

Abstract
Introduction:
Anomalous left coronary artery from the pulmonary artery (ALCAPA) is a rare but severe congenital cardiac malformation. The prognosis mainly depends on the early and accurate diagnosis and treatment. However, without a typical and specific clinical manifestation in early stage, ALCAPA has a higher rate of false initial diagnosis.
Diagnostic and therapeutic procedure:
Three infants with impaired left ventricle (LV) function, LV enlargement, mitral valve regurgitation (MR), and LV endocardium thickness were initially diagnosed as endocardial fibroelastosis (EFE). Due to the treatment effectiveness with prednisone acetate and digoxin, abnormal Q waves with T inversion, and dilated right coronary artery (RCA), the diagnosis of ALCAPA was suspected. Lastly, cardiac angiography confirmed the diagnosis. All of them were transferred to the cardiac surgery department and received a successful surgical repair. The follow-up results showed that abnormal Q waves with T waves inversion on electrocardiogram gradually regressed and disappeared, LV ejection fraction and LV dilation returned to a normal range after surgery, with alleviation of MR. Besides, endocardial thickness secondary to ischemia also returned to normal.
Conclusion:
ALCAPA should be suspected when confronted with patients with left heart enlargement, impaired left ventricular function, and signs of myocardial ischemia, particularly in infancy. EFE is an important differential diagnosis and may also arise as a result of ALCAPA. Abnormal Q waves with T waves inversion, particularly in avL, dilated RCA and increased ratio of RCA/AO are important differential key points for the identification of ALCAPA and EFE. Awareness of this condition is essential for prompt recognition and referral to a tertiary cardiac center to enable early surgical intervention and improved prognosis for these children.
Keywords: anomalous left coronary artery from the pulmonary artery, endocardial fibroelastosis, misdiagnosis
1. Introduction
Anomalous left coronary artery from the pulmonary artery (ALCAPA), as known as Bland–White–Garland syndrome, is a rare but severe congenital cardiac malformation,[1] presenting in 1 out of 300,000 neonate.[2] It is speculated occurring from either an abnormal septation of the conus arteriosus or from the persistence of the pulmonary buds combined with involution of the aortic buds that form the coronary arteries.[5] The prognosis of this syndrome depends on early diagnosis and successful cardiac surgery. The preferred method is a direct reimplantation of the left coronary artery (LCA) to the aorta. Without early and accurate diagnosis and treatment, the natural course of the disease is devastating and 90% untreated children die in the first year of life because of massive myocardial injury and following end-stage heart failure.[3,4] With appropriate diagnosis and emergency cardiac surgery, they have a much better chance to survive. However, owing to the lack of specificity in the clinical manifestations, ALCAPA has a higher rate of false initial diagnosis.[1,6] Endocardial fibroelastosis (EFE), with characteristics like hyperplasia of endocardial elastic fibers and diffuse thickening of endocardium is an important differential diagnosis as it could arise as a result of ALCAPA and they always have similar clinical features in early stage.[7] Here we reported 3 misdiagnosed cases as EFE in infancy, aiming to improve the recognition of ALCAPA and intending to provide some differential key points for the identification of ALCAPA and EFE.
