

Abstract
Accurate modeling and design of protein-ligand interactions have broad applications in cell, synthetic biology and drug discovery but remain challenging without experimental protein structures. Here we developed an integrated protein homology modeling-ligand docking-protein design approach that reconstructs protein-ligand binding sites from homolog protein structures in the presence of protein-bound ligand poses to capture conformational selection and induced fit modes of ligand binding. In structure modeling tests, we blindly predicted near-atomic accuracy ligand conformations bound to G protein-coupled receptors (GPCRs) that were rarely identified by traditional approaches. We also quantitatively predicted the binding selectivity of diverse ligands to structurally-uncharacterized GPCRs. We then applied the technique to design functional human dopamine receptors with novel ligand binding selectivity. Most blindly predicted ligand binding specificities closely agreed with experimental validations. Our method should prove useful in ligand discovery approaches and in reprogramming the ligand binding profile of membrane receptors that remain difficult to crystallize.
Graphical Abstract
Introduction
Membrane receptors such as G protein-coupled receptors (GPCRs) can typically bind and respond to distinct extracellular ligands1–3. Ligand binding promiscuity allows a single receptor to control and trigger up to several intracellular signaling pathways through distinct ligand-bound receptor conformations4–7. Achieving this property requires receptor structures to be highly flexible4,6,8. However, conformational flexibility represents a challenge for predicting the structure and energetics of receptor-ligand interactions, a critical step in rational drug screening and design approaches. Additionally, despite tremendous progress in membrane protein crystallography9, only a small fraction of ligand-bound receptors (e.g. less than 5% of all GPCRs) have been crystallized to date10.
To address this problem, computational homology modeling approaches have been developed to model receptor structures from structural homologs11–14. Ligand-bound receptor structures are then typically generated by docking the ligand onto selected ligand-free receptor homology models15–20. GPCR-DOCK blind prediction contests have been organized in recent years to assess the accuracy of these computational techniques21–23. Overall, it was concluded that, when close receptor structural homologs and experimental information on ligand-receptor interactions are available, current methods guided by expert modelers can select receptor-ligand structures with high (i.e. near-atomic) accuracy. However, very few successes were obtained on more difficult targets, highlighting the challenges associated with modeling receptors from more distant homologs and with poorly characterized pharmacology (e.g. orphan GPCRs 20). In addition to recurrent sequence-structure alignment problems in homology modeling approaches24,25, the lack of structural accuracy of the receptor ligand-binding site was cited as a major limitation of the techniques. Extracellular ligand binding site sequences and loop structures are often divergent even between GPCRs from the same family3,7,23. Consequently, de novo coarse-grained remodeling techniques11–13,26 are used to first construct ligand-free receptor structures which often result in loop conformations making non-native contacts with the receptor in absence of the ligand. Additionally, current approaches dock ligands onto receptor models without fully relaxing the receptor structure15,17–19,27. Such strategy assumes that ligand binding proceeds by selection of receptor conformations (i.e. conformational selection mechanism28) and cannot fully model effects induced by the ligand on the receptor structure (i.e. induced fit mechanism29,30). Reliably predicting ligand-bound receptor structures by mimicking both induced fit and conformational selection modes of ligand binding remains a challenge.
Since membrane receptors, particularly GPCRs, critically control intracellular signaling but remain difficult to study in their native forms due to high ligand promiscuity, there is growing interest in designing receptors with novel ligand binding selectivity. Empirical approaches have engineered very powerful GPCRs that respond only to designed drugs (e.g. RASSLs31, DREADDs32), enabling the role of particular cellular signaling pathways to be probed. GPCRs with reprogrammed native ligand binding selectivity would provide the next generation of molecules to deconstruct the role of native GPCRs in complex signaling responses and to design novel therapies. However, engineering ligand binding sites with novel binding selectivity remains a challenge in absence of high resolution receptor-ligand structures.
To address these limitations, we have developed IPHoLD, an Integrated Protein Homology modeling, Ligand Docking and protein design approach which models induced fit and conformational selection ligand binding modes from homolog receptor structures only. The method achieved unprecedented near-atomic accuracy in predicting ligand-bound GPCR conformations in a blinded fashion (i.e. without any human guidance or experimental information). We also quantitatively recapitulated the binding selectivity of diverse ligands to structurally-uncharacterized GPCRs using the predicted GPCR-ligand structural models only. Lastly, we combined IPHoLD with experimental validation to rationally engineer GPCRs with novel ligand binding selectivity. When applied to the structurally-uncharacterized dopamine D2 receptor, most designed receptors experimentally displayed binding specificities in close agreement with the predictions.
