

Abstract
Massively Parallel Signature Sequencing (MPSS), a recently developed high-throughput transcription profiling technology, has the ability to profile almost every transcript in a sample without requiring prior knowledge of the sequence of the transcribed genes. As is the case with DNA microarrays, effective data analysis depends crucially on understanding how noise affects measurements. We analyze the sources of noise in MPSS and present a quantitative model describing the variability between replicate MPSS assays. We use this model to construct statistical hypotheses that test whether an observed change in gene expression in a pair-wise comparison is significant. This analysis is then extended to the determination of the significance of changes in expression levels measured over the course of a time series of measurements. We apply these analytic techniques to the study of a time series of MPSS gene expression measurements on LPS-stimulated macrophages. To evaluate our statistical significance metrics, we compare our results with published data on macrophage activation measured by using Affymetrix GeneChips.
Keywords: transcription profiling, noise model
The last decade has witnessed a shift in molecular biology from methods that probe hypotheses a few molecules at a time toward whole-genome measurements. With the advent of global gene expression assays, data are becoming available at considerably accelerated speeds. Interestingly, the power of global methodologies brings along its own drawbacks; useful information is typically measured amid high levels of noise. Thus, the quality of analyses obtained from these data depends crucially on an understanding of how noise affects measurement.
The most mature global gene expression technology is arguably the microarray (1, 2). In all of its implementations (cDNA arrays, oligonucleotide arrays, etc.), this transcription profiling method exhibits significant technology-dependent noise. Models that characterize this noise through the study of replicate measurements have been developed (3–9) and provide a measure of security against false discoveries. An alternative gene expression profiling method uses the sequencing of short sequence tags derived from the ends of messenger RNA. This methodology encompasses the techniques of serial analysis of gene expression (SAGE) (10, 11) and Massively Parallel Signature Sequencing (MPSS) (12, 13). MPSS represents a powerful alternative to microarray technologies. Recent studies comparing gene expression measurements for the same biological samples with different probe-based microarray technologies showed considerable divergence across platforms (7). Some of these differences are due to the fact that different platforms use different probes for detection of the same genes. Because MPSS provides a sensitive measure of gene expression without requiring a priori knowledge of transcribed sequences, probe selection is not a problem for MPSS.
The MPSS process is complex; from the extraction of the total RNA to the quantification of transcripts, there are a number of steps that contribute to noise. In this paper, we develop a quantitative description of this noise. We then use this description to develop statistical hypotheses that test whether an observed change in gene expression is significant both in binary comparisons and in time course data. Finally, we apply this methodology to MPSS data from macrophages activated with LPS. We identify genes whose expression levels are significantly altered by this pathogenic challenge and compare our results with earlier data obtained by using Affymetrix GeneChips (14).
Materials and Methods
MPSS. A review of the principal stages of the MPSS protocol follows (see Overview of the Protocols Used in MPSS Transcription Profiling in Supporting Text and Fig. 5, which are published as supporting information on the PNAS web site; refs. 12 and 13; or www.lynxgen.com for more details).
cDNA signature/tag conjugate library construction. Poly(A)+ mRNA is extracted from the tissue of interest from which cDNA is synthesized. The 20 bases adjacent to the 3′-most DpnII site (GATC) of each cDNA are captured. The GATC and its contiguous 13 mer form a 17-mer sequence referred to as a signature. These signatures are PCR-amplified, and a unique identification tag is added to each signature.
Microbead loading. Multiple pools of ≈640,000 signature/tag conjugates are amplified, and the tags are hybridized with microbeads, each of which has bound to it ≈104 copies of one of the antitags. The signature/tag-containing microbeads (loaded microbeads) are isolated by using a fluorescence-activated cell sorter. Approximately 1.5 × 106 loaded microbeads are assembled in a flow cell, and the signature sequence on each bead is determined by MPSS.
MPSS. The signatures are sequenced by the parallel identification of four bases by hybridization to fluorescently labeled encoders, followed by removal of that set of four bases and exposure of the next four bases by type IIs endonuclease digestion. The process is then repeated. The imaged fluorescent data are processed to yield the number of beads that have a given signature sequence. Two types of initiating adaptors, whose type IIs restriction sites are offset by two bases, are ligated to two separate sets of microbeads containing a replicate of the same signature library. This is done to reduce signature losses from self-ligation of ends of signatures produced when digestion exposes palindromic overhangs. These two alternative sequencing reactions are referred to as two-stepper (TS) and four-stepper (FS) sequencing.
