

Abstract
Bevacizumab (Avastin®, Genentech, CA) was granted accelerated approval by the FDA for metastatic breast cancer in 2008. This occurred after the initial clinical trial, E2100, demonstrated an improvement in progression-free survival (PFS) and overall survival (OS) with the addition of bevacizumab to a standard chemotherapy. Unfortunately, the approval was rescinded in 2011 when two subsequent trials, AVADO and RIBBON-1, failed to show survival benefit. We compare and analyze the landmark trials E2100, AVADO and RIBBON-1, and suggest that the present-day clinical trial model may not be suited for the investigation of targeted therapies such as bevacizumab. The existing clinical trial model does not allow for modification of chemotherapeutic doses in a manner that maximizes the effect of biologic response modifiers and does not account for its “chemosensitizing” effect. The E2100, AVADO, and RIBBON-1 trials differed in the type and dose of chemotherapy, the dose and frequency of bevacizumab, and in the trial design, making it difficult to effectively compare and evaluate the results. The efficacy of combining bevacizumab with a maximum tolerated dose (MTD) of chemotherapy is also discussed in view of the observation that increased tumor response did not translate to an increase in survival. We suggest that even though an-giogenesis inhibitors are non-toxic as monotherapies, they increase the toxicity of standard chemotherapy, and consequently a re-design of the now classic clinical trial model should be considered. Modifying the existing clinical trial model will lead to a more accurate evaluation of the safety and efficacy of bevacizumab and other biological agents in treating metastatic cancer.
Keywords: Anti-angiogenic therapy, Angiogenesis, AVADO, Breast cancer, Avastin, Bevacizumab, E2100, RIBBON-1, Breast cancer treatment, VEGF, Vascular endothelial growth factor, Metronomic therapy
Introduction
Chemotherapy, as coined by Paul Ehrlich in the early 20th century, is the use of chemicals to treat diseases [1]. Most traditional cancer chemotherapies are cytotoxic and either alter DNA synthesis or interfere with microtubule formation [see Fig. 1]. The number of these “chemicals” has been steadily increasing since the days Sidney Farber used folate antagonists to treat childhood leukemia, but the survival curves have plateaued. In contrast, ‘targeted’ therapies inhibit specific physiological processes, and include tyrosine kinase inhibitors, immunomodulators, cytokines or cytokine inhibitors, protease inhibitors, anti-growth factor antibodies among others.
Fig. 1.
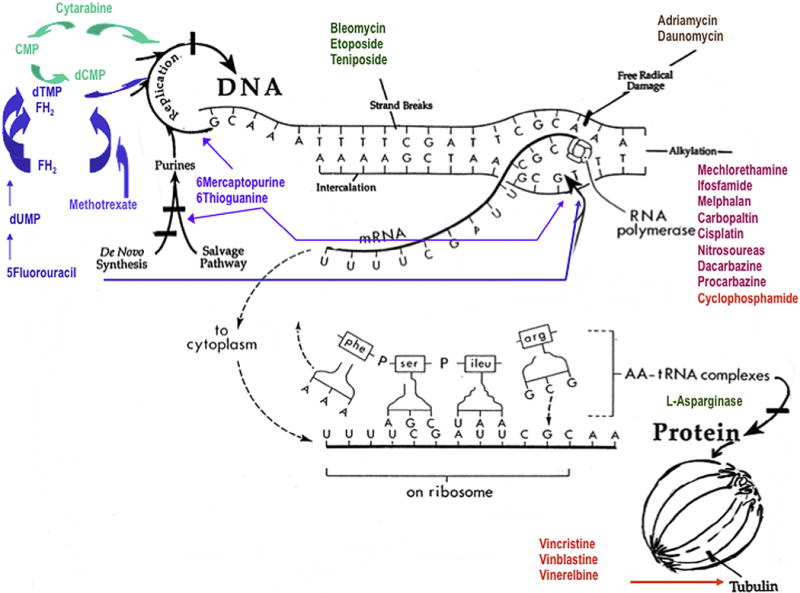
Sites of action of traditional chemotherapeutic agents. The target of traditional chemotherapeutic agents is the DNA replication (cytarabine, methotrexate, 5-fluorouracil, 6 thioguanine), mature DNA (bleomycin, etoposide, teniposide, adriamycin and daunomycin), DNA alkylation (ifosfamide, cyclophosphamide, platin based drugs etc.), translation (L-asparginase) or the mitotic spindle (vincristine, vinblastine, taxanes). This is in direct contrast to the biologic agents such as bevacizumab.