Results
Integration of homology modeling and ligand docking
So far, attempts at modeling ligand-bound GPCR conformations in absence of experimental receptor structures have adopted the following sequential approach21–23. First, the ligand-free receptor structure is modeled by homology to existing GPCR X-ray structures and, second, the ligand is docked onto selected ligand-free receptor homology models (Supplementary Results, Supplementary Fig. 1). This sequential approach presents two main caveats that can prevent the modeling and selection of accurate ligand-bound GPCR conformations.
First, because sequence and structure homology is often low between GPCR homologs in the extracellular ligand-binding region, several long loops need to be rebuilt de novo from sequence information only26. De novo loop reconstruction is often achieved using peptide fragment insertion techniques generating a large ensemble of unconstrained conformations, which, in absence of ligand, can partially occlude the ligand binding site by making energetically favorable non-native contacts with the transmembrane helices (TMH) of the receptor (i.e. “loop collapse” scenario, Supplementary Fig. 1). Since ligand docking techniques do not relax receptor structures extensively 15,16,28,33, ligand molecules cannot find their native conformations in the binding site of receptor models with collapsed loops.
Second, the sequential receptor modeling/ligand docking approach is inherently based on the assumption that ligand binding to the receptor proceeds by selection of ligand-free receptor conformations (i.e. “conformational selection” mechanism). Because ligand docking does not involve extensive receptor structure relaxation, induced fit effects (i.e. structural change induced by the ligand) cannot be fully modeled (i.e. “no induced fit” scenario, Supplementary Fig. 1).
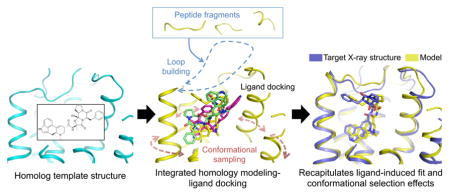
To address these limitations, we reasoned that the receptor structure should be reconstructed in the presence of the bound ligand to readily generate optimal ligand bound receptor conformations. However, because the position and conformation of the ligand in the target structure is unknown, both the optimal conformation of the receptor and that of the ligand need to be searched simultaneously. We implemented this new concept in an integrated receptor homology modeling / ligand docking approach (Fig. 1, Method). The protocol first cycles between coarse-grained ligand docking and coarse-grained de novo loop reconstruction to generate loop conformations making favorable contacts with ligand poses displaying optimal surface complementarity with the entire binding site (Fig. 1, step2). Then, ligand-bound receptor structures with closed loops are extensively relaxed using atomistic representation of both the protein and the ligand to generate a diverse ensemble of low-energy ligand-bound receptor conformations (Fig. 1, step2). The most optimal protein-bound ligand poses are refined and selected by fine-grained all-atom redocking of a large library of ligand conformers onto the low energy ensemble of receptor conformations generated in step 2 (Fig. 1, step3). In essence, the IPHoLD protocol is designed to model the effects associated with two main mechanisms of ligand binding; i.e. induced fit in step 2 where the ligand can influence the conformation of the receptor binding site being constructed, and conformational selection in step 3 where the ligand can preferentially bind to a subset of the large ensemble of receptor conformations. A detailed description and discussion of the method and its implementation is provided in Methods and Supplementary Fig. 2.
Figure 1. Integrated homology modeling and ligand docking.

Step1: generation of ligand conformers and selection of structural homolog template by sequence-structure alignment to the target sequence. Step2: target structure modeling in the presence of bound ligand involving simultaneous coarse-grained de novo reconstruction of receptor loops and sampling of ligand conformations followed by all-atom relaxation of ligand-bound receptor structures. Step3: All-atom docking of a large library of ligand conformers on the ensemble of low-energy receptor conformations generated in step 2 to refine the structure and the selection of receptor-bound ligand poses.
Blind prediction of ergotamine bound serotonin receptors
A first version of the above-described protocol was tested during the last blind GPCR-DOCK 2013 contest23. To stringently assess the accuracy of our technique, we generated and selected models of ergotamine bound serotonin receptors starting from the antagonist-bound D3DR structure without any guidance from published studies on the receptor and the ligand (e.g. mutational effects on ligand binding). This is in contrast to most other participants who extensively guided and biased their modeling selection using human expertise and experimental data. Unlike previous GPCR-DOCK contests21,22, we were able to predict using objective physical criteria alone (Method) a model of the ligand-bound GPCR where the conformation of the ergoline core of ergotamine making extensive contacts with the 5HT2B receptor was predicted with atomic accuracy (heavy atom rmsd of 0.97 Å; Supplementary Fig. 3a). Owing to extensive receptor relaxation in presence of the ligand, our technique substantially moved the extracellular conformation of several TMHs forming the ligand-binding site toward the target structure (Ca rmsd over ligand-interacting residues of 1.1 Å compared to 1.5 Å for the starting template; Supplementary Fig. 3b). The structural shifts were particularly notable in TMH6 and TMH7 which adopts a partially active state conformation in the X-ray structure of ergotamine-bound 5HT2B.
