focus locally+), CK (nasal mucosa epithelium +), Ki-67 (20%), Bcl-2 (partial+), CD3 (scattered +), CD20 (scattered +), Kappa (++), Lambda (+); no obvious pituitary.
Table 2.
LH and FSH value (basal and response to intravenous injection of 100 ug gonadotropin-releasing hormone) before treatment.

Hypophysitis is featured by inflammation and cell infiltration of the pituitary. Depending on the position involved, hypophysitis is divided into panhypophysitis, adenohypophysis, and infundibuloneurohypophysitis. In terms of histopathology, hypophysitis is divided into lymphocytic hypophysitis, granulomatous hypophysitis, xanthomatous hypophysitis, necrotizing hypophysitis, IgG4-related hypophysitis, and mixed hypophysitis. By etiology, hypophysitis is either primary or secondary.[4] We report 1 case of IgG4-related hypophysitis, which is very rare clinically. IgG4-related hypophysitis is part of the IgG4-related disease, which was first described by van der Vliet and Perenboom[5] in 2004. Wong et al[6] produced the first pathological diagnosis of this disease. The diagnostic criteria were proposed in 2011 by Leporati et al (Table 6).[7] Our patient met the criteria 2, 3, and 5.
Table 6.
Diagnostic criteria for IgG4-related hypophysitis.

IgG4-related disease involving the central nervous system is rare. According to the existing reports, the disease is confined in the pituitary. So far 46 cases of IgG4-related disease have been reported, and most of them are from Japan. There are 36 males (78.3%) and 10 females (21.7%), with the age of onset being 25 to 81 years, 63 years on average; 4 cases (8.7%) are affected in the saddle area, 8 (19.6%) in the pituitary stalk, and 33 (71.7%) in the saddle area as well as the pituitary stalk; 10 cases (22.2%) have hypofunction of anterior pituitary, 6 (13.3) have diabetes insipidus, 27 (60.0%) have hypofunction of the while pituitary, 2 (4.4%) have normal pituitary function, and 35 (76.1%) are combined with involvement of other organs; 18 cases (41.3%) are pathologically confirmed by biopsy.[8–39] Bando et al[33] carried out a retrospective analysis on 170 cases of hypofunction of anterior pituitary with and without diabetes insipidus at Kobe University Hospital from 2011 to 2012. Among them, 23 cases had hypophysitis, and only 7 cases were diagnosed as IgG4-related hypophysitis, accounting for 30.4% among all cases with hypophysitis and only 7% among all cases of hypofunction of anterior pituitary with and without diabetes insipidus. The case in our study was a 57-year-old male with ophthalmopathy as the initial presentation. He first showed hypofunction of the whole pituitary, which evolved into multiorgan involvement. MRI revealed lesions in the saddle area and the clivus, which were confirmed by biopsy. This was the first case with ophthalmopathy as the initial presentation. We reviewed 55 cases of IgG4-related disease confirmed at our hospital from January 2012 to March 2015.[40] Among them, 3 cases were combined with pituitary damage (2 males and 1 female, aged 55–67 years at onset), 1 case with diabetes insipidus, and 2 cases with hypofunction of the whole pituitary. All cases were combined with involvement of other organs, presenting as lesions in the saddle area or suprasellar region on MRI.
4. Discussion and conclusions
The distinguishing features of IgG4-related disease are the elevation of serum IgG4 level and infiltration by IgG4-positive plasma cells. For pituitary tumors of unknown etiology, we recommend detection of serum IgG4 level before the glucocorticoid therapy. Although elevated serum level of IgG4 is an important diagnostic indicator of IgG4-related hypophysitis, it is neither a necessary nor specific one. In our case, the serum IgG4 level was detected after irregular glucocorticoid therapy and the result was lower than 140 mg/dL, which did not reach the diagnostic criterion. Moreover, among 46 cases of IgG4-related hypophysitis, 10 cases had serum IgG4 level lower than the criterion after glucocorticoid therapy. This indicated the role of glucocorticoid therapy in reducing the serum level of IgG4. Tabata et al[41] detected the changes of serum IgG4 level in 44 cases of IgG4-related disease and proposed that the serum IgG4 level can be a predictor of the relapse.