In this article, we use bevacizumab, a monoclonal antibody against Vascular Endothelial Growth Factor (VEGF), as a surrogate for targeted agents, and consider tumor angiogenesis host biological process supporting cancer progression [2–4]. The attractiveness of targeting angiogenesis was ensured by lower toxicity and the absence of physiological angiogenesis after birth [5]. VEGF is an initiating signal for angiogenesis, and while it is haplotype lethal during embryogenesis [6], it is only needed for initiation of a vascular sprout in the wound or tumor microenvironment postnatally. Once a sprout (tip cell) is formed, other angiogenesis stimulators such as bFGF and PDGF support the development of stalk cells, and recruitment of smooth muscle cell, rendering the vasculature quiescent [7,8]. VPF (VEGF) was discovered in Dr. Dvorak’s laboratory in 1983 [9], and was re-named in 1989 [10] after subsequent cDNA cloning of VPF [11] and VEGF [12] proved that VPF and VEGF were the same molecule [2]. It proved to be an evolutionally well preserved protein [13], and its secretion leads to a proliferative signal when bound to VEGFR2 on endothelial cells, and to a differentiation signal when it binds to VEGFR1. Other functions of VEGF include recruitment, stimulation and differentiation of progenitor endothelial cells, promotion of monocyte chemotaxis in the bone marrow [14], induction of colony formation by mature subsets of granulocyte–macrophage progenitor cells [15], and regulation of immune and anti-inflammatory cells” [16].
When in 1997 Ferrara et al. developed bevacizumab (Genentech: Avastin®), a neutralizing antibody to VEGF, it was the first of many angiogenesis inhibitors. Early safety and efficacy trials demonstrated that bevacizumab, similar to other monoclonal antibodies, lacked traditional toxicities when used as monotherapy [17], and that most “bevacizumab-associated” toxicities develop when one receives bevacizumab combined with standard chemotherapy regimens [16]. The toxicities due to bevacizumab itself include hypertension and proteinuria. Hypertension appears to be dose dependent, because 10–15 mg/kg of bevacizumab (the typical dose in metastatic breast cancer trials) had a higher incidence of hypertension, than 7.5 mg/kg (the amount used in colorectal cancer trials). Less common but severe toxicities (Grades 3–4) are bleeding, cardiomyopathy, arterial thromboembolism and hemorrhage. The often-mentioned risk of delayed wound healing and complications after surgery remain undocumented. According to Gressett and Shah [16], wound-healing issues can be avoided if bevacizumab is not started until 28 days after a surgical procedure and is halted 60 days before a surgical procedure, but no corresponding arm of continued treatment was available. Finally, the most rare, but severe toxicities include gastrointestinal perforation and reversible posterior leukoencephalopathy syndrome (RPLS).
The focus of this manuscript is the manner in which bevacizumab-related toxicities were handled in the select Phase III randomized controlled clinical trials and whether efficacy could be assessed in this setting. Bevacizumab was the first monoclonal antibody (and the first angiogenesis inhibitors in clinics) approved by the FDA. It was approved for colorectal cancer in 2004. It was then granted accelerated approval for metastatic breast cancer in 2008 when the E2100 study demonstrated that paclitaxel with bevacizumab improved PFs from 5.9 months in the paclitaxel alone group, to 11.8 months. Unfortunately, in the subsequent trials: AVADO and RIBBON-1 did not demonstrate an increase in OS for the bevacizumab plus chemotherapy group, leading to a reversal of the FDA approval [18]. Was the reversal possibly due to an un-favorable clinical trial design?
Despite the distinctly different mechanism of action of targeted therapies there have not been changes in clinical trial design from that used traditionally. The Phase I–IV model has been designed to find the maximum tolerated dose in Phase I and assumes additive rather than synergistic activity with existing agents of the standard arm. However, while a targeted agent may not be toxic as monotherapy, its addition to an already maximally toxic chemotherapeutic regimen may have unacceptable consequences. In fact the traditional clinical trial model, which adds the targeted agent to a highly toxic arm, puts the targeted agent at a disadvantage. We compare the methods and results of three randomized controlled trials, namely E2100, AVADO and RIBBON-1, and explore the reasons for the discrepancies in their results. We show that the lack of a significant OS benefit may not have been due to the lack of efficacy or “toxicity” of bevacizumab, but rather lies in differences in types of agents used in the subsequent trials, in differences in doses and frequency of administration of the agents, and in the clinical trial design itself. We will propose that because traditional chemotherapy and anti-VEGF agents are synergistic in their action, the combination requires lower doses of chemotherapy. We propose that the “bevacizumab-associated” toxicities were actually chemotherapy-related toxicities and depended on the combination.
The three landmark trials
E2100 trial
E2100 was an open-label Phase 3 randomized controlled trial [19] of 722 patients with metastatic breast cancer enrolled from December 2001 to May 2004. Chemotherapy naive patients, heavily pretreated, or those who received hormonal and/or adjuvant therapy were all acceptable. HER2-positive patients were only allowed if they failed trastuzumab (Herceptin®) therapy. Only 8 enrolled patients were HER2-positive. Patients were randomized into two groups: paclitaxel alone (control), and paclitaxel/bevacizumab. Both groups received 90 mg/m2 of paclitaxel on Days 1, 8, 15 of a 4-week cycle. The experimental group received 10 mg/kg of bevacizumab on Days 1 and 15. When toxic effects occurred in either the experimental or the control group, the dose of paclitaxel was reduced, and if toxicities persisted paclitaxel was stopped. Notably, a much greater number of patients had to stop paclitaxel in the combination group. While 35.9% (117) of patients had their paclitaxel halted in the control group, 51.3% (178) of patients had to do so in the combination group. The dose of bevacizumab was not lowered for any of the side effects, with the exception of proteinuria. None of the patients in the paclitaxel group received bevacizumab, in distinct variance from the other trials.
