

Abstract
Mre11, Rad50, and Nbs1 form an evolutionarily conserved protein complex (Mre11–Rad50–Nbs1, MRN) that has been proposed to function as a DNA damage sensor. Hypomorphic mutations in Mre11 and Nbs1 result in the human ataxia–telangiectasia-like disorder and Nijmegen breakage syndrome (NBS), respectively. In contrast, complete inactivation of Mre11, Rad50, or Nbs1 leads to early embryonic lethality, suggesting that the hypomorphic mutations may fail to reveal some of the essential functions of MRN. Here, we use Cre–loxP-mediated recombination to restrict Nbs1 deletion to B lymphocytes. We find that disruption of Nbs1 results in the accumulation of high levels of spontaneous DNA damage, impaired proliferation, and chromosomal endoreduplication. Moreover, we show that Ig class-switch recombination (CSR) is diminished in Nbs1-deficient B cells. The CSR defect is B cell-intrinsic, independent of switch-region transcription, and a consequence of inefficient recombination at the DNA level. Our findings reveal that Nbs1 is critical for efficient Ig CSR and maintenance of the integrity of chromosomal structure and number.
In response to antigen, the mature B-cell repertoire is diversified by means of somatic hypermutation (SHM) and class-switch recombination (CSR). SHM alters Ab affinities by introducing point mutations in the variable region of both heavy- and light-chain genes (1). CSR modulates Ab effector functions by replacing the IgM constant region with a downstream CH gene, switching the Ab isotype expressed while retaining antigen-binding specificity (1, 2). SHM and CSR depend on the expression of activation-induced cytidine deaminase (AID; refs. 3 and 4). The currently available evidence strongly suggests that AID initiates CSR and SHM by direct deamination of cytidines in DNA (1, 2, 5), although this issue remains controversial (5, 6).
CSR is a region-specific recombination reaction that involves the joining of large, repetitive switch regions (S regions) located upstream of each of the constant region genes (1, 2). Transcription through S regions, which is controlled by upstream intronic promoters, makes these regions accessible to modification by AID (1, 2). The precise mechanism by which deaminated cytidines are processed to generate double-strand-break (DSB) intermediates is unclear (7, 8), but it has been proposed that base excision repair and/or mismatch repair enzymes convert AID-induced lesions into DSBs (8–11). After DSB generation and synapsis of the broken ends, CSR is completed by the ligation of donor and acceptor S regions via the nonhomologous end-joining pathway (12–15).
During CSR, AID-induced DSBs in Ig S regions are associated with the phosphorylation of histone H2AX (γ-H2AX) and the accumulation of the DNA damage-response proteins Nbs1 and 53BP1 at the IgH locus (16, 17). H2AX (15, 16, 18) and 53BP1 (19, 20) are required for efficient CSR, and it has been proposed that γ-H2AX and 53BP1 facilitate the synapsis of broken S-region DNA before repair by nonhomologous end-joining (15, 17, 19, 20).
Nbs1 is a component of the Mre11–Rad50–Nbs1 (MRN) complex, which has been implicated in many aspects of DSB detection and processing (21, 22). Because null mutations in MRN components lead to early embryonic lethality (23–26), it has been difficult to determine the precise role of these proteins in mammalian cells. Hypomorphic mutations in Nbs1 and Mre11 have been found in the rare autosomal recessive diseases Nijmegen breakage syndrome (NBS) and ataxia–telangiectasia-like disorder, respectively. NBS patients (27, 28) display a wide range of clinical features including growth retardation, radiation sensitivity, and an increased cancer risk. Also, deficiencies in both cellular and humoral immunity are found in NBS patients. Commonly reported cellular defects include a low proportion of helper CD4+ T cells, decreased CD4+/CD8+ ratio, defective T cell proliferation, and chromosomal translocations (including those involving antigen receptor genes) in mitogen stimulated T cells. NBS patients also display deficiencies in serum IgA and IgG4, suggesting a defect in CSR (29, 30). However, impaired helper T cell function could also contribute to the reduction in switched isotypes in these patients. Here, we examine the role of Nbs1 in DNA replication, genomic stability, and CSR by using a conditional mouse model in which Nbs1 is inactivated in B lymphocytes.
Materials and Methods
Mice. Nbs1Δ/- (CD19cre+Nbs1-/loxP) mice were generated as described in Fig. 4 (which is published as supporting information on the PNAS web site), and they were bred and maintained under specific pathogen-free conditions. All work was performed under protocols approved by the National Cancer Institute and The Rockefeller University Institutional Animal Care and Use Committee.
Lymphocyte Cultures, Flow Cytometry, and Cell Sorting. Lymphocyte cultures, flow cytometry, and cell sorting were performed as described (15). DNA content was analyzed by flow cytometry on fixed cells stained with propidium iodide (5 μg/ml).
S-Phase Checkpoint Assay. For intra-S-phase checkpoints, B cells were challenged with different doses of γ-irradiation after stimulation with LPS and IL-4. Cells were pulsed immediately with [3H]thymidine (5 μCi; 1 Ci = 37 GBq) for 4 h, and thymidine uptake was measured.
Chromosome Analysis. B cells stimulated with LPS and IL-4 for 48 h were arrested at mitosis with Colcemid (GIBCO/BRL) treatment at 0.1 μg/ml for 1 h. Mitotic chromosome spreads were prepared, and spectral-karyotype analysis was performed as described (18).
PCR, Mutation Analysis, and Quantitative Real-Time RT-PCR. Primers, PCR, and real-time RT-PCR conditions were as described (15).