In the absence of contraindications, glucocorticoid is the first-line drug for inducing remission for patients at early active phase of the disease.[42] The initial dose of prednisone is usually 30 to 40 mg/d (0.6 mg/kg),[43] and the dose can be increased for higher body weight or rapid progression. For milder symptoms, small initial dose can be used. After 2 to 4 weeks, the dose can be reduced to the dose of maintenance therapy (5.0–7.5 mg/d) at the rate of 5 mg every 2 weeks.[44,45] The small-dose maintenance therapy can sometimes last for 3 years.[43] Although hormone therapy is effective, relapse may be induced by drug disuse (30–45%). Therefore, immunosuppressant can be prescribed in combination, including azathioprine, methotrexate, mycophenolate mofetil, and cyclophosphamide. The immunosuppressant is necessary for the starting therapy in some patients. In case of relapse, glucocorticoid pulse therapy and maintenance therapy can be still applied.[43] The present case evolved into multiorgan involvement after irregular glucocorticoid therapy with symptom aggravation. The symptoms of eyes, lung, intestine, and headache were greatly relieved after pulsed therapy using methylprednisolone at 36 mg/d for 4 weeks. MRI revealed reduced pituitary as compared with before, but without functional recovery. Considering the long course of disease and severity of the symptoms, methylprednisolone 8 mg/d combined with azathioprine 100 mg/d was used, but the patient relapsed several months later. After methylprednisolone pulse therapy at 600 mg/d for 3 days, rituximab injection was performed and good effect was achieved. So far only 6 recurrent cases of IgG4-related hypophysitis have been reported, and all of them are males aged 40 to 75 years; 4 cases are combined with lesions of other organs. The glucocorticoid therapy for recurrent patients consists of prednisone 30 mg/d, 10 mg/d, 7.5 mg/d, 5 mg/d or hydrocortisone. Recurrent patients may suffer from headache, visual field defect, and reduced pituitary function. Five cases were relieved by the regimen of first increasing and then decreasing the dose of prednisone; another case was relieved by the use of azathioprine on the basis of prednisone. However, all 6 cases failed to achieve functional recovery of the pituitary.[39] Our case was the first case of IgG4-related hypophysitis who achieved complete remission after relapse by rituximab injection. This indicated the superiority of rituximab as a maintenance and remedial therapy compared with hormone therapy and immunosuppressants. Hart et al[46] carried out a follow-up study that lasted for 47 months. One group of recurrent patients was treated by prednisone, and the other group by hormone therapy combined with immunosuppressant. Two groups did not show significant difference at the time of remission. Besides, the failure rate of immunosuppressant was as high as 48%. In contrast, rituximab injection exhibits better effect in resistant to immunosuppressant and intolerant to hormone. Long-term remission was achieved in 83% of the patients with resistance to hormone and (or) immunosuppressant after rituximab injection. They did not relapse during the maintenance therapy, and presented with no obvious adverse events. So far we have no available data of large-sample follow-up of IgG4-related hypophysitis. Most patients can achieve rapid remission after hormone therapy, but others require long-term hormone replacement therapy with good prognosis. The major prognostic factors are the time of diagnosis and multiorgan involvement.
IgG4-related hypophysitis is presented as functional damage and space-occupying lesion in the pituitary, probably with involvement of other organs. Some patients may be first diagnosed due to symptoms of the affected organs as the initial presentation, such as pancreatitis, cholangitis, and pneumonia. We report 1 case of IgG4-related hypophysitis with ophthalmopathy as the initial presentation. This case was combined with multiorgan involvement and relieved by glucocorticoid therapy combined with immunosuppressant. Later the case relapsed and achieved complete remission by rituximab injection. For patients with IgG4-related hypophysitis, not only hypophysitis, but also the involvement of other organs deserves evaluation before the formulation of treatment regimen. Long-term follow-up is preferred.