Matching the signature to the genome. Each of the sequenced 17-mer signatures [which typically matches only one position in a complex genome (11)] is associated with a proximal gene. Depending on the position of the signature relative to its associated gene, the signature is given a category (see Classification of Signatures from Genomic Sequence in Supporting Text and Table 1, which are published as supporting information on the PNAS web site) indicative of the quality of the association.
Result of an MPSS Run and Nomenclature. The net result of an MPSS run is a list of 17-mer signatures and the count of beads having that signature. MPSS sequencing is typically done in replicate. For a given biological sample, loaded beads are taken in fixed aliquots and independently sequenced k times with the TS and FS protocol (k = 2–4). We call these the MPSS or sequencing replicates. All of these sequencing replicates correspond to the same biological sample.
From the several replicate measurements, we compute a transcripts-per-million (tpm) measure for each signature. First, for each signature i, either the TS or FS data are chosen by selecting the stepper that counted the most beads for that signature across all available experiments. Because a signature may resist sequencing by one or the other stepper protocol, the stepper with the largest count is most likely to be better suited for measuring that signature. Once the stepper is chosen for each signature, the values of the k independent sequencing replicates are combined to give an aggregate tpm value τi ≡ ((νi1 + · · · + νik)/(N1 + · · · + Nk)) × 106, where the νis and the Ns are the bead counts for the given signature i and the total number of sequenced beads in each MPSS run, respectively. If, for a given signature, νij = 0, then the MPSS replicate j is excluded from both the numerator and the denominator. The reason for this is that zero counts deserve special attention in MPSS measurements, as will be discussed later (see also Discussion on the Effects of Zero Measurements in MPSS in Supporting Text). In addition to the aggregate tpm, we also define the tpm value obtained from a single replicate measurement as τij ≡ (νij/Nj) × 106. Experimentally observed tpm values can span several orders of magnitude, and thus we find it useful to define θij ≡ log10 τij and θi ≡ log10 τi.
Inherent Noise in Replicate Measurements. In Fig. 1a, we compare two replicate MPSS runs by plotting (θij, θij′) for replicate MPSS runs j and j′. Each point corresponds to a signature i and, ideally, these points should lie along the diagonal. Deviations from the diagonal are due to noise. As is the case for DNA microarrays (9), the noise depends strongly on the expression level. Therefore, an expression-dependent distribution function is needed to characterize the variability between replicates. For two replicate values θij and θij′ for the same signature, we define the measurement error as 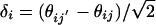 and estimate the mean expression level as
and estimate the mean expression level as 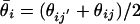 . Fig. 1c shows the dependence of measurement error on expression level by binning the data in intervals containing a fixed number k of signatures whose values of
. Fig. 1c shows the dependence of measurement error on expression level by binning the data in intervals containing a fixed number k of signatures whose values of 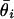 are the closest and then computing the standard deviation σ in each bin as a function of the mean μ of the
are the closest and then computing the standard deviation σ in each bin as a function of the mean μ of the 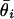 in the bin's k signatures. (Results were independent of k in the range between 100 and 500. We chose k = 250.) That is,
in the bin's k signatures. (Results were independent of k in the range between 100 and 500. We chose k = 250.) That is, 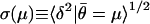 . Plots of the function σ(μ) derived from several pairs of replicate data (including those in Fig. 1a) are shown in Fig. 1c. The dominant feature of these plots is that σ decreases with increasing μ. Plots of σ(μ) can be used to characterize the variability between equivalent MPSS data runs (such as those shown in Fig. 1a), as well as the variability observed between the aggregate tpms obtained from biological replicates (such as those shown in Fig. 3).
. Plots of the function σ(μ) derived from several pairs of replicate data (including those in Fig. 1a) are shown in Fig. 1c. The dominant feature of these plots is that σ decreases with increasing μ. Plots of σ(μ) can be used to characterize the variability between equivalent MPSS data runs (such as those shown in Fig. 1a), as well as the variability observed between the aggregate tpms obtained from biological replicates (such as those shown in Fig. 3).
Fig. 1.

Noise analysis in the MPSS process. (a) Scatter plot of signature log-tpm pairs (θi,A1.a, θi,A1.b), where replicates A1.a and A1.b are separate MPSS measurements on samples taken from the same set of loaded beads (see d). (b) Scatter plot of signature log-tpm pairs (θi,A1.a, θi,B.a), where replicates A1.a and B.a are taken from distinct initial mRNA biological replicas and processed separately (see d). In both a and b, noise appears as deviations of the data points from the diagonal. Note that the noise level is higher in b than a. (c) Standard deviation of measurement noise σ as a function of signal level μ for pair-wise comparisons between replicates A1.a and A1.b (+), A1.a and A2.a (⋄), and, A1.a and B.a (•) (see text). (d) Illustration of replicate experiments setup.