Results
The median PFS was statistically significantly higher (p < 0.01) for the paclitaxel + bevacizumab group (11.8 months) than the paclitaxel-only group (5.9 months). There was also a slight increase in the median OS in the paclitaxel + bevacizumab group versus the control group (26.7 months vs. 25.2 months), although it was not statistically significant (p = 0.16). The addition of bevacizumab resulted in a 1-year survival rate of 81.2%, compared to 73.4% with paclitaxel alone (p = 0.01). For a summary see Table 1.
Table 1.
Comparison of outcomes and length of treatment. Three landmark trials using bevacizumab (E2100, AVADO, RIBBON-1) are compared here for outcomes such as overall response rate (ORR), progression free survival (PFS), 1-year survival and overall survival (OS).
| E2100
|
AVADO
|
RIBBON-1
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Endpoints | Paclitaxel alone | Paclitaxel + beva | Docetaxel + placebo | Docetaxel + 7.5 beva | Docetaxel + 15 beva | Capecitabine + placebo | Capecitabine + beva | Tax/Anthrac + placebo | Tax/Anthrac + beva |
| ORR (%) | 21.2 | 36.9 | 46.4 | 55.2 | 64.1 | 23.6 | 35.4 | 37.9 | 51.3 |
| PFS (months) | 5.9 | 11.8 | 8.1 | 9.0 | 10.0 | 5.7 | 8.6 | 8.0 | 9.2 |
| OS (months) | 25.2 | 26.7 | 31.9 | 30.8 | 30.2 | Not provided | Not provided | Not provided | Not provided |
| 1 year survival (%) | 73.4 | 81.2 | 76 | 81 | 84 | 74.4 | 81.0 | 83.2 | 80.7 |
| Length of treatment (cycles or months) | 5.1 | 7.1 | 4.9 | 5.1 | 5.5 | 8.7cycles | 10.3cycles | 10.3–10.5cycles | 11.4cycles |
| Possible to receive bevacizumab in control arm(s)? | No | Yes | Yes | Yes | |||||
Safety
A greater percentage of patients in the combination group experienced adverse events. Those included: fatigue (8.5% vs. 4.9%, p = 0.04), infection (9.3% vs. 2.9%, p < 0.001), headache and grade 3/4 neuropathy (23.6% vs. 17.6%, p = 0.03), and hypertension (Grade 3: 14.5% vs. 0%; Grade 4: 0.3% vs. 0). Three Grade 5 events occurred in this trial: 1 left ventricular dysfunction in the paclitaxel alone group, and 1 ruptured diverticulum and 1 erosion of the bowel-wall area in the combination group. All three events were considered unrelated to therapy. For a summary, see Table 2.
Table 2.
Comparison of toxicities. Three landmark trials using bevacizumab (E2100, AVADO, RIBBON-1) are compared here for generally accepted chemotherapeutic toxicities such as bone marrow suppression (neutropenia), as well as bevacizumab-specific toxicities such as proteinuria, hypertension.
| E2100
|
AVADO
|
RIBBON-1
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Adverse events [Grades 3 & 4] |
Paclitaxel only |
Paclitaxel + 10 beva |
Docetaxel + placebo |
Docetaxel + 7.5 beva |
Docetaxel + 15 beva |
Capecitabine + placebo |
Capecitabine + 15 beva |
Tax + placebo |
Tax + 15 beva |
Anthrac + placebo |
Anthrac + 15 beva |
| Hypertension (%) | 0 | 14.8 | 1.3 | 0.8 | 4.5 | 1 | 10.1 | 2 | 8.9 | 0 | 10.5 |
| Bleeding (%) | 0 | 0.5 | 0.9 | 1.2 | 0.8 | 0.5 | 0.2 | 0 | 5.4 | 0 | 1.0 |
| Proteinuria (%) | 0 | 3.5 | 0 | 0.8 | 2 | 0 | 2.2 | 0 | 3.9 | 0 | 2.9 |
| Neutropenia (%) | 0.3 | 0 | 17.3 | 19.8 | 19.8 | 1.0 | 1.2 | 4.9 | 9.4 | 4.0 | 4.3 |
| Febrile neutropenia (%) | 0 | 0.8 | 11.3 | 15.1 | 16.2 | 0 | 0 | 2.0 | 8.4 | 5.0 | 3.8 |
| Peripheral edema (%) | Not Provided | Not Provided | 2.6 | 1.2 | 0.4 | Not Provided | Not Provided | Not Provided | Not Provided | Not Provided | Not Provided |
AVADO
This Avastin and Docetaxel Study was a Phase 3, double-blind, three-armed, placebo-controlled trial [20]. The enrollment included chemotherapy naive patients with HER2-negative local recurrent or metastatic breast cancer, as well as patients with prior neoadjuvant or adjuvant therapy. Patients with hypertension or non-measurable tumor could be enrolled.
