

Supplemental Digital Content is available in the text
Keywords: C3 glomerulonephritis, complement alternative pathway, complement-mediated hemolytic uremic syndrome, gene diagnosis
Abstract
Rationale:
C3 glomerulonephritis (C3GN) and complement-mediated hemolytic uremic syndrome (HUS) both result from the abnormal regulation of the complement system. A significant number of patients with C3GN or complement-mediated HUS have mutations of more than 1 complement protein. This discovery has had a major impact on identifying the underlying cause of familial C3GN or complement-mediated HUS.
Patient concerns:
We report the cases of 2 brothers (herein referred to as patient II-1 and patient II-9), both with complement disorders that differed in their clinical and genetic features.
Diagnoses:
Patient II-1 clinically presented with nephrotic syndrome and acute kidney injury and pathologically presented with C3GN combined with thrombotic microangiopathy (TMA) and subacute tubulointerstitial nephritis. Meanwhile, patient II-9 clinically presented with HUS and pathologically presented with TMA combined with acute severe tubular injury.
Interventions:
Screenings for genetic mutations contributed to complement system dysregulation were performed on patient II-1.
Outcomes:
The genome sequencing identified that patient II-1 had a heterozygous mutation in the C3 gene (c.C1774T/p.R592W). Nine other relatives of the brothers were checked for this C3 mutation and only the daughter of patient II-1 (herein referred to as patient III-2) carried it, but so far, she does not have any clinical manifestations of kidney disease.
Lessions:
Family members with a dysregulation of the complement alternative pathway may differ in its clinical and genetic features.
1. Introduction
C3 glomerulonephritis (C3GN) is a kind of C3 glomerulopathy caused by the excessive activation of the CAP. C3GN is characterized by isolated deposits of C3 on immunofluorescence.[1] Clinical manifestations of C3GN include urinary abnormalities, renal insufficiency, hypertension, and depressed serum C3 levels. In C3GN patients, the activity of C3 convertase is enhanced by the C3 convertase stabilizing autoantibody C3 nephritic factor (C3NeF) or by the loss of a functional complement factor H (CFH) activity.[2] Mutations in CFHR5, C3, and other CFH-related proteins (ie, CFHR1-4) may also be responsible for C3GN.[3,4]
Hemolytic uremic syndrome (HUS) is characterized by the simultaneous occurrence of microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury.[5] Currently, HUS is classified into primary HUS without coexisting disease (ie, cases due to complement gene mutations and antibodies to CFH) and secondary HUS secondary to infection (ie, Shiga toxin-producing Escherichia coli), drug toxicity, pregnancy, or autoimmune disorders. Primary HUS without coexisting disease is called complement-mediated HUS or atypical HUS.[6] Mutations of the CFH, complement factor I (CFI), C3, membrane cofactor protein (MCP), complement factor B (CFB), and thrombomodulin (THBD) genes have been reported in patients with complement-mediated HUS.[7–9] Therefore, family members of affected patients should undergo genetic testing to verify a potentially causative mutation in the patient.
Both C3GN and HUS are caused by dysregulation of the complement alternative pathway (CAP). A study reported that HUS and C3GN without MPGN share common genetic risk factors and concluded that constitutional or acquired dysregulation of the alternative complement pathway is probably associated with a wide spectrum of diseases, ranging from HUS to C3GN without MPGN.[1]
Here, we report a case of 2 brothers with disorders of the alternative complement pathway, but one presents with C3GN accompanied with the C3 mutation, and the other presents with HUS. The data suggest that C3GN and HUS share a similar pathogenic pathway but present different clinical manifestations.