Fig. 3.

Nonzero null hypothesis for binary comparisons. (a) Scatter plot of signature log-tpm pairs (θi,j, θi,j′), where replicates j and j′ are biological replicates taken at t = 0 (i.e., each θ is the log of an aggregate tpm derived from two biological replicates) for all signatures i that have nonzero tpm counts in all four sequence replicates. Also plotted are equivalent points for which j and j′ are biological replicates taken at t = 4 h. (b) Standard deviation of measurement noise σ as a function of signal level μ for data shown in a. Solid line is best fit of calculated values of σ to an exponential decay function. (c) Conditional probability density function P(ξ|μ) as a function of the rescaled noise ξ ≡ δ/σ for ranges of signal level 1.25 < μ < 1.75 (□), 1.75 < μ < 2.25 (•), 2.25 < μ < 2.75 (▵), and 2.75 < μ < 3.25 (+). Note that after normalization, these distributions are independent of signal level. Fits to the data are discussed in the text. (d) P value as a function of the fraction of significant data for measurements to which the nonzero null hypothesis is applicable (♦) as well as for all measurements (⋄). (d Inset). Illustration of data in region of significance for a and P = 0.05.
Binary Comparisons. To evaluate the significance of the difference between a pair of gene expression values (θij, θij′) for the same signature but different experiments, we begin with the null hypothesis that the θij and θij′ arise from the same distribution, and that the difference between them is due to noise. We define a gene expression-dependent p value as
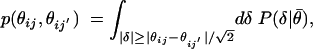 |
where 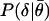 is the conditional probability of measuring a difference
is the conditional probability of measuring a difference 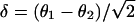 between two replicate measurements θ1 and θ2 given that
between two replicate measurements θ1 and θ2 given that 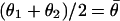 . An explicit calculation of
. An explicit calculation of 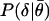 is presented in Results. The change in expression between two measurements θij and θij′ will be deemed significant when P(θij, θij′) is smaller than some threshold P0. For more details, see Statistical Significance of Differential Expression in Binary Comparisons in Supporting Text and Fig. 6, which are published as supporting information on the PNAS web site.
is presented in Results. The change in expression between two measurements θij and θij′ will be deemed significant when P(θij, θij′) is smaller than some threshold P0. For more details, see Statistical Significance of Differential Expression in Binary Comparisons in Supporting Text and Fig. 6, which are published as supporting information on the PNAS web site.
Time Traces and Multiple Comparisons. Changes in expression level as a function of time are particularly important in understanding the response of cells to a perturbation. Suppose that the aggregate tpm of a signature is measured at n time points t0 = 0 (i.e., before perturbation), t1, t2,..., tn-1, yielding a series of log-tpms (θt0, θt1,..., θtn-1). If the perturbation significantly affects the expression of that signature, then we expect a small P value for at least one of the n × (n-1)/2 pair-wise comparisons between temporal data points. We consider all pair-wise comparisons and not just those between consecutive measurements, because we have observed numerous instances (see All vs. All Comparisons for Assignment of Significance to Time Traces: An Example in Supporting Text for an example), where consecutive comparisons are not beyond the level of significance, but those between nonadjacent time points are. A significance index (SI) for the time series of a given signature is defined as the minimum P value obtained from all possible pair-wise comparisons within the series. (For more details, see All vs. All Comparisons for Assignment of Significance to Time Traces: An Example in Supporting Text and Fig. 7, which are published as supporting information on the PNAS web site.) An SI is considered significant if it is smaller than some chosen threshold P0. Note that the most significant P value does not necessarily correspond to the largest fold change, because the significance of a fold change depends on the expression level.
Data Sets Used in This Study. Human breast cancer cells. Estrogen receptor-negative BT-20 cell lines (15) were grown. Two distinct poly(A)+ mRNA samples (A and B in Fig. 1d) were collected from plated cells and used to generate two signature/tag libraries. One of these two libraries was split in two parts and used to generate two sets (A1 and A2 in Fig. 1d) of loaded microbeads. The other library was used to generate one set of loaded microbeads (B in Fig. 1d). After loading, each set of beads was independently processed in multiple MPSS runs.