From March 2006 to April 2007, 736 patients were randomized in a 1:1:1 ratio into 3 groups: i) placebo group (N = 241), ii) experimental group (N = 247) receiving 7.5 mg/kg of bevacizumab on Day 1 of every 3 week cycle (bevacizumab dose approved in colorectal cancer), and iii) experimental group (N = 248) receiving 15 mg/kg of bevacizumab on Day 1 of every 3-week cycle (dose used in metastatic breast cancer). Note that this dose differs from the E2100 trial, where 10 mg/kg was given every two weeks. All three groups received 100 mg/m2 of docetaxel on Day 1 of every 3-week cycle. In this study the doses of placebo and bevacizumab were never altered, rather docetaxel was withheld with any grade 3/4 toxicities, and if the toxicity persisted docetaxel treatment was stopped completely.
Results
The primary endpoint for this study was the PFS. The median PFS was 8.1 months for the placebo group, 9.0 months for the 7.5 mg/kg bevacizumab (p = 0.045), and 10 months (p < 0.001) for the 15 mg/kg bevacizumab group. Of note is that the median PFS for all groups were lower in this study than in the E2100 trial [see Table 1]. The improvement in PFS in the 15 mg/kg group was statistically significant despite a lower number of patients. The study was not powered to reveal differences in OS, a secondary endpoint in this study, and OS was similar (31 months) in all three groups. Yet, there was a statistically significant increase in the 1-year survival rate for the 15 mg/kg bevacizumab group compared to placebo (84% vs. 76%; p = 0.02). Of procedural concern is that 83/241 patients in the placebo group received bevacizumab. This included fourteen patients (unblinded) in the placebo-controlled group who received bevacizumab prior to disease progression. These patients were included in the safety and efficacy analysis of the placebo arm, even though adverse events from this cohort were not included upon crossover. For a summary see Table 1.
Safety
Majority of patients in the AVADO trial had at least one adverse event, most docetaxel-related. Hypertension was dose dependent [14.3% in 7.5 mg/kg and 21.9% in 15 mg/kg bevacizumab], and more frequent in combination groups [10% in the placebo-controlled group]. Bleeding was also more frequent in the combination groups [48.4% and 49.4% respectively] vs. the placebo group [19.5%]. Grade 3/4 neutropenia, as well as Grade 3/4 peripheral edema occurred more frequently in the placebo group. For a summary, see Table 2.
RIBBON-1
Regimens in Bevacizumab for Breast Oncology-1 (RIBBON-1) was a Phase 3 randomized clinical trial that studied bevacizumab addition to a number of different types of chemotherapy [21]. From December 2005 to April 2007, a total of 1237 patients were randomized in a 2:1 ratio to either chemotherapy + bevacizumab or chemotherapy + placebo groups. The trial assumed equivalency between the various chemotherapy regimens. In addition to the two placebo groups for each of the chemotherapy regimens, there were two types of chemotherapy: A) Capecitabine 1000 mg/m2 orally twice a day from Days 1 to 14 of the 3 week cycle, or B) A combination of taxane and/or anthracycline as listed below:
Docetaxel (75–100 mg/m2) or paclitaxel protein-bound particles (260 mg/m2) every 3 weeks
Fluorouracil (500 mg/m2), epirubicin (90–100 mg/m2), and cyclophosphamide (500 mg/m2)
Fluorouracil (500 mg/m2), doxorubicin (50 mg/m2) and cyclophosphamide (500 mg/m2)
Doxorubicin (50–60 mg/m2) and cyclophosphamide (500–600 mg/m2)
Epirubicin (90–100 mg/m2) and cyclophosphamide (500–600 mg/m2)
All patients receiving bevacizumab were given 15 mg/kg of bevacizumab every 3 weeks. The type of chemotherapy each patient received was assigned before randomization.
Results
The median PFS, the primary endpoint, was again higher for the bevacizumab groups than for the placebo groups. There was a statistically significant difference (p < 0.001) in PFS of the capecitabine/placebo group [5.7 months] compared to the capecitabine/bevacizumab group [8.6 months]. Similarly, in the taxane/anthracycline cohort, addition of bevacizumab resulted in an improvement in PFS from 8 to 9.2 months. There was no significant difference among the study groups in OS, but over half of the patients in the placebo groups received bevacizumab [50.7% of the patients in the taxane/anthracycline/placebo group, and 54.4% in the capecitabine/placebo group]. While the differences in the 1-year survival rates among the groups were not statistically significant, there were improvements from 74% in the capecitabine/placebo group compared to 81.0% rate in the capecitabine/bevacizumab group (p = 0.076). The taxane/anthracycline/bevacizumab group showed a high overall response rate (ORR) and had a 1-year survival rate of 83.2% compared to 80.7% in the taxane/anthracycline/placebo (p = 0.44). There are clear improvements in ORR between the chemotherapy arms [capecitabine alone 23.6%, taxane/anthracycline 37.9%] and bevacizumab arms [35.4% capecitabine/bevacizumab, 51.3% taxane/anthracycline/bevacizumab]. See Table 1.
Safety
The incidence of Grades 3–5 adverse events and serious adverse events was again higher for all bevacizumab/chemotherapy groups, with hypertension and proteinuria being most common. The percentage of patients experiencing fatal adverse events was higher in the placebo groups [2.5% in capecitabine/placebo vs. 1.5% capecitabine/bevacizumab, and 3% anthracycline/placebo vs. 1.4% anthracycline/bevacizumab]. The exception was taxane including regimens where fatal events were identical [taxane/placebo: 2.9% vs. taxane/bevacizumab: 2.5%]. None of the differences in fatal events were statistically significant. For a summary see Table 2.