2. Case presentation
Patient II-1 (Fig. 1) was a 54-year-old Chinese male. One week before admission to our hospital, the patient presented with bilateral lower extremity pitting edema and increased serum creatinine. His son was diagnosed with nephrotic syndrome at the age of 7 years and died of renal failure at the age of 8 years. When the patient was admitted to our hospital on July 1, 2014, his urine volume was 850 mL/d and his blood pressure was 160/100 mm Hg. Blood analyses revealed a decrease in renal function, hypoalbuminemia, mild elevation in lactic dehydrogenase (LDH), and low C3 levels. The white blood cell count was 10.1 × 109/L; hemoglobin concentration, 128 g/L; platelet count, 172 × 109/L; serum creatinine, 186 μmol/L; blood urea nitrogen, 12.93 mmol/L; albumin (ALB), 17.8 g/L; LDH, 286 U/L; and C3, 0.52 g/L. He had normal levels of reticulocyte, alanine aminotransferase, aspartate aminotransferase, creatine kinase (CK), MB isoenzyme of CK, complement 4 (C4), CFH, CFI, and immunoglobulins A, G, and M. He was negative for antinuclear antibodies, antidouble-stranded-DNA antibodies, anti-CFH antibodies, and anti-CFI antibodies. The ADAMTS13 activity was normal. Fragmented red blood cells were absent in the peripheral blood smear. Urinalysis revealed the urine protein score to be 3+ and RBC to be 0 to 2/HP. The 24-hour urine protein level was 5.83 g. Chest radiograph and electrocardiogram were normal. Echocardiography showed moderate pericardial effusion, thickening of the ventricular septum, and mild diastolic dysfunction. Abdominal ultrasonography revealed the size of kidney to be normal. Fundoscopy did not show retinal hemorrhages or papilledema. We performed kidney biopsy in order to identify the diagnosis of patient II-1. Immunofluorescence microscopy showed deposits of C3 (3+). Light microscopy showed mesangial proliferative, thickening and hyalinization of the renal arteriolar wall, and “onion skinning” of small renal arteries. Electron microscopy found electron-dense material in the mesangium. The pathological diagnosis was C3GN combined with thrombotic microangiopathy (TMA) and subacute tubulointerstitial nephritis (Fig. 2).
Figure 1.
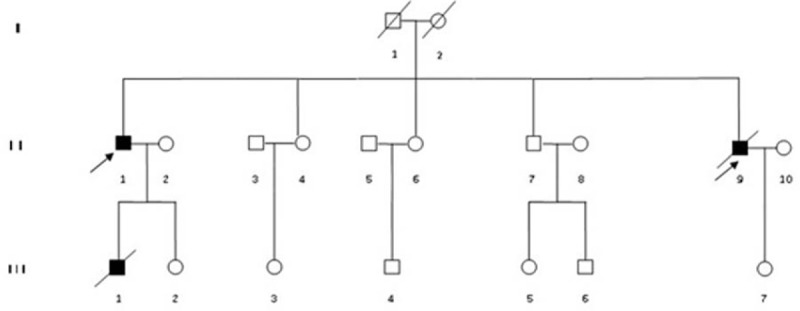
Pedigree of the family with complement-mediated diseases. Squares denote male family members, circles denote female family members, solid symbols denote affected members, and slashes denote deceased family members. The index patients are indicated with an arrow.
Figure 2.
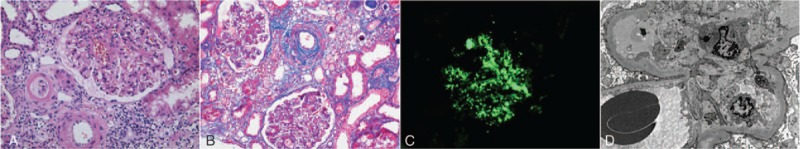
Kidney biopsy findings from patient II-2. (A, B) Light microscopy shows mesangial proliferative, thickening and hyalinization of the renal arteriolar wall, and “onion skinning” of small renal arteries. (Hematoxylin and eosin stain, original magnification: ×400; Masson stain, original magnification: ×200). (C) Immunofluorescence microscopy shows bright granular C3 staining in the mesangium and along capillary walls. Staining for all Igs and C1q was negative. (Original magnification: ×400). (D) Electron microscopy shows electron-dense material in the mesangium and diffuse foot-process effacement. (Original magnification: ×8000).