2. Case 1
The first case was of a 2-year and 4-month-old male child, who was misdiagnosed as EFE since he was 9 months old. At the age of 9 months, he was admitted into our hospital because of cough and dyspnea for 1 week, we heard a lot of rough and wheezing sounds on his chest wall, and an II–III degree systolic murmur was heard at the cardiac apex. Chest x-rays showed an increased cardiothoracic ratio. The electrocardiogram (ECG) showed abnormal Q waves with T waves inversion (I avL and V6) and high voltage in the left ventricle (LV). The echocardiography revealed LV enlargement (left ventricular end-diastolic dimension [LVEDD] = 40 mm), LV endocardium thickness (4 mm), decreased left ventricular ejection fraction (LVEF) (23%), decreased amplitude of ventricular septum and posterior wall of LV, and a moderate to severe mitral valve regurgitation (MR). Due to the lack of knowledge about ALCAPA, this baby was initially misdiagnosed as EFE, which is the most common myocardial disease in infancy. Besides relieving respiratory symptoms, he was treated by prednisone acetate and digoxin for over 1 year, but it was proved to be noneffective. At the age of 2-year and 4 months, he was hospitalized in our pediatric intensive care unit (PICU) because of more serious cough, dyspnea, and oral cyanosis. At this time, the abnormal Q waves with T waves inversion on ECG (I avL and V6) remained unchanged. For the echocardiography, except for the diminished LVEF (31%), LV dilation (LVEDD = 53 mm), more severe MR, and LV endocardial thickness (5 mm), a dilated RCA (3.2 mm) was noted (Fig. 1). Due to the treatment ineffectiveness and dilated RCA, the diagnosis of ALCAPA was suspected. Then we carefully took the echocardiography examination for several times. Encouragingly, connection of LCA to pulmonary artery (PA) was visualized through 2-dimensional echocardiography (Fig. 1) and thin narrow red shunting flow toward the probe was visualized at the left or posterior wall of the PA. Lastly, cardiac catheterization and angiography confirmed the diagnosis showing a dilated and tortuous right coronary artery (RCA) with right to left collaterals filling the left coronary system, and ultimate shunting from the LCA to PA (Fig. 2A and B). Immediately, this baby was transferred to the cardiac surgery department and received a successful surgical repair. After a follow-up of 15 months, the abnormal Q waves with T waves inversion on ECG gradually regressed and disappeared, and LVEF (62%) and LV dilation (LVEDD = 31 mm) returned to a normal range, with alleviation of MR (mild). Besides, endocardial thickness also returned to normal.
Figure 1.
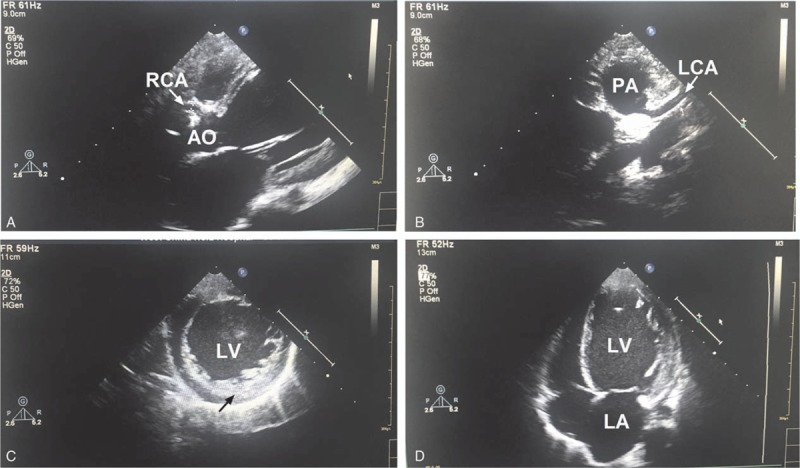
Echocardiography reveals dilated RCA (A), anomalous LCA originating from the PA (B), LV endocardium thickness (C), and LV enlargement (D) in case 1. LCA = left coronary artery, LV = left ventricle, PA = pulmonary artery, RCA = right coronary artery.
Figure 2.