Discussion
There are significant differences between the E2100, AVADO and RIBBON-1 trials, and a deeper scrutiny may provide explanation for at least some of the variability in results. The comparison of the three trials reveals that the type and dose of chemotherapy used in combination with a biological agent matter. Most biologically active agents, and angiogenesis inhibitors in particular, sensitize to chemotherapy [22] and/or radiation. It was therefore not surprising to see increased toxicity of alkylating agents when used in combination with bevacizumab. Even though the addition of bevacizumab to the taxane/anthracycline combination improved the overall response rate from 37.9% to 51.3% and the PFS from 8 months to 9.2 months, these improvements did not translate to improvement in survival. In fact the 1-year survival seemed worse (80.7%) in the bevacizumab group than in the taxane/anthracycline/placebo group (83.2%), suggesting that doxorubicin, an anthracycline with significant cardiac toxicity, may not be the best agent to combine with an angiogenesis inhibitor. Taxanes showed a much better therapeutic index in the E2100 trial, where ORR improved from 21.2% to 36.9% by the addition of bevacizumab to paclitaxel, and in the AVADO trial where the addition of bevacizumab to docetaxel increased ORR from 46.4% to 64.1% in the docetaxel/15 mg bevacizumab group. It should be noted that in the AVADO trial, only the 15 mg/kg of bevacizumab Q3weeks is comparable to the 10 mg/kg bevacizumab Q2weeks in the E2100 trial. The congruence in PFS, ORR and OS improvement across the three trials when bevacizumab was added to chemotherapy [Table 1] underscores the synergism between angiogenesis inhibitors and chemotherapy, and suggests the possibility to harness this for future therapy.
Neither the approval of bevacizumab for metastatic breast cancer nor its retraction had taken into account the biological nature of this therapy, and the suitability of the classical double-arm trial for showing its efficacy. If angiogenesis inhibitors are chemotherapy sensitizers [22] should becizumab be added to maximum tolerated doses (MTD) of chemotherapy? Similarly, no consideration was given to the differences in toxicities between the various chemotherapeutic agents. The RIBBON-1 used an array of combinations of chemotherapeutic agents, which were considered equivalent despite the previously published evidence that anthracyclines and tubulin inhibitors differ significantly in their toxicities when used with VEGF pathway inhibitors [23,24]. The use of MTD or high doses of alkylating agents may not only be unnecessary, and it may be in fact harmful, as the results of the 3 trials explored for this article suggest.
In Table 2 we review that the gains in response rates were coupled with increased toxicities. It should also be pointed out that the toxicities were handled differently in each trial. In the E2100 trial, paclitaxel dose was reduced when an adverse effect occurred, whereas in the AVADO trial, docetaxel was withheld with the first occurrence of toxicity and stopped completely if the toxicity recurred following re-introduction. In RIBBON-1, when toxicities occurred chemotherapy could be modified “at the investigator’s discretion”, making the specific changes untraceable. While we do not know what was done, considering the common reaction to toxicities “investigator discretion” is more likely to have led to lowering the dose of the biological agent (the new addition to the protocol) while keeping the standard doses of chemotherapy unchanged because for the traditionally trained oncologist, the relationship of chemotherapy dose and cancer cell kill shows a linear relationship and preserving MTD is of importance [25,26]. In the case of biologic agents however, the dose depends on an optimal inhibition of the target [Fig. 2], rendering a lower (or higher) dose of bevacizumab ineffective. In E2100, more patients in the bevacizumab/paclitaxel group had reductions of paclitaxel when paclitaxel-related toxicities occurred, thus preserving paclitaxel effect with bevacizumab sensitization. Taxanes [27,28] and other tubulin inhibitors [29] affect vascular endothelium, and synergize with bevacizumab at picomolar doses to effectively prevent vascular sprouting [28]. Bevacizumab can also modify the delivery of chemotherapeutic agents to tumors by decreasing the tumor intestinal pressure and altering tumor vascularity [30]. This bevacizumab-facilitated increase in chemotherapeutic delivery would have decreased the toxicities while maintaining anti-tumor efficacy with reduced doses of chemotherapy.
Fig. 2.
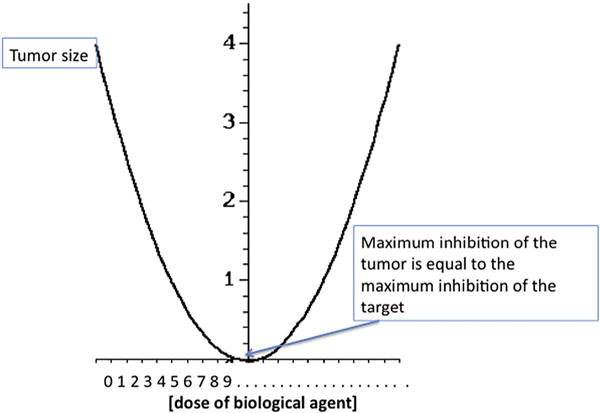
The U-shaped curve associated with the effect of BRMs. Unlike the linear relationship between dose and effect assumed in the initial experiments with chemotherapy in leukemia done by Skipper and Schabel [25,26], biologic agents lose effect at low or at high doses, creating a U-shaped curve of activity. The optimal biologically effective dose falls in the middle where the majority of the receptor is inhibited, but no off-site effects are induced. This may facilitate the up and down regulation of physiological biological processes during stress response setting with a linear increase in the initial effect, but turning off the effect in the presence of excess ligand.