The patient received venoclysis of methylprednisolone, 60 mg/d, since July 7. Ten days later, blood analysis showed a serum creatinine of 97 μmol/L and an ALB of 27.8 g/L. The 24-hour urine protein level was 0.74 g/d. His urine volume increased to 3400 mL/d. At the same time, he lost weight by 15 kg and his edema was relieved significantly. Then, the patient took methylprednisolone 48 mg/d orally and was discharged from hospital. During the follow-up, his 24-hour urine protein level decreased to 0.25 g, and the dosage of methylprednisolone was reduced gradually. Nineteen months later, the patient continues to be in remission, with easily controlled hypertension and normal kidney function.
Patient II-9, a 32-year-old male, was the younger brother of patient II-1. He was admitted to our hospital with complaints of fatigue, oliguria, edema, and dyspnea on March 22, 1999. One week before admission to our hospital, the patient had nocturnal dyspnea, oliguria (800 mL/d), and bilateral lower extremity pitting edema. Three days before admission to our hospital, the patient's urine volume decreased to 200 mL/d. When we assessed his vital signs, his blood pressure was 180/112 mm Hg and his pulse rate was 102 beats per minute. Blood analysis revealed severe anemia, a notable decrease in renal function, a dramatic elevation in LDH, and low C3 levels. The white blood cell count was 15.4 × 109 /L; hemoglobin concentration, 44 g/L; platelet count, 175 × 109/L; serum creatinine, 1140 μmol/L; blood urea nitrogen, 46.78 mmol/L; ALB, 32 g/L; LDH, 1991U/L; and C3, 0.29 g/L. The levels of reticulocyte, alanine aminotransferase, aspartate aminotransferase, CK, MB isoenzyme of CK, C4, CFH, and CFI and immunoglobulins A, G, and M were all normal. He was negative for antinuclear antibodies, antidouble-stranded-DNA antibodies, antineutrophil cytoplasmic antibody, and antiglomerular basement membrane antibody. He had normal ADAMTS13 activity. Fragmented red blood cells were less than 2% in the peripheral blood smear. Urinalysis revealed the urine protein score to be 3+ and RBC to be 16 to 28/HP. The 24-hour urine protein level was 6.0 g. The chest radiograph showed an enlarged cardiac silhouette. Electrocardiogram showed sinus tachyarrhythmia. Echocardiography showed thickening of the ventricular septum and posterior wall of the left ventricular, mild pericardial effusion, and pleural effusion. Abdominal ultrasonography revealed hepatomegaly and a normal kidney size. Kidney biopsy was performed on April 9 and the result was TMA combined with acute severe tubular injury. Immunofluorescence microscopy showed deposits of C3(+). Light microscopy showed a thickening and an “onion skinning” of small renal arteries (Fig. 3).
Figure 3.
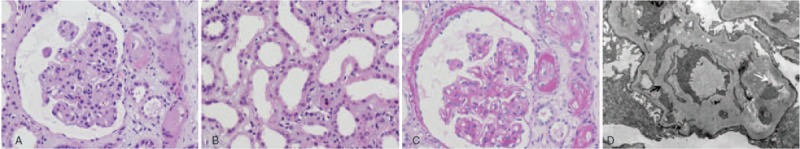
Kidney biopsy findings from patient II-9. (A) Light microscopy shows an increased mesangial cellularity and matrix, thickened glomerular basement membrane, and double-contour formation. (Hematoxylin and eosin stain, original magnification: ×400). (B) Light microscopy shows tubular epithelium vacuolar degeneration, defuse loss of the tubule brush border, and tubule dilatation. (Hematoxylin and eosin stain, original magnification: ×400). (C) Light microscopy shows capillary wall mesangial interposition and double-contour formation. (Periodic acid–Schiff stain, original magnification: ×400). (D) Electron microscopy shows a thickened glomerular basement membrane (black arrow) and expansion of the lamina rara interna (white arrow) (original magnification: ×10,000).