We also identified key limitations in the protocol preventing near-atomic accuracy prediction of the entire ligand conformation bound to the serotonin receptors. First, we noticed that no ligand conformation in our initial library was close to that identified in the X-ray structure which prompted us to systematically increase the size of the ligand rotamer ensemble (Method). Additionally, we identified a suboptimal sequence alignment in TMH4 and in the extracellular loop 2 between the serotonin 5HT1B and the closest homolog template which could be corrected by automatically aligning conserved proline and cysteine residues in these regions (Method). Increasing the diversity of ligand conformers used in the simulation and correcting the sequence alignments allowed for generating and selecting ergotamine-bound 5HT1B and 5HT2B models where the conformation of the entire ligand was predicted with near-atomic accuracy (Supplementary Fig. 3c,d, Supplementary Table 1). Thanks to the method improvements, the accuracy of the ligand binding site region increased for 5HT1B, with a Ca RMSD to the X-ray structure which decreased from 2.0 Å to 1.5 Å when calculated over the ligand-interacting residues (Supplementary Table 2).
Accurate prediction of ligand-bound GPCR conformations
Taking into account the lessons gained from the blind prediction, we tested IPHoLD on a representative subset of 22 ligand-bound GPCR structures sampling a wide diversity of ligand binding site location, structures and ligand sizes (Table 1). Since we were most interested in developing the technique for rational design applications which rely on near-atomic accuracy structure predictions, we did not attempt to model receptors from distant homologs sharing very low sequence identity (i.e. < 20%) with the target sequence. Such modeling requires extensive de novo reconstruction of both loops and TMH core regions and may not consistently generate near-atomic accuracy structures of the receptor13. We also excluded close homolog pairs which likely do not necessitate the structural relaxation provided by our approach. As an example, IPHoLD performed similarly well to other homology modeling and ligand docking techniques when modeling the beta2 from the closely related beta1 adrenergic receptor (Supplementary Table 1). The sequence identities between all other targets and templates in our benchmark ranged from 20% to 37% and from 20% to 42% over the full length and the ligand binding region, respectively (Supplementary Table 1).
Table 1. Ligands docked onto GPCRs in the structure prediction benchmark.
The pdb code for each ligand-bound GPCR structure (target and starting homolog template) is indicated under parentheses. M.W.: ligand molecular weight (Da).
| Ligand Synonym | Ligand | Target protein (PDB code) | Template protein (PDB code) | Ligand type | M.W. |
|---|---|---|---|---|---|
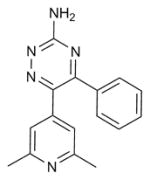
| 6-(2,6-dimethylpyridin-4-yl)-5-phenyl-1,2,4-triazin-3-amine |
|
A2aR (3UZA) | B1AR (2VT4) | Antagonist | 277.3 |
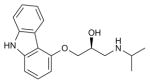
| (S)-Carazolol |
|
B2AR (2RH1) | DRD3 (3PBL) | Inverse agonist | 298.4 |
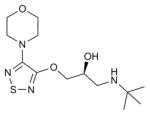
| Timolol |
|
B2AR (3D4S) | H1R (3RZE) | Inverse agonist | 316.4 |
| Cyanopindolol |
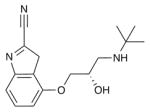
|
B1AR (2VT4) | H1R (3RZE) | Antagonist | 287.4 |
| Doxepin |
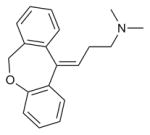
|
H1R (3RZE) | M2R (3UON) | Antagonist | 279.4 |
| 3-quinuclidinyl-benzilate |
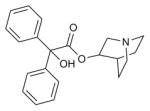
|
M2R (3UON) | H1R (3RZE) | Antagonist | 337.4 |
| Eticlopride |
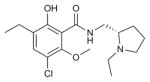
|
DRD3 (3PBL) | B2AR (2RH1) | Antagonist | 340.1 |
| JDTic |
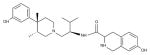
|
KOR (4DJH) | H1R (3RZE) | Antagonist | 465.6 |
| Maraviroc |
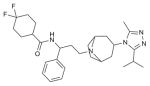
|
CCR5 (4MBS) | CXCR4 (3ODU) | Antagonist | 513.7 |
| Ergotamine |
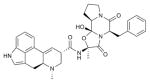
|
5HT1B (4IAR), 5HT2B (4IB4) | DRD3 (3PBL) | Agonist | 581.66 |
| IT1t |
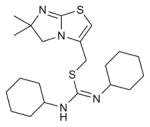
|
CXCR4 (3ODU) | CCR5 (4MBS) | Antagonist | 406.6 |
| ZM241385 |
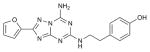
|
A2aR (3EML) | B1AR (2VT4) | Antagonist | 337.3 |
| ML056 |