Another important confounding variable, and the reason why the final OS analysis revealed little difference between the three study arms in the AVADO trial, was the use of bevacizumab in the control groups. In both AVADO and RIBBON-1 trials, the patients in the control arms were allowed to receive bevacizumab after tumor progression. In the AVADO trial, 83 patients out of 241 in the placebo/docetaxel group received bevacizumab, including the 14 unblinded patients in the placebo-controlled group who received bevacizumab prior to disease progression. These 14 patients were included in the safety and efficacy analysis of the placebo arm, but adverse events from this cohort were not included upon crossover. It is more than likely that the use of bevacizumab (the experimental intervention) in the control group would have negatively impacted on the OS calculation. This was not the case for patients in the control arm of the E2100 study, where an increase in OS with the use of bevacizumab was seen.
The doses of bevacizumab and the frequency of its use also significantly differed between the three trials. The AVADO and RIBBON-1 trial used three-week intervals for the bevacizumab infusions, whereas E2100 used 10 mg/kg every two weeks. This is an important change considering the average half-life of a monoclonal antibody is 21 days, and a tri-weekly dosing allows for a significant drop in bevacizumab plasma levels in the last week when most of the bone marrow recovery and angiogenesis occurs. The dose of bevacizumab, 10 mg/kg Q2weeks in the E2100 trial, is therefore only marginally comparable to the 15 mg/kg bevacizumab Q3weeks dosing in AVADO and RIBBON-1.
Similarly, the doses of paclitaxel are difficult to compare between the three trials because in E2100 they used 90 mg/m2 of paclitaxel whereas in RIBBON-1 the taxane cohort could have received either: docetaxel 75–100 mg/m2 or 260 mg/m2 paclitaxel protein-bound particles (Abraxane®). In the AVADO trial, each group received 100 mg/m2 of docetaxel. The variations in dose and frequency of chemotherapy administration invariably had an effect on PFS. In the E2100 trial, 90 mg/m2 of paclitaxel was administered on days 1, 8, and 15 every 4 weeks (a more frequent and lower dose than the standard MTD of 175 mg/m2 given every 3 weeks [31]). The 90 mg/m2 of paclitaxel, while not a metronomic dose [32] was lower, and could be administered more frequently than the MTD. The AVADO study administrated 100 mg/m2 of docetaxel every 3 weeks. The most difficult to interpret were the chemotherapeutic regimens utilized in the RIBBON-1 study, because these varied even within the study itself. RIBBON-1 consisted of a variety of combinations of taxanes and anthracyclines or capecitabine, and it is very difficult to determine whether standard or lower than standard doses were used. The capecitabine cohort of this trial received 1000 mg/m2 of capecitabine twice a day on Days 1–14 of a 3-week cycle, which is within the MTD dose of 825–1250 mg/m2 twice daily on days 1–14 [33]. Within the taxane/anthracycline cohort, patients could receive 75–100 mg/m2 of docetaxel (within the standard amount), or 260 mg/m2 of nab-paclitaxel (equal to the standard amount) [34]. Within the taxane/anthracycline cohort, patients could also receive 50–60 mg of doxorubicin in combination with 500–600 mg/m2 of cyclophosphamide or 50 mg of doxorubicin along with 500 mg/m2 flurouracil and 500 mg/m2 cyclophosphamide. A dose of 40– 60 mg/m2 of doxorubicin is often recommended as the highest dose when used in addition with other chemotherapeutic agents [35]. But in this study patients could receive 90–100 mg/m2 of epirubicin along with 500–600 mg/m2 of cyclophosphamide or with 500 mg/m2 cyclophosphamide and 500 mg/m2 fluorouracil. The recommended starting dose of epirubicin is 100–120 mg/m2. When epirubicin is administered in combination with fluorouracil and cyclophosphamide, the standard dose is 100 mg/m2 [36]. Anthracyclines such as doxorubicin and epirubicin are especially toxic when combined with angiogenesis inhibitors [24] and various studies have demonstrated the increased frequency of cardiotoxic effects ranging from acute arrhythmias to chronic dysfunction such as left ventricular dysfunction and congestive heart failure. Thus, the differences in the administration frequencies and the type of chemotherapeutic agent may have significantly impacted on how bevacizumab modified the toxicity of MTD, and the variability in doses, frequencies and types of agents in RIBBON-1 may make it impossible to compare and derive meaningful conclusions about why the subsequent trials failed.
The doses and frequencies of bevacizumab differed as well, and these variations highlight the fact that AVADO and RIBBON-1 may not necessarily disprove the PFS and OS gains that occurred with the addition of bevacizumab in E2100. By confirming the gains in ORR and PFS observed in E2100, the two subsequent trials highlight the need to change the existing clinical trial model. The clinical studies of angiogenesis inhibitors and other biological agents should use an “optimal” (i.e. biologically effective) dose of the biological agent and study biological agents/chemotherapy combinations in settings that maximize synergistic action. The optimal dosage may need to be specific for each class of biological agents and each agent/chemotherapy combination, and only once an optimal regimen is determined can a randomized double-blind controlled clinical trial be used to assess the efficacy of bevacizumab. It is the expectation, on the basis of the available literature, that the combinations of chemotherapy and biological agents may require modifications in the dose and frequency of chemotherapy, leading to new clinical paradigms such as “metronomic” dosing.