Macrophage samples and data. Plastic-adherent monocytes were isolated from peripheral blood mononuclear cells collected from buffy coats from five healthy humans and cultured for ≈10 days in RPMI medium 1640, supplemented with 20% FBS/l-glutamine/20 mM Hepes/penicillin/streptomycin/50 ng/ml macrophage colony-stimulating factor to generate monocyte-derived macrophages. Macrophages were stimulated with 100 ng/ml LPS (Salmonella minnesota R595 ultrapure LPS, List Biological Laboratories, Campbell, CA) and sampled at time points 0 (i.e., before stimulation), 2, 4, 8, and 24 h. For each of these time points, total RNA was isolated with the Trizol reagent (Invitrogen), the total RNA from the individual donors was pooled, and poly(A)+ RNA was isolated with a MicroPoly(A-)Pure kit (Ambion, Austin, TX). Culture supernatants were tested to confirm appropriate induction of cytokines (tumor necrosis factor, IL-6, and IL-12), and an aliquot of total RNA was tested by using real-time PCR to ensure appropriate induction of selected genes. The poly(A)+ RNA was processed through the signature library generation and assayed by using MPSS. Duplicate samples at 0 and 4 h were generated by using independent cultures of macrophages and independent pools of RNA for the purpose of replicate noise modeling. We summarize some characteristics of the signature library in Summary of Signature Libraries Obtained from Our MPSS Measurements in Supporting Text and Table 2, which are published as supporting information on the PNAS web site.
Results and Discussion
Analysis of Noise Inherent in MPSS. To separate the sources of measurement noise in MPSS, we have carried out multiple replicate experiments (9) where, at different stages of the MPSS process, the sample is divided into multiple aliquots, and subsequent steps of the experiments are carried out independently. The experimental design (shown schematically in Fig. 1d) allows us to separate the measurement variances resulting from signature library generation, bead loading, and sequencing. Total RNA from estrogen receptor-negative breast cancer cell lines (see Materials and Methods) was divided into two aliquots (A and B in Fig. 1d). Each of these aliquots was processed independently, generating separate signature/tag libraries. The signature library (A) was subdivided into two equal parts and each part, along with the signature library (B), was independently loaded onto beads, giving rise to loaded bead groups denoted by A1, A2, and B. Finally, each of these loaded bead groups was processed with three MPSS FS runs (FS a, b, and c in Fig. 1d) and three MPSS TS runs (TS a, b, and c in Fig. 1d). Assuming that each stage of the process is independent of the others, these data sets enable us to estimate the noise introduced at each stage of the process.
In Fig. 1 a and b, we compare two replicate MPSS runs by plotting (θij, θij′) for replicate MPSS runs j and j′. The spread around the diagonal is a measure of noise. In Fig. 1 a and b, the x axes are the log-tpm value of the signatures in experiment FS.A1.a (see Fig. 1d), whereas the y axes correspond, respectively, to the log-tpm of the signatures in experiments FS.A1.b (Fig. 1a) and FS.B.a (Fig. 1b). The spread due to the combined variance introduced by library creation, bead loading, and sequencing (Fig. 1b) is much larger than that due to sequencing alone (Fig. 1a). Plots of the standard deviations σ(μ) (see Materials and Methods) derived from the data of Fig. 1 a and b show that σ decreases with the expression intensity μ, with the overall σ (filled circles, Fig. 1c) being about twice as large as the σ arising from the bead loading and sequencing (diamonds, Fig. 1c). Note that the noise from the combination of bead loading and sequencing is almost indistinguishable from that of sequencing alone (plus signs, Fig. 1c), demonstrating that noise stemming from bead loading is negligible.
The Statistics of the Zero Counts. Thus far, our analysis has dealt only with signatures whose bead counts are at least unity in each of the replicate experiments under consideration. Many signatures, however, have a finite bead count for one replicate experiment and zero for the other. These appear in Fig. 1 a and b as the sets of points forming linear structures at the left and bottom of Fig. 1 a and b. (The value of zero counts, i.e., 0 tpm, has been arbitrarily given a log-tpm value of 0.) This figure shows that the statistics of the signatures with low but positive counts in both runs are quite different from the statistics of the signatures measured as zero in one of the replicates. Similar results have been observed in other MPSS experiments (16).
To investigate the significance of zero count measurements, we studied expression data taken on macrophages 8 h after LPS stimulation. (These data were chosen because four TS and four FS MPSS runs were taken on this sample.) We identified the signatures with exactly four, three, and two nonzero bead counts within the four replicates (see Materials and Methods for the method of determining whether the TS or FS data were used for a given signature). We compute the function σ(μ) [in computing σ(μ), two nonzero replicate measurements are chosen at random when dealing with signatures with more than two nonzero values] separately for the data sets in which zero, one, or two of the four sequencing replicates yielded a zero count. The results are shown in Fig. 2. It can be seen that μ for the nonzero statistics (circles) reaches values of more than one order of magnitude larger than those for the one- or two-zero statistics (squares and diamonds). Further, the observed noise strength is considerably smaller for the nonzero statistics than for the other two, which are of similar magnitude.