Abs, Immunofluorescence, Immunocytochemistry FISH, and Western Blotting. We used the following Abs: rabbit polyclonal anti-Nbs1 (31), mouse anti-β-actin (Sigma, clone AC-74), rabbit anti-Mre11 (a gift from J. H. Petrini, Memorial Sloan–Kettering Cancer Center, New York), and goat anti-rabbit Alexa Fluor 568. Immunofluorescence and immunocytochemstry FISH were performed as described (16). Western blot analyses were performed by using standard techniques.
ELISA. Serum samples from 5- to 8-week-old mice were collected. Total serum Ig was assayed by ELISA as described (18). Titers are expressed as the serum dilution that gave half-maximum optical density.
Results
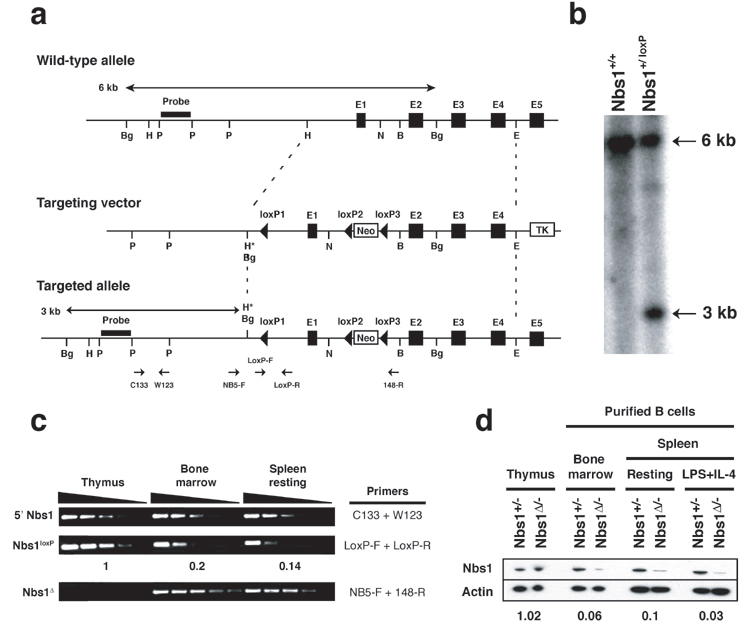
Conditional Inactivation of Nbs1 in B Cells. To explore the role of Nbs1 in somatic tissues, we produced a conditional allele of Nbs1 by flanking exon 1 and regulatory elements with loxP sites (Fig. 4). Nbs1 was deleted from the genome in B cells by expressing Cre recombinase under the control of the CD19 promoter in mice that carried one inactivated and one loxP-flanked Nbs1 allele (CD19cre+Nbs1-/loxP, designated Nbs1Δ/-; Fig. 4). CD19 is expressed initially among early B cell precursors, and it continues to be expressed throughout the B cell lineage. Consistent with this pattern of expression, we found a high rate of Cre-mediated deletion (Fig. 4) and a significant decrease in Nbs1 protein levels in mature B cells, as measured by Western blotting (Fig. 4). Despite Nbs1 deletion in most developing B cells, we found normal B cell development and equivalent representation of all B cell types in Nbs1Δ/- and control mice, as determined by flow cytometry (see Fig. 5, which is published as supporting information on the PNAS web site). Thus, although Nbs1 is essential for embryogenesis (25, 26), there is no specific block in B cell development in Nbs1Δ/- mice.
Intra-S-Phase Checkpoint and Proliferation Defects in Nbs1Δ/- Cells. In NBS patient cells and murine models of NBS, the intra-S-phase checkpoint is partially defective, and there is a reduction in the nuclear levels of Mre11 (22). To determine the effect of loss of Nbs1 on the S phase checkpoint, we irradiated B cells after stimulation in vitro with LPS and IL-4. Like the Nbs1 hypomorphic mutations, Nbs1Δ/- B cells exhibited radioresistant DNA synthesis indicative of an S-phase checkpoint defect (Fig. 1a). Moreover, in the absence of damage, Mre11 exhibited a predominantly cytoplasmic distribution (Fig. 1b). Thus, Nbs1 null mutation resembles the hypomorphic mutation in terms of the intra-S-phase-checkpoint defect and redistribution of Mre11 to the cytoplasm.
Fig. 1.

Abnormal cell-cycle progression, proliferation, and increased mortality in Nbs1Δ/- B cells. (a) Thymidine incorporation after γ-irradiation of Nbs1+/- (▪) and Nbs1Δ/- (□) B cells stimulated for 48 h with LPS and IL-4. Data from triplicates are normalized to the counts in unirradiated samples of the same genotype. (b) Distribution of Mre11 in Nbs1Δ/- and Nbs1+/- B cells stimulated for 48 h with LPS and IL-4. Confocal images were optically sectioned at 0.5-μm intervals and merged into a maximum projection. The percentage of cells with nuclear or cytoplasmic staining is indicated. (c) Flow-cytometric analysis of cell division as measured by CFSE dye dilution in Nbs1Δ/- and Nbs1+/- B cells stimulated for 72 h with LPS and IL-4. Percentage of cells having undergone 0, 1, and 2 divisions is indicated. (d) Flow-cytometric analysis of DNA content in Nbs1Δ/- and Nbs1+/- B cells stimulated for 48 h with LPS and IL-4. Percentage of cells in G1, S, and G2/M phases of the cell cycle and with a DNA content of 4n is indicated. Representative histograms from five independent experiments are shown. The percentage of cells with 4n DNA content was 1 ± 0.11% in Nbs1Δ/- and 0.06 ± 0.01% in Nbs1+/-cells. (e) Cell viability of CFSE-labeled Nbs1Δ/- and Nbs1+/- B cells after 60 h in culture with LPS and IL-4 as determined by flow cytometry. Percentage of live (Topro-3-negative) CFSE+ cells is indicated.