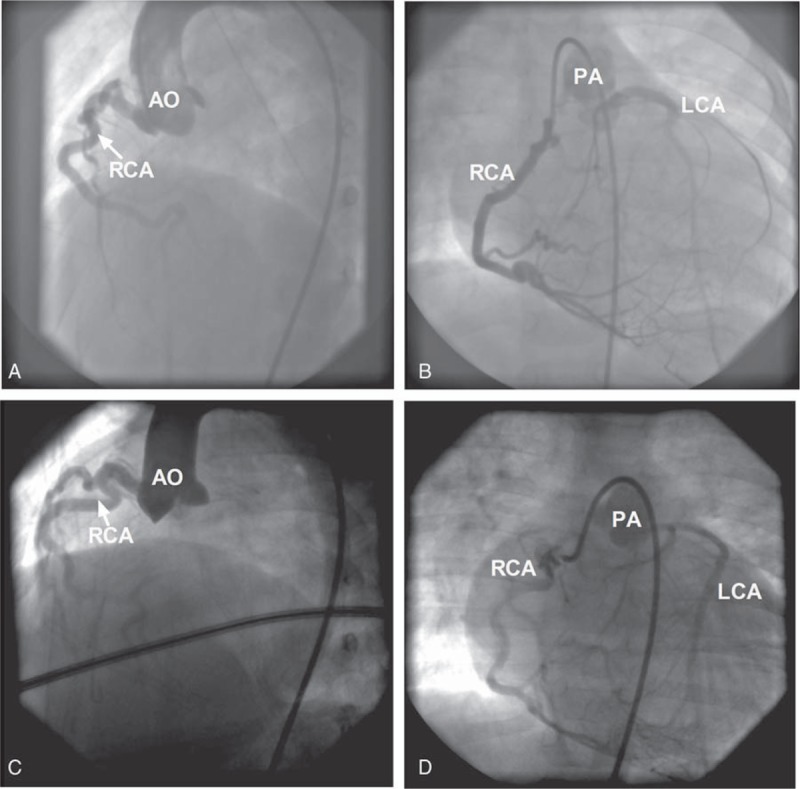
Cardiac catheterization and aortic root injections showing dilated and tortuous RCA origins from AO but no LCA (A,C), selective RCA injection showing RCA with right-to-left collaterals filling the left coronary system and ultimate shunting from the LCA to PA (B,D) in the case 1 patient (A,B) and case 3 patient (C,D). AO = aorta, LCA = left coronary artery, PA = pulmonary artery, RCA = right coronary artery.
3. Case 2
The second case was of a 3-month and 22 days female child, who was admitted into our hospital because of being found to have tachycardia for more than 2 months and cough and dyspnea for 1 month. We heard a rough sound and a gallop rhythm on her chest wall, no heart murmur was heard. Her liver was found enlarged (>4 cm under the rib, and >3 cm under the processus xiphoideus). Chest x-rays showed a lot of inflammatory changes in double lungs and increased cardiothoracic ratio. The ECG showed pathologic Q waves in leads I, avL, and V5, and inverted T waves in leads I, avL, V3 and V5 (Fig. 3). The echocardiography revealed LV enlargement (LVEDD = 38 mm), decreased LV systolic function (LVEF = 34%), LV endocardium thickness (3.0 mm), and a moderate to severe MR. This baby was also initially misdiagnosed as EFE and treated by digoxin, prednisone acetate, spironolactone, and captopril. However, 1 month later, the baby was followed up in our outpatient department. Her parents told us that she was finally diagnosed as the ALCAPA through echocardiography and received a successful surgical repair in another medical center. Compared with the echocardiography report in our hospital, we found that the dilated RCA (2.1 mm) and increased ratio of RCA/AO (0.19, ratio of the proximal RCA diameter to the aortic root diameter) were particularly addressed. Besides, many connections between the branches of RCA and LCA were found. After the heart surgery in that hospital, the condition of this baby became better, and our follow-up showed a recovering heart size and systolic function.
Figure 3.
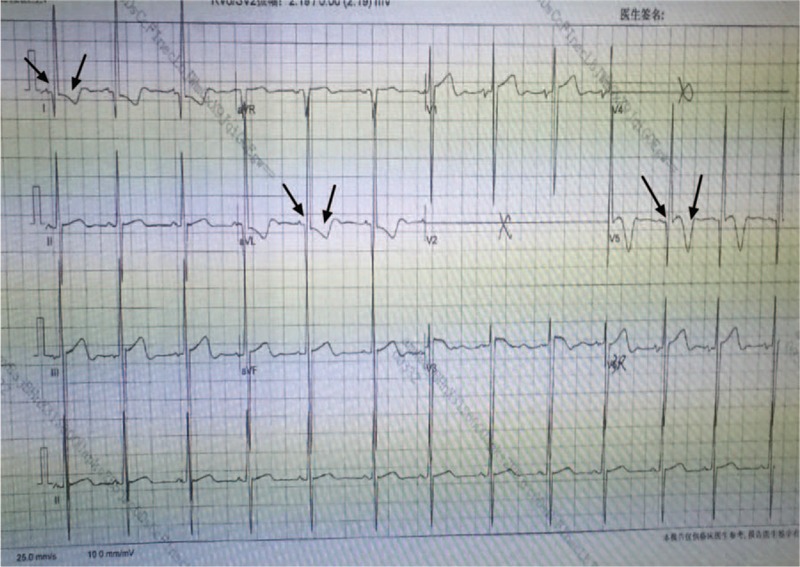
Electrocardiography showing abnormal Q waves in leads I, avL, and V5 and T waves inversion in leads I, avL, V3, and V5 in the case 2 patient.