Fig. 2.

Standard deviation of measurement noise σ as a function of signal level μ (see text) for MPSS measurements on LPS-activated macrophages at 8 h after activation. Four replicate MPSS runs were taken, and the noise level was calculated separately for signatures with no (o), one (▪), and two (♦) zero measurements (see text). Note that those signatures with nonzero measurements exhibit significantly lower noise at higher expression levels.
It is clear from Fig. 2 that data on signatures for which some of the sequencing replicates are zero exhibit significantly higher variability, suggesting that the absence of a signature in one of the sequencing replicates indicates the need for statistical modeling different from that used when the signature is present in all replicate measurements. See Discussion on the Effects of Zero Measurements in MPSS in Supporting Text and Fig. 8, which are published as supporting information on the PNAS web site. for more discussion of zero measurements in MPSS.
Three Null Hypotheses Are Required for Binary Comparisons. To determine the significance of changes in tpm value observed for different signatures in the LPS-activated macrophage data, it is first necessary to formulate null hypotheses by using biological replicates (see Materials and Methods). Both before activation and at 4 h after activation, biological replicate data were taken (each biological replicate data set is comprised of two sequencing replicates), and it is from these data sets that we formulate our null hypotheses. Each signature that was measured at least once in a pair of biological replicates yields two aggregate (i.e., determined from two or more sequencing replicates) tpm values, τ1 and τ2. Three possibilities can arise in these measurements: (i) none of the counts (νis) used to compute τ1 and τ2 were zero. (ii) At least one of the counts was zero, but neither τ1 nor τ2 is zero. (iii) Either τ1 = 0 and τ2 > 0 or τ2 = 0 and τ1 > 0. As shown above, the statistics characterizing the expression of signatures when measurements of zero counts are observed are fundamentally different from those resulting when no zeros are observed. Thus, it is necessary to formulate three distinct conditional hypotheses, one for each of the three conditions above. That is, given two samples and their respective MPSS measurements, we inspect the pattern of zeros obtained in the different sequencing replicas and use the appropriate null hypothesis signature by signature.
We begin formulating the null hypothesis for signatures with nonzero count measurements (case 1) by plotting in Fig. 3a all (θij, θij′), where j and j′ are biological replicates taken at t = 0 (where each θ is the log of an aggregate tpm count) for all signatures i that have nonzero tpm values in all replicate MPSS runs. Also plotted are equivalent points for which j and j′ are biological replicates taken at t = 4 h. A plot of σ(μ) derived from these data [along with a fit of the calculated values of σ(μ) to an exponential] is shown in Fig. 3b. For a given μ, we can define a distribution for the rescaled noise ξ ≡ δ/σ(μ) and obtain the conditional distribution function P(ξ|μ). We plot this distribution for several ranges of μ in Fig. 3c. These ranges of values for μ correspond to the regions delimited by the dashed lines in Fig. 3a, and the symbols drawn in each region correspond to the symbols in Fig. 3c. Notice that once normalized by its standard deviation, the distribution of the spread away from the diagonal (measured by δ) is independent of the expression strength, because all of the distributions coalesce approximately to the same curve. The tails of these distributions decrease more slowly than a Gaussian (dashed line in Fig. 3c) and are well described by the function (9) 0.5 exp{-x2/(0.5 + 0.6|x|)} (thick solid line in Fig. 3c).
The noise distribution plotted in Fig. 3c can be used to formulate the null hypothesis testing whether a difference in expression in a binary comparison is beyond measurement error (9). Given a positive value of ξ, say ξ0, the area under the distribution of Fig. 3c for |ξ| > ξ0 is the P value corresponding to a normalized differential expression of magnitude ξ0. Likewise, for a chosen P value, we can find a corresponding x0. For example, a P value of 0.05 corresponds to a ξ0 of 2.13. That is, all points with |δ| > 2.13σ(μ) will have a P value <0.05. These points are plotted in Fig. 3d Inset, along with the two delimiting curves corresponding to the equation |δ| = 2.13σ(μ). If our parameterization of the distribution is correct, then the fraction of points outside of those curves should be close to 0.05. Indeed, it is 0.04.