To examine the effects of the null mutation on B cell proliferation, we tracked cell division by labeling cells with carboxyfluorescein diacetate succinimidyl ester (CFSE), a vital dye whose fluorescence intensity is reduced by one-half with each generation. Cell-cycle distribution was assessed by f low-cytometric analysis of cellular DNA content. After 72 h, 86% of the control cells had completed three or more cell divisions, as compared with 39% of the Nbs1Δ/- cells (Fig. 1c). Consistent with decreased cell division, we found accumulation of Nbs1Δ/- B cells in the G2/M phase of the cell cycle (Fig. 1d) and a relative increase in cell death (Fig. 1e). Whereas cells in control cultures doubled in the first 72 h of stimulation, the number of live B lymphocytes in Nbs1Δ/- cultures was reduced by 70%. Also, there was a significant increase in the number of cells with 4n DNA content (Fig. 1d; 1 ± 0.11% in Nbs1Δ/- vs. 0.06 ± 0.01% in control B cells in five experiments). We conclude that Nbs1 is required for normal levels of B cell proliferation in vitro.
Genomic Instability in Nbs1Δ/- Cells. DSBs accumulate during DNA replication in Mre11-depleted Xenopus cell-free extracts (32), and in Mre11 knockout DT40 cells (33). To determine whether Nbs1 is required for genomic stability in mammalian cells, we examined metaphase spreads from B lymphocytes stimulated for 48 h with LPS and IL-4. Whereas 1% of wild type and 8% of CD19cre+Nbs1+/- B cells showed aberrant metaphases, mainly limited to chromatid breaks, 76% of the Nbs1Δ/- B cells contained diverse structural abnormalities, including chromatid breaks, radial-type structures that involved exchanges between nonhomologous chromosomes, and dicentric chromosomes (Fig. 2 and Table 1). Furthermore, spectral-karyotype analysis revealed that 16.7% of the Nbs1Δ/- B cells showed chromosomal translocations, whereas none were observed in control B cells (Nbs1Δ/-: 5/30; Nbs1+/-: 0/15). In addition to these structural aberrations, there were tetraploid Nbs1Δ/- cells including metaphases in which all four sister chromatids were paired (Fig. 2b and Table 1), confirming the increase in B cells with 4n DNA content detected by flow cytometry (Fig. 1d). Thus, loss of Nbs1 allows cells to undergo more than a single round of DNA replication per cell cycle. Consistent with this finding, immortalized human NBS fibroblasts display endoreduplication and tetraploidy when a dominant interfering N-terminal Nbs1 fragment is ectopically expressed in these cells in vitro (34). We conclude that physiological levels of Nbs1 are required to maintain the integrity of chromosome structure as well as chromosome number.
Fig. 2.

Chromosomal instability in Nbs1Δ/- B cells. (a) Representative metaphase prepared from Nbs1Δ/- (CD19cre+Nbs1-/loxP) B cells after 2 days of stimulation with LPS and IL-4. Arrows indicate chromatid breaks; arrowhead indicates a radial structure. (b) An example of endoreduplication in Nbs1Δ/- cells. Spectral-karyotype analysis shows that all four sister chromatids are paired (Inset). Arrows indicate chromatid breaks. (c) Examples of radial-type chromosome structures in which genetic material is exchanged between nonhomologous chromosomes. Results are derived from two independent experiments.
Table 1. Summary of cytogenic changes observed in Nbs1Δ/- and control (CD19cre-Nbs+/+ and CD19cre+Nbs+/-) B cells.
| Genotype | Metaphases analyzed | Tetraploid cells, % | Radial structures | Chromatid breaks | Other aberrations* | % aberrant metaphases | Mean no. of aberrations per cell |
|---|---|---|---|---|---|---|---|
| CD19cre-Nbs1+/+ | 103 | 0 | 0 | 1 (0.01/cell) | 0 | 1 | 0.01 |
| CD19cre+Nbs1+/- | 100 | 1 (1.0) | 0 | 8 (0.08/cell) | 0 | 8 | 0.08 |
| CD19cre+Nbs1Δ/- | 204 | 14 (6.9)† | 65 (0.32/cell) | 366 (1.79/cell) | 21 (0.1/cell) | 76 | 2.22 |
Includes dicentrics and chromosome breaks
Includes metaphase spreads with endoreduplication
Defective CSR in Nbs1Δ/- B Cells. Human patients with hypomorphic mutations in Nbs1 have lower levels of serum Igs (29, 30). However, similar Nbs1 mutations in mice revealed no intrinsic defect in CSR (35, 36), whereas complete inactivation of Nbs1 resulted in embryonic lethality (25, 26). To determine whether Nbs1 deficiency affects CSR, we measured serum Ig levels in Nbs1Δ/- mice. Sera from 5- to 8-week old Nbs1Δ/- and littermate control mice were analyzed for IgM, IgG, IgG1, IgG2b, and IgG3 by ELISA. We found that IgM serum levels were similar in Nbs1Δ/- and control mice, but all other analyzed isotypes were reduced in Nbs1Δ/- mice (Fig. 3a). We conclude that Nbs1 is required for the accumulation of switched isotypes in vivo.
Fig. 3.