4. Case 3
The third case was of a 9-month-old male child, who experienced cough and fever for 5 days, with cyanosis and an exertional dyspnea. Rough breathe sounds, phlegm sounds, and stridor sounds were heard on his chest wall, and the inspiratory 3 concave sign was positive. No heart murmur was heard. The ECG showed abnormal Q wave, inverted T wave, and ST-T segment depression at leads I, II, avL, avF, V5, and V6. The echocardiography revealed LV dilation (LVEDD = 43 mm), reduced amplitude of most parts of left ventricular wall, LV endocardium thickness (6 mm), a moderate to severe MR, and diminished LV systolic function (LVEF = 39%). The patient was diagnosed and treated as an EFE at the first, but with the experience of last 2 patients and increasing knowledge of ALCAPA, we took times of echocardiography to focus on the RCA diameter, ratio of RCA/AO, origin of LCA, and connections between the branches of RCA and LCA. Besides, when a cardiac MRI examination was also performed, myocardial ischemia and a suspiciously abnormal origin of LCA from the PA were found. On the basis of those findings, the diagnosis of ALCAPA was amended soon and finally confirmed by cardiac cauterization and angiography (Fig. 2C and D). Lastly, this patient was cured by surgical operation. After 6-month follow-up, the LV systolic function significantly improved (LVEF = 52%) and the size of LV was diminished (LVEDD = 39 mm), with alleviation of MR (mild to moderate).
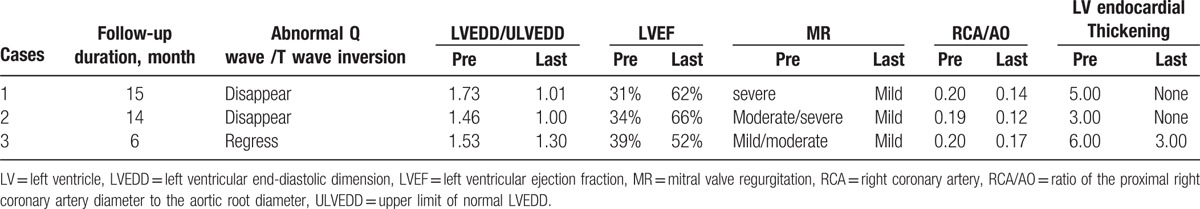
In summary, the basic and clinical characteristics and the follow-up results of the 3 cases are shown in Tables 1 and 2 in detail, respectively. All of them received successful surgical repair and were followed up with a range of 6 to 15 months. We could notice that the abnormal Q waves with T waves inversion on ECG gradually disappeared in cases 1 and 2 and regressed in case 3. LVEF and LV dilation of all 3 cases almost returned to a normal range, with alleviation of MR. Besides, RCA/AO decreased significantly and endocardial thickness secondary to ischemia disappeared 1 year later after surgery in cases 1 and 2. The study was approved by the University Committee on Human Subjects at Sichuan University.
Table 1.
The basic and clinical characteristics of 3 cases.