The one-zero null hypothesis (case 2) is formulated in a manner similar to the nonzero hypothesis. That is, we begin by considering all replicate points (θij, θij′) where j and j′ are biological replicates taken at t = 0 as well as at t = 4 h. However, in this instance, only those signatures i for which at least one of the pair of biological replicates is comprised of one zero and one nonzero sequencing replicate are considered. The variation between replicates in these data is significantly greater than that observed for the nonzero data. However, upon computing σ(μ), we find that the conditional distribution function P(ξ|μ) as a function of the rescaled noise distribution ξ ≡ δ/σ independent of μ for these data as well. Thus, we can follow the procedure outlined above to determine P values by using this one-zero null hypothesis data set (see Further Details on the Three Null Hypothesis in Supporting Text for more details).
The two-zero hypothesis (case 3) is formulated from data for which one of the biological replicates shows zero counts for a given signature in both sequencing replicates, whereas the other biological replicate shows at least one nonzero measurement for the signature. The probability distribution of the aggregate tpm values of the nonzero replicate measurements is computed, and the significance region for a particular P value is defined as the region under the high signal tail of the distribution whose area equals the desired P value (see Further Details on the Three Null Hypothesis in Supporting Text for more details).
Fig. 3d shows a plot of the fraction of points left out of the delimiting curves given by |δ| = ξ0σ(μ) as a function of the P value (ξ0 depends on the P value). The curve with solid diamonds corresponds to the subset of signatures for which the nonzero hypothesis applies. The open diamonds indicate all of the measured signatures and the corresponding null hypotheses. Both the nonzero hypothesis and the all-hypothesis curves show that the fraction of points left out of the delimiting curves is very well estimated by the P value calculation over four orders of magnitude. The precipitous drop-off of the curves at the small P value range is due to the two outliers indicated with arrows in Fig. 3d Inset.
We have developed our P value formalism in the context of binary comparisons (typically case/control studies) and time traces. By using the same formalism, it is also possible to estimate error bars for the log-tpm for a given signature. To do this, simply notice that if the log-tpm value of a signature yields a value θ, then [θ -2.13σ(θ), θ +2.13σ(θ)] is an estimate of the 95% confidence interval. In other words, the probability that a subsequent measurement of that signature falls outside that interval is <0.05. This confidence interval interpretation of our analysis is especially useful when data from only a single MPSS run of a given condition are extant, and error bars need to be assigned to these measurements. The calculation of a 95% confidence interval requires an estimate of σ(θ). When replicate measurements are not available, σ(θ) can be estimated from studies such as the one presented in this paper. Computational tools to analyze MPSS data for confidence intervals, as well as P values in case/control measurements and time traces, can be obtained at www.research.ibm.com/FunGen.
The Macrophage Data. We measured the gene expression of macrophages before LPS stimulation and at times t = 2, 4, 8, and 24 h after it. For each observed signature, we computed the SI of its time series (see Materials and Methods). Often, multiple signatures were found to correspond to a single gene (in the National Center for Biotechnology Information database). In such cases, the signature with the lowest SI values was associated with the gene. Following this protocol, we identified 12,657 signatures, of which 2,356 (20%) are statistically significant with SI < 0.05 (see Statistics of Significant Signatures in the Signature Library in Supporting Text and Fig. 10, which are published as supporting information on the PNAS web site. for more details). Significant signatures corresponded to well characterized genes in greater proportion than nonsignificant signatures (see Statistics of Significant Signatures in the Signature Library in Supporting Text), an indication that the SI allows the identification of meaningful signals from massive amounts of signature data.
Comparison with Earlier Experiments. We compare our results on LPS-activated macrophages with measurements reported by Nau et al. (14), in which gene expression is measured by using Affymetrix 6800 GeneChips. In both studies, the biology is similar: cultures of human monocyte-derived macrophages were stimulated with LPS, and the gene expression was measured over the course of 24 h. However, Nau et al. (14) measured gene expression at 0, 1, 2, 6, 12, and 24 h, as opposed to our measurements at 0, 2, 4, 8, and 24 h. In addition, Nau et al. (14) reported the expression levels of only 132 genes after exposure to LPS, where these genes were selected because they were induced by exposure to at least six of eight different bacteria (as defined by a fold change-based significance criterion). By using Nau et al.'s (14) significance criterion, we find that 107 of these 132 genes are also significant in their pure-LPS stimulation time-series measurements.
We studied the same 132 genes in the context of our MPSS measurements. We identified signatures corresponding to 127 of the 132 genes. Of the 127 genes identified in both data sets, 26 (20%) were nonsignificant with SI > 0.05. The remaining 101 significant genes have values of SI that range from 10-13 to 0.05. These 101 genes, although statistically significant, are not the most significant genes identified by using our LPS time-series data.