Impaired CSR in Nbs1Δ/- B cells. (a) Sera from 5- to 8-week-old Nbs1Δ/- (CD19cre+Nbs1-/loxP, □) and age-matched Nbs1+/- (CD19cre+Nbs1+/-, ▪) mice were collected, and total IgM, IgG, IgG1, IgG2b, and IgG3 levels were determined by ELISA. Data are plotted as the average dilution that gave half-maximum optical density in five mice of each genotype. (b) Flow-cytometric analysis of Nbs1Δ/- and Nbs1+/- B cells stimulated with LPS and IL-4 for 3 days. Cell division as measured by CFSE dye dilution is shown in Upper. The percentage of cells expressing IgG1 after a specific number of cell divisions is indicated in Lower. (c) Western blot analysis of murine Nbs1 in whole-cell extracts prepared from purified splenic B cells before stimulation and sorted for two or three cell divisions after stimulation with LPS and IL-4 (2D and 3D, respectively). Numbers reflect the intensity of bands representing Nbs1 protein in Nbs1Δ/- relative to Nbs1+/- after normalization to actin. Data from two independent experiments are shown. (d) Real-time RT-PCR for IgM sterile transcript (IgM ST), IgG1 sterile transcript (IgG1 ST), and IgG1 circle transcript after switch (IgG1 CT) in Nbs1+/- (filled bars) and Nbs1Δ/- (open bars) B cells stimulated with LPS and IL-4 and sorted after two or three cell divisions (2D and 3D, respectively). Results from two independent experiments are expressed as fold change relative to Nbs1+/- after normalization to GAPDH. (e) Mutations in Sμ determined in Nbs1+/- and Nbs1Δ/- IgM+ B cells sorted for five cell divisions. Segment sizes in the pie charts are proportional to the number of sequences carrying the number of mutations indicated in the periphery of the charts. The mutation frequency and the total number of independent sequences that were analyzed are indicated below and in the center of each chart, respectively. Statistical significance was determined by a two-tailed t test assuming unequal variance and by comparing with resting B cells from wild-type mice (Nbs1+/-, P = 0.01; Nbs1Δ/-, P = 0.69) or comparing Nbs1+/- with Nbs1Δ/-(P = 0.007). The number of mutations was as follows: Nbs1+/-, 20 mutations, 51,304 bp; Nbs1Δ/-, five mutations, 58,800 bp. (f) Histogram depicting the percentage of sequences with the indicated length of microhomologies at Sμ/Sγ1 junctions in Nbs1+/- (filled bars) and Nbs1Δ/- (open bars) B cells. Overlap was determined by identifying the longest region at the switch junction of perfect uninterrupted donor/acceptor identity. Statistical significance was determined by Student's one-tailed t test (P = 0.07). *, Includes junction sequences with small 2- to 4-bp insertions (Nbs1+/-,2/34; Nbs1Δ/-,4/29). (g) Mutations in the vicinity of the junctions obtained from Nbs1+/- and Nbs1Δ/- B cells. Statistical significance was determined by a two-tailed t test assuming unequal variance and comparing Nbs1+/- with Nbs1Δ/-(P = 0.28).
To determine whether the decrease in serum isotype levels in Nbs1Δ/- mice is due to a B cell-intrinsic defect in CSR, we induced CSR to IgG1 in vitro by stimulating B cells from Nbs1Δ/- and control mice with LPS and IL-4. To rule out potential effects of abnormal cell proliferation and survival on the switch reaction, we assessed IgG1 surface expression by flow cytometry on live CFSE labeled B cells. The percentage of B cells expressing IgG1 after 3 days in culture was determined for cell populations having undergone different numbers of cell divisions by gating on CFSE intensity (Fig. 3b). We found that CSR to IgG1 in B cells from Nbs1Δ/- was impaired (Fig. 3b). The decrease in CSR varied with the number of cell divisions, with an increased rate of reduction in cells that had undergone fewer divisions and ranging from 87% after two divisions to 44% after six divisions (Fig. 3b). The decrease was most apparent in cells that had undergone only two or three cell divisions, suggesting that cells with residual Nbs1 protein had a selective advantage in survival, cell division, and CSR (Fig. 3b).
To determine whether Nbs1 protein levels varied among Nbs1Δ/- B cells undergoing cell division, we purified B cells that had completed two or three cell divisions by cell sorting, and we measured Nbs1 protein levels by Western blotting. We found that Nbs1 protein levels dropped by 20- to 50-fold after two cell divisions but that cells that survived three cell divisions had levels of Nbs1 similar to unstimulated Nbs1Δ/- B cells (Fig. 3c, two experiments). Thus, Nbs1Δ/- B cells that have lost both Nbs1 alleles dilute residual protein during cell division, but these cells are preferentially lost from the population, whereas the few B cells that retain Nbs1 continue to divide and can also undergo CSR.
Sterile transcription through Ig S regions precedes CSR and is essential for recombination (1, 2). To determine whether Nbs1 deficiency affects S-region transcription, we measured Sμ and Sγ1 sterile transcripts by real-time RT-PCR from Nbs1Δ/- and control B cells. To rule out potential effects of abnormal cell proliferation and survival, we performed this experiment on sorted live-cell populations that had completed two or three cell divisions (Fig. 3d, two experiments). We found that Sμ and Sγ1 sterile transcripts were similar in Nbs1Δ/- and control B cells (Fig. 3d). Thus, impaired CSR in Nbs1Δ/- B cells is not due to alterations in S-region transcription.
To determine whether the defect in CSR is at the level of DNA recombination, we measured the IgG1 postswitch transcript produced by the circular episomes excised from the genome after successful CSR (37). We found that purified Nbs1Δ/- B-cell populations that had completed two or three cell divisions had an average 3-fold reduction in IgG1 circle transcripts, compared with controls (Fig. 3d, two experiments). This reduction in circle transcripts is similar to the decrease in B-cell surface IgG1 expression in Nbs1Δ/- cells (Fig. 3b). We conclude that Nbs1 is required for efficient DNA recombination during CSR.