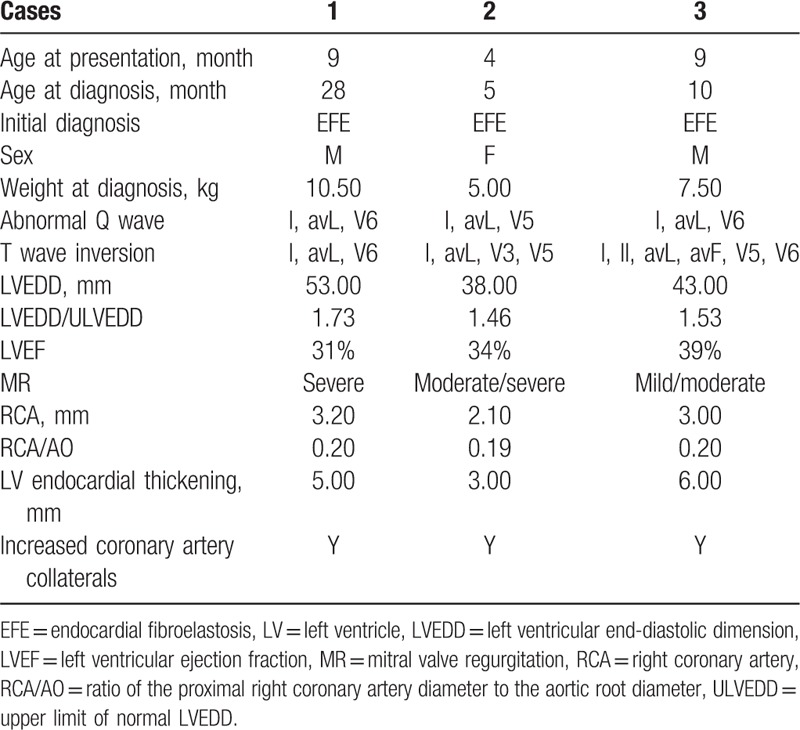
Table 2.
The follow-up results of 3 cases.
5. Discussion
ALCAPA is a very rare congenital anomaly.[2] During the neonatal period, the anomalous LCA could be supplied by the PA because the pulmonary vascular resistance is high. Despite relatively low oxygen saturation, the effective perfusion pressure is high enough to exclude overt myocardial ischemia. However, from the seventh week of life, after the PA pressure decreased, the LCA is no longer supplied by the PA but by the coronary artery collaterals from the RCA, with “coronary steal” into the PA, which resulted in diminished perfusion of the left ventricular muscle, extensive myocardial ischemia, necrosis, and fibrosis. These changes could lead to LV dilation, MR, and impaired cardiac function. Infants usually experienced pneumonia and heart failure within the first few months of life, which is in accordance with the conditions of 3 cases reported in the present study. Dyspnea, feeding intolerance, diaphoresis, and failure to thrive were the most common presentations.
However, without a typical clinical manifestation and specific features in echocardiography, ALCAPA has a higher rate of false initial diagnosis. In our 3 cases, all infants were hospitalized because of cough, feeding intolerance, yspnea, etc. Besides, all the echocardiography results revealed LV enlargement, decreased LV systolic function, MR, and LV endocardium thickness. From those clinical presentations, it is quite easily to misdiagnose all of them as EFE initially because the EFE is the most common myocardial disease in infancy. Those mistakes, particularly in first 2 cases, reminded us that EFE is an important differential diagnosis in infancy and may also arise as a result of ALCAPA. The accumulating experience helped us amend our diagnosis in third case in time. From our experiences and after a review of the literature,[8–11] there were some signs that may guide the clinicians to have an early and accurate diagnosis of ALCAPA and are helpful in distinguishing ALCAPA with EFE. Firstly, in ECG, we should focus on the abnormal Q waves with T waves inversion in leads I, avL, and V4–V6, especially in lead avL, where almost all children with ALCAPA had deep and wide Q waves. Secondly, in echocardiography, signs like dilated RCA, RCA/AO ≥1.4, coronary artery collateral circulation, and unable to show the clear origin of LCA strongly indicate the presence of ALCAPA. Moreover, our third case reminds us that cardiac MRI can also play a important role in distinguishing ALCAPA with EFE, in which ALCAPA could present with myocardial infarction changes. Although we amended the diagnosis of ALCAPA in all 3 cases by cardiac catheterization and angiography at last and all of them received successful operation, the ALCAPA should be considered at first if we had enough experience and combined all those relatively specific signs, particularly for the first case that was misdiagnosed for nearly 2 years.