By performing a hierarchical cluster analysis (17) of the time-series data for these 101 genes, we find that the time course of these genes is similar in both data sets. As shown in Fig. 4, the measurements done with these two expression assaying techniques (after normalizing the expression of each gene relative to its value at t = 0 to zero mean and unit variance within each platform) are highly consistent. This remarkable consistency between techniques is observed in the correct ordering of the temporal conditions: the two pairs of conditions at 24 and at 2 h in both platforms are the closest, whereas the MPSS 8-h results interpolate between the Affymetrix 6 and 12 h, and the MPSS 4-h results interpolate between the Affymetrix 6 and 2 h. This consistency is much less clear when the remaining 26 nonsignificant genes are considered (see Eisen Plot of the MPSS Statistically Nonsignificant Genes Among the Macrophage Activation Program Genes in Supporting Text and Fig. 11, which are published as supporting information on the PNAS web site.).
Fig. 4.

Hierarchical cluster analysis of LPS-activated macrophage expression data. Columns are individual genes, and rows are MPSS measurements taken at 2, 4, 8, and 24 h after activation as well as previously published (14) Affymetrix GeneChip measurements taken at 1, 2, 6, 12, and 24 h after activation. See text for details and Table 4 for gene names.
Fig. 4 shows two clear gene clusters and two condition clusters. We interpret this structure as a group of 55 early responder genes (genes mostly active between 1 and 6 h) and 46 late responder genes (genes mostly active between 8 and 24 h). A number of interesting features of this set of genes follow from this early and late response interpretation (see Categories of Earlier and Later Responders Among Genes Significant in Both Nau et al. and MPSS Measurements in Supporting Text and Tables 3 and 4, which are published as supporting information on the PNAS web site.). Antiapoptotic, adhesion, cytokines, chemokines, transcription-related, and signaling genes tend to be expressed soon after stimulation, whereas enzymes (mostly associated with metabolism) and receptors are transcribed later. One of the earlier responder genes is the transcription factor NFKB1, which itself is regulated by the NFκB signaling pathway. Indeed, at least 20 of the 101 genes under consideration have been previously reported as NFκB targets (K. Basso, personal communication). The categories that are strongly associated with an early LPS response are composed mostly of NFκB targets; 8 of the 10 early responding cytokines and chemokines are known NFκB targets, as are 4 of the 8 early responding transcription-related genes and 4 of the 5 early responding antiapoptotic genes. Although Affymetrix arrays are necessarily biased toward previously identified genes (such as NFκB targets), we believe this bias alone is insufficient to explain these observations.
Conclusion
Given the digital nature of the MPSS measurements, it is natural to put our work in the context of prior ideas developed for SAGE analysis. Previous statistical analyses of SAGE used Fisher's exact test (18) and the χ2 or Z test for comparison of proportions (19) to model SAGE as a process of sampling signatures from signature libraries. In MPSS, signature-sampling fluctuations alone cannot account for the involved biochemical manipulations taking place in the process of signature library production. More elaborate Bayesian approaches represent the distribution of SAGE signature counts as mixtures of binomial (20) or Poisson (18) statistics whose parameters are weighted with prior distributions (chosen using heuristic criteria or mathematical convenience), yielding an enhanced variance when compared with plain Bernoulli or Poisson statistics. It is unlikely, however, that the basic process at play is a Bernoulli-type process, because there are many manipulations done to the signatures before they are counted that inherently contribute to its sample-to-sample count variability. Our approach was to empirically estimate the distribution of the differences in replicate data for MPSS measurements. Given that SAGE and MPSS differ considerably in the biochemical manipulations and in the sequencing of the signatures, a determination of empirical distributions of replicate fluctuations would be worthwhile in SAGE.
In conclusion, we have seen that, despite the complexity involved in the measurement of gene expression using MPSS, it is possible to formulate reasonable statistical tests to determine the extent to which the differential expression between two conditions could be masked by measurement noise. These statistical tests provide a valuable means of identifying significant genes from the vast number of signatures yielded by this technology.
Supplementary Material
Acknowledgments
We thank Yuhai Tu and Jeremy Rice for useful discussions. We are also grateful to Katia Basso for sharing her list of NFκB targets. Two anonymous reviewers provided valuable comments.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: FS, four stepper; TS, two stepper; MPSS, Massively Parallel Signature Sequencing; tpm, transcripts per million; SAGE, serial analysis of gene expression; SI, significance index.