Ig S regions and the DNA sequences upstream of the S regions are mutated in B cells undergoing CSR by an AID- and Ku80-dependent mechanism (15–18, 38, 39). To determine whether Nbs1 is required for mutation in the 5′ region of Sμ, we analyzed IgM+ B cells that had completed five cell divisions after stimulation with LPS and IL-4 in vitro. We found that Nbs1+/- IgM+ B cells that had completed five cell divisions had a mutation frequency in the 5′ of Sμ of 3.9 × 10-4 mutations per bp sequenced, similar to what has been reported for wild-type B cells (Fig. 3e; refs. 15–18, 38, and 39). In contrast, the mutation frequency in Nbs1Δ/- B cells was indistinguishable from PCR background error (Fig. 3e;0.85 × 10-4; P = 0.69 vs. background, and P = 0.007 vs. Nbs1+/-). We conclude that Sμ mutation is Nbs1-dependent.
Although CSR is inefficient in the absence of Nbs1, some cells do switch to IgG1 in response to LPS and IL-4 (Fig. 3b). To determine whether the CSR junctions are normal, we cloned and sequenced IgG1 CSR junctions from Nbs1Δ/- (n = 29) and Nbs1+/- (n = 34) B cells. Analysis of the Nbs1Δ/- switch junctions revealed no statistically significant differences in the extent of donor/acceptor homology at the junctions or the average length of overlap (Nbs1Δ/-, 0.85 bp, vs. Nbs1Δ/-, 1.44 bp; P = 0.07; Fig. 3f). Also, the mutation frequency in the vicinity of the junctions (±50 bp) was similar between Nbs1Δ/- and Nbs1+/- B cells (Nbs1+/-, 3.4 × 10-2, vs. Nbs1Δ/-, 4.1 × 10-2; P = 0.28; Fig. 3f). We conclude that S-region joining is not altered significantly in Nbs1Δ/- B cells.
Colocalization of Mre11 at the IgH Locus Depends on AID. In B cells undergoing CSR, S-region DSBs are associated with γ-H2AX and Nbs1 in an AID-dependent manner (16). We used immunocytochemistry and FISH to determine whether Mre11 forms AID-dependent CSR-associated foci. In wild-type B cells, 50% of the cells containing IgH signals and Mre11 foci showed colocalization (n = 54; Fig. 6, which is published as supporting information on the PNAS web site). In contrast, colocalization of IgH and Mre11 foci was not significant in AID-/- B cells (6%; n = 92; Fig. 6). We conclude that Mre11 accumulation at sites of DNA damage during CSR is AID-dependent.
Discussion
AID-induced DSBs are intermediates in CSR, but they can also lead to chromosome translocations involving switch loci (40). This type of self-inflicted DNA damage is normally processed by H2AX, 53BP1, and ATM, as well as components of the nonhomologous end-joining pathway, such as Ku70, Ku80, and DNA-PKcs (12–20, 41). Although the precise role of H2AX, 53BP1, and ATM in CSR is not defined, each of these factors is required for efficient CSR, possibly by facilitating S-region synapsis (15, 17, 19, 20). Structural and biochemical studies have shown that the MRN complex oligomerizes on linear DNA, bridges DNA ends together, and stimulates intermolecular joining by nonhomologous end-joining (42–44). Similarly, MRN might promote joining of DSBs in S regions by tethering DNA ends and stimulating the ligation of synapsed termini.
Ig S regions are mutated in an AID- and Ku80-dependent manner in B cells undergoing CSR (15–18, 38, 39). Although the exact mechanism by which these mutations are generated is not known, it is believed that they are introduced during the end-processing step of the reaction before joining (15–18, 38, 39). Nbs1Δ/- B cells differ from H2AX, 53BP1, and ATM, and they resemble Ku80-knockout B cells in that mutations in Sμ do not accumulate in the absence of Nbs1. Nevertheless, Nbs1Δ/- B cells show normal CSR junctions and normal mutations in the regions surrounding the junctions. One explanation for this phenomenon would be that Nbs1Δ/- B cells that survived multiple rounds of cell division and remained IgM+ are enriched in cells that did not suffer DSBs in S regions. In this scenario, Nbs1Δ/- B cells that are unable to divide or repair DSBs in S regions would be preferentially removed from the population by cell death. It follows that Nbs1Δ/- B cells that do undergo CSR express some residual Nbs1. The finding that CSR junctions and mutations surrounding the junctions are normal in such cells is consistent with this hypothesis, as is the relative increase in Nbs1 protein in Nbs1Δ/- B cells undergoing several rounds of cell division. However, our experiments do not rule out the possibility that the Nbs1Δ/- B cells that undergo CSR are lost from the population because of selective defects in cellular proliferation and survival.
MRN is rapidly recruited to sites of DSBs, where it is involved in activating cell-cycle checkpoints (21, 22). ATM is the prototype signal transducer in the DNA damage response and hypomorphic mutations in Mre11 or Nbs1 result in disorders resembling ataxia–telangiectasia, suggesting that MRN and ATM function in similar pathways in vivo. Indeed, Nbs1 and Mre11 are required for the efficient autophosphorylation of ATM on serine 1981, the recruitment of activated ATM into irradiation-induced foci, and for the phosphorylation of downstream ATM targets (45–51). Interestingly, the reduction in CSR in Nbs1Δ/- B cells is of similar magnitude to that observed in ATM-/- mice (17, 41, 52). Thus, it is possible that during CSR, in addition to tethering DNA ends, Nbs1 may regulate ATM activation, its accumulation at S-region breaks, and the phosphorylation of downstream substrates required for CSR.