Fortunately, all those 3 cases received successful surgical repair and the follow-up results showed that abnormal Q waves with T waves inversion on ECG could gradually regress and disappear, and LVEF and LV dilation could return to a normal range after surgery, with alleviation of MR. Besides, endocardial thickness secondary to ischemia also could return to normal.
6. Conclusion
Although ALCAPA are relatively rare among all the congenital heart diseases, clinical pediatric cardiologists and ultrasound doctors still should pay attention to this cardiac anomaly when confronted with patients with LV enlargement, impaired left ventricular function, and signs of myocardial ischemia, particularly in infancy. EFE is an important differential diagnosis and may also arise as a result of ALCAPA. Abnormal Q waves with T waves inversion, particularly in avL, dilated RCA, increased ratio of RCA/AO, and more connections between RCA and LCA are important differential key points for the identification of ALCAPA and EFE. Awareness of this condition is essential for prompt recognition and referral to a tertiary cardiac center to enable early surgical intervention and improved prognosis for these children.
Footnotes
Abbreviations: ALCAPA = anomalous left coronary artery from the pulmonary artery, ECG = electrocardiography, EFE = endocardial fibroelastosis, LCA = left coronary artery, LV = left ventricle, LVEDD = left ventricular end-diastolic dimension, LVEF = left ventricular ejection fraction, MR = mitral valve regurgitation, PA = pulmonary artery, RCA = right coronary artery.
FM and KZ contributed equally to this article.
The authors have no conflicts of interest to disclose.
References
- [1].Molaei A, Rastkar Hemmati B, Khosroshahi H, et al. Misdiagnosis of Bland–white–Garland syndrome: report of two cases with different presentations. J Cardiovasc Thorac Res 2014;6:65–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Szmigielska A, Roszkowska-Blaim M, Golabek-Dylewska M, et al. Bland–White–Garland syndrome—a rare and serious cause of failure to thrive. Am J Case Rep 2013;14:370–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lee AC, Foster E, Yeghiazarians Y. Anomalous origin of the left coronary artery from the pulmonary artery: a case series and brief review. Congenital Heart Dis 2006;1:111–5. [DOI] [PubMed] [Google Scholar]
- [4].Vila Mollinedo LG, Jaime Uribe A, Aceves Chimal JL, et al. Case Report: ALCAPA syndrome: successful repair with an anatomical and physiological alternative surgical technique. F1000Research 2016;5:1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kothari J, Lakhia K, Solanki P, et al. Anomalous origin of the left coronary artery from the pulmonary artery in adulthood: challenges and outcomes. Korean J Thorac Cardiovasc Surg 2016;49:383–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Levitas A, Krymko H, Ioffe V, et al. Anomalous left coronary artery from the pulmonary artery in infants and toddlers misdiagnosed as myocarditis. Pediatr Emerg Care 2016;32:232–4. [DOI] [PubMed] [Google Scholar]
- [7].Seki A, Patel S, Ashraf S, et al. Primary endocardial fibroelastosis: an underappreciated cause of cardiomyopathy in children. Cardiovasc Pathol 2013;22:345–50. [DOI] [PubMed] [Google Scholar]
- [8].Zheng J, Ding W, Xiao Y, et al. Anomalous origin of the left coronary artery from the pulmonary artery in children: 15 years experience. Pediatr Cardiol 2011;32:24–31. [DOI] [PubMed] [Google Scholar]
- [9].Xiao Y, Jin M, Han L, et al. Two congenital coronary abnormalities affecting heart function: anomalous origin of the left coronary artery from the pulmonary artery and congenital left main coronary artery atresia. Chin Med J 2014;127:3724–31. [PubMed] [Google Scholar]
- [10].Silverman NH. Echocardiographic presentation of anomalous origin of the left coronary artery from the pulmonary artery. Cardiol Young 2015;25:1512–23. [DOI] [PubMed] [Google Scholar]
- [11].Aliku TO, Lubega S, Lwabi P. A case of anomalous origin of the left coronary artery presenting with acute myocardial infarction and cardiovascular collapse. African Health Sci 2014;14:223–7. [DOI] [PMC free article] [PubMed] [Google Scholar]