Metronomic dosing is the continuous administration of low-dose chemotherapy without long periods of rest [32]. It takes advantage of the fact that at low, frequent doses conventional chemotherapeutic agents have angiogenesis inhibitory [37], immunomodulatory [38,39] and anti-inflammatory [40] activities with minimal toxic effects [41]. In 2000, Klement et al. administered low doses of vinblastine and a monoclonal VEGF-R2 antibody (DC101) to human neuroblastoma xenografts in SCID mice [37]. The results showed that while vinblastine alone may reduce tumor growth for a brief period, it is the combination of low-dose vinblastine and DC101 that leads to sustained inhibition [37]. The same year Browder et al. [42]. demonstrated that the combination of low dose metronomic regimen of cyclophosphamide with an angiogenesis inhibitor was effective in eradicating cyclophosphamide-resistant xenografts in immuno-deficient mice, where standard MTD of cyclophosphamide had failed. Because in both cases the combination of metronomic chemotherapy and angiogenesis inhibitor effectively eradicated chemotherapy-resistant tumor cells, it had to be effective on other cells of the tumor microenvironment.
Conclusion
The use of biologic agents is changing the horizons of today’s oncology. Patients are less accepting of cancer therapy related toxicities and cancer treatment therapies are more and more frequently tailored to molecular changes in the tumor. However, the introduction of biological therapies into the clinical setting may be hindered by an antiquated system of clinical evaluation of these therapies. Because we no longer need to derive a maximally tolerated dose of a biologic agent, but rather find an optimal dose a Phase 1 trial may be unnecessary. Similarly, because biologic agents have minimal effect as monotherapy and act more as chemotherapy sensitizers, they are likely to be used in combination with low-dose chemotherapy and should be evaluated a priori in these combinations. The present clinical trial approach may not be providing the best modality for evaluating bevacizumab and other biologic agents and a new approach incorporating a systems biology approach to therapy should be considered.
Acknowledgments
We gratefully acknowledge the editorial and administrative assistance of Allison Jenks. The work was supported by the NIH/NIGMS GM093050 (G.L.K.), Binational Science Foundation BSF 2009241 (G.L.K.), and the Newman Lakka Cancer Foundation (G.L.K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute, or the other funding sources.
Footnotes
Authors’ contributions
JAA carried out the research and wrote the first draft of the manuscript. GLK provided oversight as well as expertise in cancer biology, clinical trials and cancer therapeutics. Both authors contributed to writing the manuscript.
Conflict of interest
None.
References
- 1.DeVita VT, Jr, Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68:8643–8653. doi: 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer. 2002;2:795–803. doi: 10.1038/nrc909. [DOI] [PubMed] [Google Scholar]
- 3.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 4.Virchow R. Cellular Pathology: As Based upon Physiological and Pathological Histology, John Churchill, London. 1860 doi: 10.1111/j.1753-4887.1989.tb02747.x. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 6.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 7.Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445:776–780. doi: 10.1038/nature05571. [DOI] [PubMed] [Google Scholar]
- 8.Jacobsson K, Gerhardt W, Skude G, Tryding N. S-GT and alcoholism-soberly considered. Lakartidningen. 1980;77:987–990. [PubMed] [Google Scholar]
- 9.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–985. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 10.Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun. 1989;161:851–858. doi: 10.1016/0006-291x(89)92678-8. [DOI] [PubMed] [Google Scholar]
- 11.Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, et al. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246:1309–1312. doi: 10.1126/science.2479987. [DOI] [PubMed] [Google Scholar]
- 12.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 13.Senger DR, Perruzzi CA, Feder J, Dvorak HF. A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines. Cancer Res. 1986;46:5629–5632. [PubMed] [Google Scholar]
- 14.Clauss M, Gerlach M, Gerlach H, Brett J, Wang F, Familletti PC, et al. Vascular permeability factor: a tumor-derived polypeptide that induces endothelial cell and monocyte procoagulant activity, and promotes monocyte migration. J Exp Med. 1990;172:1535–1545. doi: 10.1084/jem.172.6.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broxmeyer HE, Cooper S, Li ZH, Lu L, Song HY, Kwon BS, et al. Myeloid progenitor cell regulatory effects of vascular endothelial cell growth factor. Int J Hematol. 1995;62:203–215. doi: 10.1016/0925-5710(95)00412-2. [DOI] [PubMed] [Google Scholar]
- 16.Gressett SM, Shah SR. Intricacies of bevacizumab-induced toxicities and their management. Ann Pharmacother. 2009;43:490–501. doi: 10.1345/aph.1L426. [DOI] [PubMed] [Google Scholar]