Footnotes
Abbreviations: CRP = C-reactive protein, CT = computed tomography, IgG4 = immunoglobulin G4, IgG4-RD = IgG4-related disease, MRI = magnetic resonance imaging, PET-MRI = positron emission tomography-magnetic resonance imaging.
W-JG and QZ contributed equally to the work.
This case report did not receive any specific grant from any funding agency in the public, commercial, or not-for-profit sector.
Written informed consent was obtained by the patient for publication of this report and any accompanying images.
The authors have no conflicts of interest to disclose.
References
- [1].Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001;344:732–8. [DOI] [PubMed] [Google Scholar]
- [2].Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001;344:732–8. [DOI] [PubMed] [Google Scholar]
- [3].Cheuk W, Chan JK. IgG4-related sclerosing disease: a critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol 2010;17:303–32. [DOI] [PubMed] [Google Scholar]
- [4].Caturegli P, Newschaffer C, Olivi A, et al. Autoimmune hypophysitis. Endocr Rev 2005;26:599–614. [DOI] [PubMed] [Google Scholar]
- [5].van der Vliet HJ, Perenboom RM. Multiple pseudotumors in IgG4-associated multifocal systemic fibrosis. Ann Intern Med 2004;141:896–7. [DOI] [PubMed] [Google Scholar]
- [6].Wong S, Lam WY, Wong WK, et al. Hypophysitis presented as inflammatory pseudotumor in immunoglobulin G4-related systemic disease. Hum Pathol 2007;38:1720–3. [DOI] [PubMed] [Google Scholar]
- [7].Leporati P, Landek-Salgado MA, Lupi I, et al. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab 2011;96:1971–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yamamoto M, Takahashi H, Ohara M, et al. A case of Mikulicz's disease (IgG4-related plasmacytic disease) complicated by autoimmune hypophysitis. Scand J Rheumatol 2006;35:410–1. [DOI] [PubMed] [Google Scholar]
- [9].Tanabe T, Tsushima K, Yasuo M, et al. IgG4-associated multifocal systemic fibrosis complicating sclerosing sialadenitis, hypophysitis, and retroperitoneal fibrosis, but lacking pancreatic involvement. Intern Med 2006;45:1243–7. [DOI] [PubMed] [Google Scholar]
- [10].Taniguchi T, Hamasaki A, Okamoto M. A case of suspected lymphocytic hypophysitis and organizing pneumonia during maintenance therapy for autoimmune pancreatitis associated with autoimmune thrombocytopenia. Endocr J 2006;53:563–6. [DOI] [PubMed] [Google Scholar]
- [11].Wong S, Lam WY, Wong WK, et al. Hypophysitis presented as inflammatory pseudotumor in immunoglobulin G4-related systemic disease. Hum Pathol 2007;38:1720–3. [DOI] [PubMed] [Google Scholar]
- [12].Ralli S, Lin J, Farrell J. Autoimmune pancreatitis. N Engl J Med 2007;356:1586. [DOI] [PubMed] [Google Scholar]
- [13].Tsuboi H, Inokuma S, Setoguchi K, et al. Inflammatory pseudotumors in multiple organs associated with elevated serum IgG4 level: recovery by only a small replacement dose of steroid. Intern Med 2008;47:1139–42. [DOI] [PubMed] [Google Scholar]
- [14].Isaka Y, Yoshioka K, Nishio M, et al. A case of IgG4-related multifocal fibrosclerosis complicated by central diabetes insipidus. Endocr J 2008;55:723–8. [DOI] [PubMed] [Google Scholar]
- [15].Taji H, Takamura T, Mohri K, et al. A male case of lymphocytic hypophysitis accompanied by IgG4- related disease. Folia Endocrinol Jpn 2009;85suppl:42–4. [Google Scholar]