Patient II-9 was diagnosed with HUS and received hemodialysis 3 times per week. After receiving hemodialysis for 58 days, his serum creatinine decreased to 260 μmol/L, hemoglobin concentration increased to 91 g/L, and urine volume increased to 2000 mL/d. The patient ceased hemodialysis and was discharged from hospital. At the 6-month follow-up, he had a hemoglobin concentration of 92 g/L, serum creatinine of 310 μmol/L, C3 of 0.72 g/L, and 24-hour urine protein of 0.22 g/d (1450 mL). In September 2005, the patient had a recurrence of fatigue, oliguria, and dyspnea. His hemoglobin concentration level was 68 g/L, serum creatinine was 950 μmol/L, and potassium was 6.9 mmol/L. Without access to hemodialysis, the patient died of acute decompensated heart failure and hyperkalemia. Clinical data of patient II-1 and II-9 are summarized in Table 1.
Table 1.
Clinical characteristic of patient II-1 and patient II-9.
Screenings for genetic mutations contributed to complement system dysregulation were performed on patient II-1 using the target enrichment method and next-generation sequencing. Targeted genomic enrichment was performed using the Agilent HaloPlex Target Enrichment System custom panel (Agilent Technologies Inc., Santa Clara, CA), in which coding exons, the intronic flanking regions, the 5′ and 3′ untranslated regions (UTR) of 84 genes in the complement and coagulation pathways, and endothelial cells were targeted (Supplemental Table 1). The following next-generation sequencing identified that patient II-1 had a heterozygous mutation in C3 gene (c.C1774T/p.R592W) (Fig. 4). Additionally, multiplex ligation-dependent probe amplification (MLPA) using the SALSA MLPA KIT P236-A2 ARMD mix-1 (MRC-Holland, Amsterdam, Netherlands) showed that patient II-9 had 2 copies at the CFH-CFHR genomic region. Then, another 9 relatives of patient II-1 were checked by Sanger sequencing to validate whether they carried the C3 mutation (c.C1774T/p.R592W). The results showed that only the daughter of patient II-1 (patient III-2) carried this mutation and that the other relatives were homozygous for CC at position c.1774 in the C3 gene.
Figure 4.
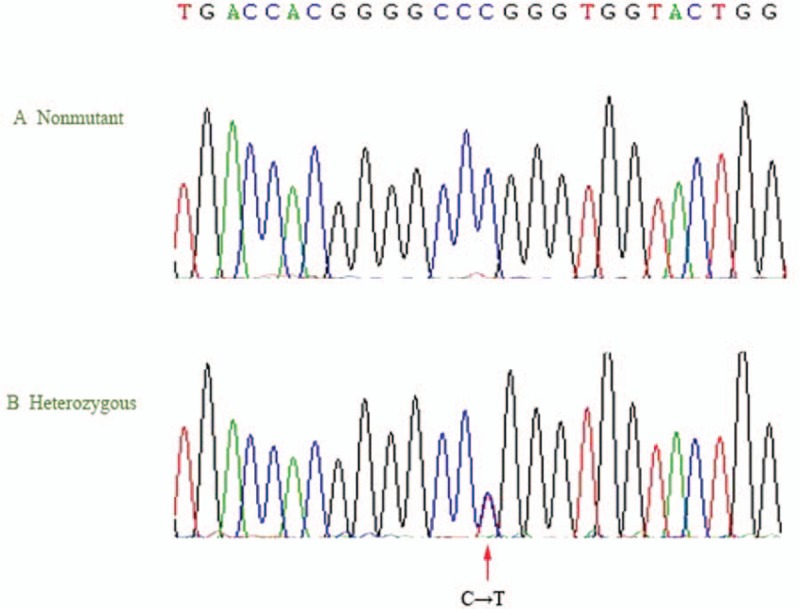
Identification of the c.C1774T/p.R592W mutation in a family with complement-mediated diseases. Panel A shows a normal sequencing profile. Panel B shows the sequence identification of a C-to-T substitution (arrow) of 1 R592W allele at position 1774 in patient II-1 and patient III-2.
3. Discussion
In this study, we have reported the cases of 2 brothers with disorders in the CAP who had different clinical and pathological presentations. The presence of the C3 mutation in 2 members of this family reveals the underlying mechanism of this kind of disease. This case therefore highlights the importance of using genetic testing in the family members of affected patients.