We have found that Nbs1 is required for the control of DNA replication and for efficient CSR in B lymphocytes, in agreement with the cytological observation that Nbs1 is deposited on sister chromatids in S phase (53) and at the Ig heavy-chain locus during CSR in the G1 phase of the cell cycle (16). Both the CSR and replication defects incurred by null mutations in Nbs1 are more severe and qualitatively different from that associated with hypomorphic mutations in the MRN complex. For example, mice harboring N-terminally truncated forms of Nbs1 are viable and show background levels of spontaneous DNA damage and no intrinsic defects in lymphocyte replication or CSR (35, 36). Similarly, B cells from mice harboring a hypomorphic mutation in Mre11 display relatively low levels of chromosomal aberrations (54). Although partial-function mutations in MRN affect cell-cycle checkpoint responses, they do not affect DNA repair (55, 56). The finding that NBS hypomorphic alleles are incompletely penetrant is consistent with the observation that NBS cells express a truncated Nbs1 protein that associates with Mre11 to produce a complex that retains some of the activities of authentic MRN (47, 57). In contrast, null mutations in components of the MRN complex in Saccharomyces cerevisiae (58) or in DT40 cells (33, 59) show profound defects in DNA repair by interfering with homologous recombination. The resulting block in replication is associated with high levels of spontaneous DNA damage and accumulation of radial-type chromosomal structures similar to those found in Nbs1Δ/- cells. We propose that Nbs1 deletion reveals an essential role for MRN in coordinating DNA repair in mammalian cells during G1 and S phases of the cell cycle.
In addition to high levels of structural aberrations, Nbs1Δ/- cells display alterations in chromosome number. In contrast, changes in DNA ploidy have not been observed in mice carrying hypomorphic mutations in Nbs1 or Mre11 (22). Significantly, we have found that complete loss of Nbs1 allowed cells to undergo cycles of endoreduplication. Although the mechanism for ensuring that DNA is replicated once each cell cycle is not understood fully, it is evident that rereplication is normally inhibited by blocking the loading of minichromosome maintenance proteins (MCMs) to the initiation complex after the origins of DNA replication have fired, a process which is regulated by the activity of cyclin-dependent kinases. Recently, MCM subunits have also been shown to be substrates for ATM and ATR (ATM- and Rad3-related) kinases, and disruption of MCM, like Nbs1, causes intra-S-phase-checkpoints defects (60). Because Nbs1 functions both upstream and downstream of checkpoint signaling pathways mediated by ATM/ATR, Nbs1 could also have a regulatory role in the activity of the MCM proteins. Further analysis of Nbs1Δ/- cells will be needed to determine precisely how Nbs1 contributes to the proper control of DNA replication.
Supplementary Material
Acknowledgments
We thank K. Rajewsky for providing the CD19-Cre transgenic mice; J. Petrini for providing Mre11 Abs; L. Tessarollo for assistance in gene targeting of Nbs1; Thomas Ried (Genetics Branch, National Cancer Institute) for providing spectral-karyotype kits; J. Gautier and A. Venkitaraman for stimulating discussions; M. Difilippantonio, O. F. Capetillo, E. Besmer, and M. Gellert for helpful suggestions on the manuscript; and H. Chen, K. Norris, M. Difilippantonio, and M. Kruhlak for genotyping and image analysis. This work was supported in part by grants from National Institutes of Health (to M.C.N.). M.C.N. is an Investigator of the Howard Hughes Medical Institute. B.R.-S.-M. is an Howard Hughes Medical Institute Postdoctoral Associate.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: CSR, class-switch recombination; MRN, Mre11–Rad50–Nbs1; NBS, Nijmegen breakage syndrome; AID, activation-induced cytidine deaminase; S region, switch region; DSB, double strand break; CFSE, carboxyfluorescein diacetate succinimidyl ester.
References
- 1.Li, Z., Woo, C. J., Iglesias-Ussel, M. D., Ronai, D. & Scharff, M. D. (2004) Genes Dev. 18, 1-11. [DOI] [PubMed] [Google Scholar]
- 2.Chaudhuri, J. & Alt, F. W. (2004) Nat. Rev. Immunol. 4, 541-552. [DOI] [PubMed] [Google Scholar]
- 3.Muramatsu, M., Kinoshita, K., Fagarasan, S., Yamada, S., Shinkai, Y. & Honjo, T. (2000) Cell 102, 553-563. [DOI] [PubMed] [Google Scholar]
- 4.Revy, P., Muto, T., Levy, Y., Geissmann, F., Plebani, A., Sanal, O., Catalan, N., Forveille, M., Dufourcq-Labelouse, R., Gennery, A., et al. (2000) Cell 102, 565-575. [DOI] [PubMed] [Google Scholar]