- [16].Osawa S, Ogawa Y, Watanabe M, et al. Hypophysitis presenting with atypical rapid deterioration: with special reference to immunoglobulin G4-related disease-case report. Neurol Med Chir (Tokyo) 2009;49:622–5. [DOI] [PubMed] [Google Scholar]
- [17].Fujisawa I. Diagnostic imaging for lymphocytic hypophysitis and IgG4-related disease in hypothalammus and pituitary. Folia Endocrinol Jpn 2009;85suppl:54–5. [Google Scholar]
- [18].Hori M, Makita N, Andoh T, et al. Long-term clinical course of IgG4-related systemic disease accompanied by hypophysitis. Endocr J 2010;57:485–92. [DOI] [PubMed] [Google Scholar]
- [19].Haraguchi A, Era A, Yasui J, et al. Putative IgG4-related pituitary disease with hypopituitarism and/or diabetes insipidus accompanied with elevated serum levels of IgG4. Endocr J 2010;57:719–25. [DOI] [PubMed] [Google Scholar]
- [20].Yukawa E, Nitta N, Taoka T, et al. A patient with IgG4-related systemic disease detected due to the presence of optic disc edema. Ganka Rinsho Kiyo 2010;3:1207–10. [Google Scholar]
- [21].Ohyama K1, Koike H, Takahashi M, et al. Clinicopathological analysis of hypophysitis in IgG4-related disease. Rinsho Shinkeigaku 2014;54:1047–9. [DOI] [PubMed] [Google Scholar]
- [22].Nagai K, Hara Y, Shinkai M, et al. A case of IgG4-related disease with deterioration in pulmonary and pituitary involvements during a 10-year clinical course of inflammatory pseudotumor. Nihon Kokyuki Gakkai Zasshi 2011;49:922–8. [PubMed] [Google Scholar]
- [23].Kaneda M, Sagara A, Hosokawa M, et al. Case report: a case of IgG4-related multifocal fibrosclerosis complicating central diabetes insipidus, sclerosing sialadenitis, and retroperitoneal fibrosis. Naika 2011;107:175–9. [Google Scholar]
- [24].Kotera N, Isogawa A, Uchida L, et al. Case report: IgG4-related hypophysitis presenting with secondary adrenal insufficiency and central diabetes insipidus in a type 1 diabetes patient. Nihon Naika Gakkai Zasshi 2011;100:1044–7. [DOI] [PubMed] [Google Scholar]
- [25].Patel SM, Szostek JH. IgG4-related systemic disease in a Native American man. Intern Med 2011;50:931–4. [DOI] [PubMed] [Google Scholar]
- [26].Nishina N, Kaneko Y, Kuwana M, et al. IgG4-related disease without overexpression of IgG4: pathogenesis implications. Case Rep Rheumatol 2012;2012:754935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Takata M, Miyoshi M, Kohno M, et al. Two cases of IgG4-related systemic disease arising from urinary tract. Hinyokika Kiyo 2012;58:613–6. [PubMed] [Google Scholar]
- [28].Shimatsu A, Nanba K, Kuwabara T. Clinical features and diagnosis of “lymphocytic” infundibulo-hypophysitis. Folia Endocrinol Jpn 2012;88suppl:68–71. [Google Scholar]
- [29].Hattori Y, Tahara S, Ishii Y, et al. A case of IgG4-related hypophysitis without pituitary insufficiency. J Clin Endocrinol Metab 2013;98:1808–11. [DOI] [PubMed] [Google Scholar]
- [30].Hsing MT, Hsu HT, Cheng CY, et al. IgG4-related hypophysitis presenting as a pituitary adenoma with systemic disease. Asian J Surg 2013;36:93–7. [DOI] [PubMed] [Google Scholar]
- [31].Chiba K, Kamisawa T, Tabata T, et al. Clinical features of 10 patients with IgG4-related retroperitoneal fibrosis. Intern Med 2013;52:1545–51. [DOI] [PubMed] [Google Scholar]