References
- 1.Brown, P. O. & Botstein, D. (1999) Nat. Genet. 21, 33-37. [DOI] [PubMed] [Google Scholar]
- 2.Lockhart, D. J., Dong, H., Byrne, M. C., Follettie, M. T., Gallo, M. V., Chee, M. S., Mittmann, M., Wang, C., Kobayashi, M., Horton, H., et al. (1996) Nat. Biotechnol. 14, 1675-1680. [DOI] [PubMed] [Google Scholar]
- 3.Huang, S., Qian, H. R., Geringer, C., Love, C., Gelbert, L. & Bemis, K. (2003) Am. J. Pharmacogenom. 3, 279-290. [DOI] [PubMed] [Google Scholar]
- 4.Mariani, T. J., Budhraja, V., Mecham, B. H., Gu, C. C., Watson, M. A. & Sadovsky, Y. (2003) FASEB J. 17, 321-323. [DOI] [PubMed] [Google Scholar]
- 5.Bakay, M., Chen, Y. W., Borup, R., Zhao, P., Nagaraju, K. & Hoffman, E. P. (2002) BMC Bioinformatics 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nygaard, V., Loland, A., Holden, M., Langaas, M., Rue, H., Liu, F., Myklebost, O., Fodstad, O., Hovig, E. & Smith-Sorensen, B. (2003) BMC Genomics 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan, P. K., Downey, T. J., Spitznagel, E. L., Jr., Xu, P., Fu, D., Dimitrov, D. S., Lempicki, R. A., Raaka, B. M. & Cam, M. C. (2003) Nucleic Acids Res. 31, 5676-5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang, I. V., Chen, E., Hasseman, J. P., Liang, W., Frank, B. C., Wang, S., Sharov, V., Saeed, A. I., White, J., Li, J., et al. (2002) Genome Biol. 3, research0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tu, Y., Stolovitzky, G. & Klein, U. (2002) Proc. Natl. Acad. Sci. USA 99, 14031-14036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Velculescu, V. E., Zhang, L., Vogelstein, B. & Kinzler, K. W. (1995) Science 270, 484-487. [DOI] [PubMed] [Google Scholar]
- 11.Saha, S., Sparks, A. B., Rago, C., Akmaev, V., Wang, C. J., Vogelstein, B., Kinzler, K. W. & Velculescu, V. E. (2002) Nat. Biotechnol. 20, 508-512. [DOI] [PubMed] [Google Scholar]
- 12.Brenner, S., Johnson, M., Bridgham, J., Golda, G., Lloyd, D. H., Johnson, D., Luo, S., McCurdy, S., Foy, M., Ewan, M., et al. (2000) Nat. Biotechnol. 18, 630-634. [DOI] [PubMed] [Google Scholar]
- 13.Brenner, S., Williams, S. R., Vermaas, E. H., Storck, T., Moon, K., McCollum, C., Mao, J. I., Luo, S., Kirchner, J. J., Eletr, S., et al. (2000) Proc. Natl. Acad. Sci. USA 97, 1665-1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nau, G. J., Richmond, J. F., Schlesinger, A., Jennings, E. G., Lander, E. S. & Young, R. A. (2002) Proc. Natl. Acad. Sci. USA 99, 1503-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugarman, B. J., Aggarwal, B. B., Hass, P. E., Figari, I. S., Palladino, M. A., Jr., & Shepard, H. M. (1985) Science 230, 943-945. [DOI] [PubMed] [Google Scholar]
- 16.Hoth, S., Ikeda, Y., Morgante, M., Wang, X., Zuo, J., Hanafey, M. K., Gaasterland, T., Tingey, S. V. & Chua, N. H. (2003) FEBS Lett. 554, 373-380. [DOI] [PubMed] [Google Scholar]
- 17.Eisen, M. B., Spellman, P. T., Brown, P. O. & Botstein, D. (1998) Proc. Natl. Acad. Sci. USA 95, 14863-14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Audic, S. & Claverie, J. M. (1997) Genome Res. 7, 986-995. [DOI] [PubMed] [Google Scholar]
- 19.Kal, A. J., van Zonneveld, A. J., Benes, V., van den Berg, M., Koerkamp, M. G., Albermann, K., Strack, N., Ruijter, J. M., Richter, A., Dujon, B., et al. (1999) Mol. Biol. Cell 10, 1859-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vencio, R. Z., Brentani, H., Patrao, D. F. & Pereira, C. A. (2004) BMC Bioinformatics 5, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.