- 5.Neuberger, M. S., Harris, R. S., Di Noia, J. & Petersen-Mahrt, S. K. (2003) Trends Biochem. Sci. 28, 305-312. [DOI] [PubMed] [Google Scholar]
- 6.Honjo, T., Muramatsu, M. & Fagarasan, S. (2004) Immunity 20, 659-668. [DOI] [PubMed] [Google Scholar]
- 7.Begum, N. A., Kinoshita, K., Kakazu, N., Muramatsu, M., Nagaoka, H., Shinkura, R., Biniszkiewicz, D., Boyer, L. A., Jaenisch, R. & Honjo, T. (2004) Science 305, 1160-1163. [DOI] [PubMed] [Google Scholar]
- 8.Rada, C., Di Noia, J. M. & Neuberger, M. S. (2004) Mol. Cell 16, 163-171. [DOI] [PubMed] [Google Scholar]
- 9.Petersen-Mahrt, S. K., Harris, R. S. & Neuberger, M. S. (2002) Nature 418, 99-103. [DOI] [PubMed] [Google Scholar]
- 10.Rada, C., Williams, G. T., Nilsen, H., Barnes, D. E., Lindahl, T. & Neuberger, M. S. (2002) Curr. Biol. 12, 1748-1755. [DOI] [PubMed] [Google Scholar]
- 11.Di Noia, J. & Neuberger, M. S. (2002) Nature 419, 43-48. [DOI] [PubMed] [Google Scholar]
- 12.Rolink, A., Melchers, F. & Andersson, J. (1996) Immunity 5, 319-330. [DOI] [PubMed] [Google Scholar]
- 13.Manis, J. P., Gu, Y., Lansford, R., Sonoda, E., Ferrini, R., Davidson, L., Rajewsky, K. & Alt, F. W. (1998) J. Exp. Med. 187, 2081-2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casellas, R., Nussenzweig, A., Wuerffel, R., Pelanda, R., Reichlin, A., Suh, H., Qin, X. F., Besmer, E., Kenter, A., Rajewsky, K. & Nussenzweig, M. C. (1998) EMBO J. 17, 2404-2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reina-San-Martin, B., Difilippantonio, S., Hanitsch, L., Masilamani, R. F., Nussenzweig, A. & Nussenzweig, M. C. (2003) J. Exp. Med. 197, 1767-1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petersen, S., Casellas, R., Reina-San-Martin, B., Chen, H. T., Difilippantonio, M. J., Wilson, P. C., Hanitsch, L., Celeste, A., Muramatsu, M., Pilch, D. R., et al. (2001) Nature 414, 660-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reina-San-Martin, B., Chen, H. T., Nussenzweig, A. & Nussenzweig, M. C. (2004) J. Exp. Med. 200, 1103-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Celeste, A., Petersen, S., Romanienko, P. J., Fernandez-Capetillo, O., Chen, H. T., Sedelnikova, O. A., Reina-San-Martin, B., Coppola, V., Meffre, E., Difilippantonio, M. J., et al. (2002) Science 296, 922-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ward, I. M., Reina-San-Martin, B., Olaru, A., Minn, K., Tamada, K., Lau, J. S., Cascalho, M., Chen, L., Nussenzweig, A., Livak, F., et al. (2004) J. Cell Biol. 165, 459-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manis, J. P., Morales, J. C., Xia, Z., Kutok, J. L., Alt, F. W. & Carpenter, P. B. (2004) Nat. Immunol. 5, 481-487. [DOI] [PubMed] [Google Scholar]
- 21.D'Amours, D. & Jackson, S. P. (2002) Nat. Rev. Mol. Cell Biol. 3, 317-327. [DOI] [PubMed] [Google Scholar]
- 22.Stracker, T. H., Theunissen, J. W., Morales, M. & Petrini, J. H. (2004) DNA Repair 3, 845-854. [DOI] [PubMed] [Google Scholar]
- 23.Xiao, Y. & Weaver, D. T. (1997) Nucleic Acids Res. 25, 2985-2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo, G., Yao, M. S., Bender, C. F., Mills, M., Bladl, A. R., Bradley, A. & Petrini, J. H. (1999) Proc. Natl. Acad. Sci. USA 96, 7376-7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu, J., Petersen, S., Tessarollo, L. & Nussenzweig, A. (2001) Curr. Biol. 11, 105-109. [DOI] [PubMed] [Google Scholar]
- 26.Dumon-Jones, V., Frappart, P. O., Tong, W. M., Sajithlal, G., Hulla, W., Schmid, G., Herceg, Z., Digweed, M. & Wang, Z. Q. (2003) Cancer Res. 63, 7263-7269. [PubMed] [Google Scholar]
- 27.Varon, R., Vissinga, C., Platzer, M., Cerosaletti, K. M., Chrzanowska, K. H., Saar, K., Beckmann, G., Seemanova, E., Cooper, P. R., Nowak, N. J., et al. (1998) Cell 93, 467-476. [DOI] [PubMed] [Google Scholar]
- 28.Carney, J. P., Maser, R. S., Olivares, H., Davis, E. M., Le Beau, M., Yates, J. R., III, Hays, L., Morgan, W. F. & Petrini, J. H. (1998) Cell 93, 477-486. [DOI] [PubMed] [Google Scholar]
- 29.Pan, Q., Petit-Frere, C., Lahdesmaki, A., Gregorek, H., Chrzanowska, K. H. & Hammarstrom, L. (2002) Eur. J. Immunol. 32, 1300-1308. [DOI] [PubMed] [Google Scholar]
- 30.Hiel, J. A., Weemaes, C. M., van den Heuvel, L. P., van Engelen, G. G., Gabreels, F. J., Smeets, D. F., van der Burgt, I., Chrzanovska, K. H., Bernatowska, E., Krajewska-Walasek, M., et al. (2000) Arch. Dis. Child 82, 400-406.10799436 [Google Scholar]
- 31.Chen, H. T., Bhandoola, A., Difilippantonio, M. J., Zhu, J., Brown, M. J., Tai, X., Rogakou, E. P., Brotz, T. M., Bonner, W. M., Ried, T. & Nussenzweig, A. (2000) Science 290, 1962-1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Costanzo, V., Robertson, K., Bibikova, M., Kim, E., Grieco, D., Gottesman, M., Carroll, D. & Gautier, J. (2001) Mol. Cell 8, 137-147. [DOI] [PubMed] [Google Scholar]