- [32].Caputo C, Bazargan A, McKelvie PA, et al. Hypophysitis due to IgG4-related disease responding to treatment with azathioprine: an alternative to corticosteroid therapy. Pituitary 2014;17:251–6. [DOI] [PubMed] [Google Scholar]
- [33].Bando H, Iguchi G, Fukuoka H, et al. The prevalence of IgG4-related hypophysitis in 170 consecutive patients with hypopituitarism and/or central diabetes insipidus and review of the literature. Eur J Endocrinol 2013;170:161–72. [DOI] [PubMed] [Google Scholar]
- [34].Iseda I, Hida K, Tone A, et al. Prednisolone markedly reduced serum IgG4 levels along with the improvement of pituitary mass and anterior pituitary function in a patient with IgG4-related infundibulo-hypophysitis. Endocr J 2014;61:195–203. [DOI] [PubMed] [Google Scholar]
- [35].Ohkubo Y, Sekido T, Takeshige K, et al. Occurrence of IgG4-related hypophysitis lacking IgG4-bearing plasma cell infiltration during steroid therapy. Intern Med 2014;53:753–7. [DOI] [PubMed] [Google Scholar]
- [36].Sosa GA, Bell S, Christiansen SB, et al. Histologically confirmed isolated IgG4-related hypophysitis: two case reports in young women. Endocrinol Diabetes Metab Case Rep 2014;2014:140062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Harano Y, Honda K, Akiyama Y, et al. A case of IgG4-related hypophysitis presented with hypopituitarism and diabetes insipidus. Clin Med Insights Case Rep 2015;8:23–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Batista RL, Ramos LS, Cescato VA, et al. Thickened pituitary stalk associated with a mass in the sphenoidal sinus: an alarm to suspect hypophysitis by immunoglobulin G4? Int Arch Otorhinolaryngol 2015;19:273–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ngaosuwan K, Trongwongsa T, Shuangshoti S. Clinical course of IgG4-related hypophysitis presenting with focal seizure and relapsing lymphocytic hypophysitis. BMC Endocr Disord 2015;15:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zeng RR, Cai ST, Sun G, et al. Analysis on clinical features of IgG4-related diseases. Chin J Pract Intern Med 2015;35:242–5. [Google Scholar]
- [41].Tabata T, Kamisawa T, Takuma K, et al. Serial changes of elevated serum IgG4 levels in IgG4-related systemic disease. Intern Med 2011;50:69–75. [DOI] [PubMed] [Google Scholar]
- [42].Khosroshahi A, Wallace ZS, Crowe JL, et al. Second International Symposium on IgG4-Related Disease. International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease. Arthritis Rheumatol 2015;67:1688–99. [DOI] [PubMed] [Google Scholar]
- [43].Kamisawa T, Okazaki K, Kawa S, et al. Working Committee of the Japan Pancreas Society and the Research Committee for Intractable Pancreatic Disease supported by the Ministry of Health, Labour and Welfare of Japan. Amendment of the Japanese Consensus Guidelines for Autoimmune Pancreatitis, 2013 III. Treatment and prognosis of autoimmune pancreatitis. J Gastroenterol 2014;49:961–70. [DOI] [PubMed] [Google Scholar]
- [44].Kamisawa T, Shimosegawa T, Okazaki K, et al. Standard steroid treatment for autoimmune pancreatitis. Gut 2009;58:1504–7. [DOI] [PubMed] [Google Scholar]
- [45].Ghazale A, Chari ST. Optimising corticosteroid treatment for autoimmune pancreatitis. Gut 2007;56:1650–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hart PA, Topazian MD, Witzig TE, et al. Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: the Mayo Clinic experience. Gut 2013;62:1607–15. [DOI] [PubMed] [Google Scholar]