- 17.Keizer RJ, Huitema ADR, Schellens JHM, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:493–507. doi: 10.2165/11531280-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 18.FDA. Proposal to withdraw approval for the breast cancer indication for AVASTIN (bevacizumab) 2011. [Google Scholar]
- 19.Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666–2676. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 20.Miles DW, Chan A, Dirix LY, Cortes J, Pivot X, Tomczak P, et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol. 2010;28:3239–3247. doi: 10.1200/JCO.2008.21.6457. [DOI] [PubMed] [Google Scholar]
- 21.Robert NJ, Dieras V, Glaspy J, Brufsky AM, Bondarenko I, Lipatov ON, et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J Clin Oncol. 2011;29:1252–1260. doi: 10.1200/JCO.2010.28.0982. [DOI] [PubMed] [Google Scholar]
- 22.Wen W, Moses MA, Wiederschain D, Arbiser JL, Folkman J. The generation of endostatin is mediated by elastase. Cancer Res. 1999;59:6052–6056. [PubMed] [Google Scholar]
- 23.Klement G, Huang P, Mayer B, Green SK, Man S, Bohlen P, et al. Differences in therapeutic indexes of combination metronomic chemotherapy and an anti-VEGFR-2 antibody in multidrug-resistant human breast cancer xenografts. Clin Cancer Res. 2002;8:221–232. [PubMed] [Google Scholar]
- 24.Scheuerer B, Ernst M, Durrbaum-Landmann I, Fleischer J, Grage-Griebenow E, Brandt E, et al. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood. 2000;95:1158–1166. [PubMed] [Google Scholar]
- 25.Skipper HE, Schabel FM, Jr, Bell M, Thomson JR, Johnson S. On the curability of experimental neoplasms. I. Amethopterin and mouse leukemias. Cancer Res. 1957;17:717–726. [PubMed] [Google Scholar]
- 26.Skipper HE, Schabel FM, Jr, Mellett LB, Montgomery JA, Wilkoff LJ, Lloyd HH, et al. Implications of biochemical, cytokinetic, pharmacologic, and toxicologic relationships in the design of optimal therapeutic schedules. Cancer Chemother Rep. 1970;54:431–450. [PubMed] [Google Scholar]
- 27.Vacca A, Ribatti D, Iurlaro M, Merchionne F, Nico B, Ria R, et al. Docetaxel versus paclitaxel for antiangiogenesis. J Hematother Stem Cell Res. 2002;11:103–118. doi: 10.1089/152581602753448577. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Lou P, Lesniewski R, Henkin J. Paclitaxel at ultra low concentrations inhibits angiogenesis without affecting cellular microtubule assembly. Anticancer Drugs. 2003;14:13–19. doi: 10.1097/00001813-200301000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Vacca A, Iurlaro M, Ribatti D, Minischetti M, Nico B, Ria R, et al. Antiangiogenesis is produced by nontoxic doses of vinblastine. Blood. 1999;94:4143–4155. [PubMed] [Google Scholar]
- 30.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 31.TAXOL®(paclitaxel) INJECTION [package insert] Bristol-Myers Squibb Company Princeton; NJ: 2011. [Google Scholar]
- 32.Hanahan D, Bergers G, Bergsland E. Less is more, regularly: metronomic dosing of cytotoxic drugs can target tumor angiogenesis in mice. J Clin Invest. 2000;105:1045–1047. doi: 10.1172/JCI9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.XELODA (capecitabine) [package insert] Genentech USA, Inc.; South San Franciso, CA: 2011. [Google Scholar]
- 34.ABRAXANE (paclitaxel protein-bound particles for injectable suspension) [package insert] Abraxis BioScience, LLC; New Jersey: 2013. [Google Scholar]
- 35.Doxorubicin Hydrochloride for Injection, USP [package insert] Pfizer Labs; New York: 2011. [Google Scholar]
- 36.ELLENCE [package insert] Pharmacia & Upjohn Co – Division of Pfizer Inc.; New York, NY: 2011. [Google Scholar]
- 37.Klement G, Baruchel S, Rak J, Man S, Clark K, Hicklin DJ, et al. Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest. 2000;105:R15–R24. doi: 10.1172/JCI8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen CS, Doloff JC, Waxman DJ. Intermittent metronomic drug schedule is essential for activating antitumor innate immunity and tumor xenograft regression. Neoplasia. 2014;16:84–96. doi: 10.1593/neo.131910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doloff JC, Waxman DJ. VEGF receptor inhibitors block the ability of metronomically dosed cyclophosphamide to activate innate immunity-induced tumor regression. Cancer Res. 2012;72:1103–1115. doi: 10.1158/0008-5472.CAN-11-3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montagna E, Cancello G, Torrisi R, Rizzo S, Scarano E, Colleoni M. Lapatinib and metronomic capecitabine combination in an HER2-positive inflammatory breast cancer patient: a case report. Ann Oncol. 2010;21:667–668. doi: 10.1093/annonc/mdp563. [DOI] [PubMed] [Google Scholar]
- 41.Scharovsky OG, Mainetti LE, Rozados VR. Metronomic chemotherapy: changing the paradigm that more is better. Curr Oncol. 2009;16:7–15. doi: 10.3747/co.v16i2.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Browder T, Butterfield CE, Kraling BM, Shi B, Marshall B, O’Reilly MS, et al. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60:1878–1886. [PubMed] [Google Scholar]