- 33.Yamaguchi-Iwai, Y., Sonoda, E., Sasaki, M. S., Morrison, C., Haraguchi, T., Hiraoka, Y., Yamashita, Y. M., Yagi, T., Takata, M., Price, C., et al. (1999) EMBO J. 18, 6619-6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu, X., Avni, D., Chiba, T., Yan, F., Zhao, Q., Lin, Y., Heng, H. & Livingston, D. (2004) Genes Dev. 18, 1305-1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams, B. R., Mirzoeva, O. K., Morgan, W. F., Lin, J., Dunnick, W. & Petrini, J. H. (2002) Curr. Biol. 12, 648-653. [DOI] [PubMed] [Google Scholar]
- 36.Kang, J., Bronson, R. T. & Xu, Y. (2002) EMBO J. 21, 1447-1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kinoshita, K., Harigai, M., Fagarasan, S., Muramatsu, M. & Honjo, T. (2001) Proc. Natl. Acad. Sci. USA 98, 12620-12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagaoka, H., Muramatsu, M., Yamamura, N., Kinoshita, K. & Honjo, T. (2002) J. Exp. Med. 195, 529-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schrader, C. E., Bradley, S. P., Vardo, J., Mochegova, S. N., Flanagan, E. & Stavnezer, J. (2003) EMBO J. 22, 5893-5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramiro, A. R., Jankovic, M., Eisenreich, T., Difilippantonio, S., Chen-Kiang, S., Muramatsu, M., Honjo, T., Nussenzweig, A. & Nussenzweig, M. C. (2004) Cell 118, 431-438. [DOI] [PubMed] [Google Scholar]
- 41.Lumsden, J. M., McCarty, T., Petiniot, L. K., Shen, R., Barlow, C., Wynn, T. A., Morse, H. C., III, Gearhart, P. J., Wynshaw-Boris, A., Max, E. E. & Hodes, R. J. (2004) J. Exp. Med. 200, 1111-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Jager, M., van Noort, J., van Gent, D. C., Dekker, C., Kanaar, R. & Wyman, C. (2001) Mol. Cell 8, 1129-1135. [DOI] [PubMed] [Google Scholar]
- 43.Chen, L., Trujillo, K., Ramos, W., Sung, P. & Tomkinson, A. E. (2001) Mol. Cell 8, 1105-1115. [DOI] [PubMed] [Google Scholar]
- 44.Paull, T. T. & Gellert, M. (2000) Proc. Natl. Acad. Sci. USA 97, 6409-6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carson, C. T., Schwartz, R. A., Stracker, T. H., Lilley, C. E., Lee, D. V. & Weitzman, M. D. (2003) EMBO J. 22, 6610-6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Horejsi, Z., Falck, J., Bakkenist, C. J., Kastan, M. B., Lukas, J. & Bartek, J. (2004) Oncogene, in press. [DOI] [PubMed]
- 47.Lee, J. H. & Paull, T. T. (2004) Science 304, 93-96. [DOI] [PubMed] [Google Scholar]
- 48.Uziel, T., Lerenthal, Y., Moyal, L., Andegeko, Y., Mittelman, L. & Shiloh, Y. (2003) EMBO J. 22, 5612-5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kitagawa, R., Bakkenist, C. J., McKinnon, P. J. & Kastan, M. B. (2004) Genes Dev. 18, 1423-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cerosaletti, K. & Concannon, P. (2004) J. Biol. Chem. 279, 38813-38819. [DOI] [PubMed] [Google Scholar]
- 51.Buscemi, G., Savio, C., Zannini, L., Micciche, F., Masnada, D., Nakanishi, M., Tauchi, H., Komatsu, K., Mizutani, S., Khanna, K., et al. (2001) Mol. Cell. Biol. 21, 5214-5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu, Y., Ashley, T., Brainerd, E. E., Bronson, R. T., Meyn, M. S. & Baltimore, D. (1996) Genes Dev. 10, 2411-2422. [DOI] [PubMed] [Google Scholar]
- 53.Maser, R. S., Mirzoeva, O. K., Wells, J., Olivares, H., Williams, B. R., Zinkel, R. A., Farnham, P. J. & Petrini, J. H. (2001) Mol. Cell. Biol. 21, 6006-6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Theunissen, J. W., Kaplan, M. I., Hunt, P. A., Williams, B. R., Ferguson, D. O., Alt, F. W. & Petrini, J. H. (2003) Mol. Cell 12, 1511-1523. [DOI] [PubMed] [Google Scholar]
- 55.Stewart, G. S., Maser, R. S., Stankovic, T., Bressan, D. A., Kaplan, M. I., Jaspers, N. G., Raams, A., Byrd, P. J., Petrini, J. H. & Taylor, A. M. (1999) Cell 99, 577-587. [DOI] [PubMed] [Google Scholar]
- 56.Kraakman-van der Zwet, M., Overkamp, W. J., Friedl, A. A., Klein, B., Verhaegh, G. W., Jaspers, N. G., Midro, A. T., Eckardt-Schupp, F., Lohman, P. H. & Zdzienicka, M. Z. (1999) Mut. Res. 434, 17-27. [DOI] [PubMed] [Google Scholar]
- 57.Maser, R. S., Zinkel, R. & Petrini, J. H. (2001) Nat. Genet. 27, 417-421. [DOI] [PubMed] [Google Scholar]
- 58.Haber, J. E. (1998) Cell 95, 583-586. [DOI] [PubMed] [Google Scholar]
- 59.Tauchi, H., Matsuura, S., Kobayashi, J., Sakamoto, S. & Komatsu, K. (2002) Oncogene 21, 8967-8980. [DOI] [PubMed] [Google Scholar]
- 60.Cortez, D., Glick, G. & Elledge, S. J. (2004) Proc. Natl. Acad. Sci. USA 101, 10078-10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}