The CAP is capable of autoactivation due to the slow spontaneous tick over of C3, generating the C3 convertase. The spontaneous activating requires rigorous control to prevent excessive alternative pathway activation. This control depends on a complex system of circulating and membrane bound factors, including CFH, CFI, and MCP.[11] CFH downregulates the activity of the CAP by increasing the rate of dissociation of the CAP convertase C3bBb and inactivate membrane bound C3b through its binding to endothelial cells. CFI is a serine protease, which cleaves C3b and C4b in the presence of CFH and MCP. MCP is a cofactor of CFI in the degradation of C3b and C4b. Diminished function of these regulatory proteins can result in dysregulation of the complement alternative pathway.[10,11] Kidney diseases caused by dysfunction of the CAP comprise complement-mediated HUS, C3 glomerulopathies, and atypical postinfectious glomerulonephritis.[11] Complement-mediated HUS is a thrombotic microangiopathy typified by the triad of AKI, macroangiopathic hemolytic anemia, and thrombocytopenia. C3GN is characterized by isolated deposits of C3 on immunofluorescence, while its clinical presentation is variable. Atypical postinfectious glomerulonephritis refers to a clinical course where the diagnosis of postinfectious GN is not followed by resolution but rather by signs of persisting glomerular damage.[12] Complement-mediated HUS, C3GN, and atypical postinfectious glomerulonephritis are all characterized by an inappropriate activation of the CAP, eventually resulting in renal damage. The microvascular endothelium is generally targeted in complement-mediated HUS, and the renal microvascular endothelium is specifically targeted in complement-mediated HUS, thereby leading to a TMA. C3GN is typically characterized by the uncontrolled activation of the CAP in the fluid phase and/or at tissue surfaces that lack membrane-anchored complement, which results in glomerular injury and a proliferative response.[11]
Transitions between glomerulopathies included in this spectrum can occur during the disease course, after kidney transplantation, or among members of the same family, which adds another layer of complexity to the CAP pathophysiology. The variation may result from differences in CAP activation patterns. Several studies have established that uncontrolled activation of the CAP in a patient with HUS is a result of CFH, CFI, or MCP gene defects.[6] Huzmeli et al[13] reported a case of C3 mesangial proliferative glomerulonephritis that initially presented with atypical HUS. The transition implied that C3GN and complement-mediated HUS may share a common pathogenic pathway. In addition, the clinical diversity of these disorders may be a result of different pathophysiologic categories of CAP dysfunction, including differences in the triggers, sites, and intensities of involvement and in the outcome of the CAP dysregulation.[1]
The genetic change c.C1774T/p.R592W is one of the genetic susceptibility factors that had already been found to be associated with complement-mediated HUS.[14]C3 mutation would reduce the efficiency of factor I-mediated C3b cleavage when catalyzed by membrane cofactor protein (MCP) but not when catalyzed by CFH. Our data showed a C3 mutation (c. C1774T/p.R592W) in patient II-1, who was diagnosed with C3GN with TMA, implying that C3GN and HUS share common genetic susceptibility factors and that the activation of the CAP leads to various diseases, ranging from C3GN to HUS. In contrast, patient II-9, who was diagnosed with HUS, was negative for the C3 mutation (c.C1774T/p.R592W). Since the patient passed away 11 years ago, we can only get the tissue from his kidney as a specimen. The accuracy of his genetic sequencing could not compare with the other family members. Furthermore, rather than a specific single defect, more than 1 genetic predisposition commonly underlies CAP dysregulation. Perhaps patient II-9 presented with genetic mutations that we did not screen for. It is worth noting that patient III-2 (patient II-1's daughter) also presented with this C3 mutation (c.C1774T/p.R592W), but she has not shown any clinical manifestations of kidney disease so far. First, the penetrance of HUS is low, as less than half of family members carrying the same mutation as the patient with complement-mediated HUS will be affected with the disease. Second, initial clinical manifestations of C3GN and HUS may be preceded by infection, vaccinations, immunosuppressive or antineoplastic drugs, oral contraceptives, pregnancy, or childbirth. In other words, the presence of these diseases requires a trigger to elicit clinical manifestations. Patient III-2 should keep monitoring C3 level, renal function, and urinalysis. It is necessary to evaluate the presence of hypocomplementemia, an abnormal urinalysis, or elevated serum creatinine in clinically unaffected relatives who have genetic abnormalities.
Patients with the diagnosis of atypical HUS should receive eculizumab as the 1st-line treatment.[15,16] Plasma therapy was the 1st-line therapy for patients during the acute episode of atypical HUS before eculizumab was introduced.[17] C3GN is uncommon and there are no randomized trials to determine informed therapeutic decisions.[18] Both eculizumab and plasma exchange were not available to patient II-9 because of their prohibitive cost.
Our study has several limitations. First, the details of patient III-7's medical history are not available. He died of an unknown cause before kidney biopsy was performed. Second, we can only get tissue from patient II-9's previous renal biopsy specimen, so the accuracy of his genetic sequencing could not compare with that of the other family members.
4. Conclusions
In summary, kidney diseases caused by the dysfunction of the CAP may differ in their clinical and pathological presentations. Patients with dysfunction of the CAP may show genetic mutations of complement proteins. It is necessary to screen for mutations of complement proteins in the family members of affected patients.
Supplementary Material
Footnotes
Abbreviations: ALB = albumin, C4 = complement 4, CK = creatine kinase, LDH = lactic dehydrogenase.
Ethics Statement: The study was approved by the institutional review board of the First Affiliated Hospital of Chinese PLA General Hospital, Beijing, China.
The authors have no funding and conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- [1].Servais A, Fremeaux-Bacchi V, Lequintrec M, et al. Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet 2007;44:193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Servais A, Noel LH, Roumenina LT, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int 2012;82:454–64. [DOI] [PubMed] [Google Scholar]
- [3].Gale DP, de Jorge EG, Cook HT, et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet 2010;376:794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Malik TH, Lavin PJ, Goicoechea de Jorge E, et al. A hybrid CFHR3-1 gene causes familial C3 glomerulopathy. J Am Soc Nephrol 2012;23:1155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol 2005;16:1035–50. [DOI] [PubMed] [Google Scholar]
- [6].Loirat C, Fremeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis 2011;6:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Geerdink LM, Westra D, van Wijk JA, et al. Atypical hemolytic uremic syndrome in children: complement mutations and clinical characteristics. Pediatr Nephrol 2012;27:1283–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 2010;5:1844–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 2013;8:554–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zipfel PF, Heinen S, Jozsi M, et al. Complement and diseases: defective alternative pathway control results in kidney and eye diseases. Mol Immunol 2006;43:97–106. [DOI] [PubMed] [Google Scholar]
- [11].De Vriese AS, Sethi S, Van Praet J, et al. Kidney disease caused by dysregulation of the complement alternative pathway: an etiologic approach. J Am Soc Nephrol 2015;26:2917–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sethi S, Fervenza FC, Zhang Y, et al. Atypical postinfectious glomerulonephritis is associated with abnormalities in the alternative pathway of complement. Kidney Int 2013;83:293–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Huzmeli C, Candan F, Seker A, et al. C3 mesangial proliferative glomerulonephritis initially presenting with atypical hemolytic uremic syndrome: a case report. J Med Case Rep 2016;10:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Martinez-Barricarte R, Heurich M, Lopez-Perrote A, et al. The molecular and structural bases for the association of complement C3 mutations with atypical hemolytic uremic syndrome. Mol Immunol 2015;66:263–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 2013;368:2169–81. [DOI] [PubMed] [Google Scholar]
- [16].Christmann M, Hansen M, Bergmann C, et al. Eculizumab as first-line therapy for atypical hemolytic uremic syndrome. Pediatrics 2014;133:e1759–63. [DOI] [PubMed] [Google Scholar]
- [17].Loirat C, Saland J, Bitzan M. Management of hemolytic uremic syndrome. Presse Med 2012;41(3 Pt 2):e115–35. [DOI] [PubMed] [Google Scholar]
- [18].Pickering MC, D’Agati VD, Nester CM, et al. C3 glomerulopathy: consensus report. Kidney Int 2013;84:1079–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.