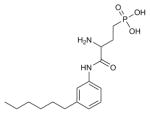
|
SPH1 (3V2W) | B1AR (2VT4) | Antagonist | 342.4 |
| C-24 |
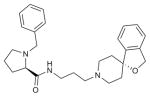
|
OPRL1 (4EA3) | DRD3 (3PBL) | Antagonist | 433.6 |
| CP-376395 |
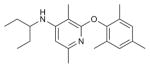
|
CRF1R (4K5Y) | GCGR (5EE7) | Antagonist | 326.5 |
| TAK-875 |
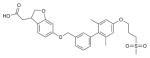
|
FFAR1 (4PHU) | PAR1 (3VW7) | Agonist | 524.62 |
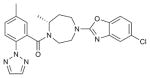
| SB-674042 |
|
OX1R (4ZJ8) | DRD3 (3PBL) | Antagonist | 450.92 |
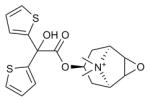
| Tiotropium |
|
M3R (4DAJ) | H1R (3RZE) | Inverse agonist | 392.5 |
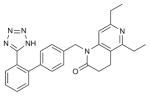
| ZD7155 |
|
AT1R (4YAY) | CCR5 (4MBS) | Antagonist | 438.5 |
| Naltrindole |
|
δ-OR (4N6H) | AT1R (4ZUD) | Antagonist | 414.5 |
We compared the results of our integrated approach to those obtained by two sequential approaches: the original Rosetta modeling and ligand docking protocol15,21,22,27 and the popular combination of receptor modeling by Modeller11 and ligand docking by Glide16 which were widely used by expert modelers in GPCR-DOCK predictions21–23. The simulations were performed with default parameters, as in a blind prediction without prior knowledge of the X-ray structures and the models were selected using the same objective physical criteria only (model population and energy). Importantly, since we aimed to test the method capabilities and not those of the human experts, the three approaches were tested objectively as if they were run automatically by computer servers.
Except for six structures, our technique generated and selected near-atomic accuracy receptor bound ligand conformations for all targets (heavy atom rmsd < 2.5 Å, average heavy atom rmsd = 2.1 Å over all targets, Fig. 2a, Supplementary Table 1). Conversely, only three ligand conformations were predicted accurately by the sequential approaches, leading to high average rmsd of 4.9 Å and 6.2 Å for Rosetta and Modeller/Glide, respectively (Fig. 2a, Supplementary Table 1). Reflecting the high accuracy predictions of IPHoLD, our models recapitulated on average 60.4% of all native atomic contacts between the ligand and the receptor compared to only 32.4% and 29.4% for Rosetta and Modeller/Glide, respectively (Fig. 2b, Supplementary Table 1). Despite these considerable improvements, we noticed a few remaining limitations. For example, the Marvin and Omega softwares could not generate CXCR4 antagonist ligand conformations close to that observed in the X-ray structure (heavy atom RMSD > 1.5 Å). Therefore, while IPHoLD predicted the CXCR4 ligand binding structure with high accuracy (interacting residues Cα RMSD = 1.2 Å, Supplementary Table 2), the ligand bound poses never reached the native conformation (ligand RMSD = 4 Å, Supplementary Table 1).
Figure 2. Accurate prediction of ligand conformations bound to GPCRs using IPHoLD.

Comparison between the integrated method IPHoLD and two traditional sequential homology modeling and ligand docking approaches: Rosetta “sequential” protocol and a combination of Modeller and Glide. a. Ligand heavy atom rmsd (Å) between the predicted and the experimentally determined ligand poses. b. Percent native atomic contacts between receptor residues and ligand recovered by the modeling approaches. Dotted lines: high accuracy ligand pose prediction is defined by Ligand heavy atom rmsd < 2.5 Å and percent native atomic contact recovered > 50%. IPHoLD outperforms Rosetta and Modeller/Glide: paired two-sided t-test P values < 0.0001 for both ligand RMSD and native atomic contact.
Accurate prediction of ligand-binding region structures
In principle, a higher rate of near-native ligand conformation predictions should partly result from a more accurate modeling of the receptor ligand binding site structure as intended in the implementation of our integrated IPHoLD approach. We systematically compared the receptor extracellular ligand binding structure of the models selected by the sequential Rosetta, Modeller/Glide and IPHoLD approaches. Whether the entire extracellular region (including the ECL2 loop) or just the receptor residues interacting with the ligand were considered, our models were substantially closer to the X-ray structure than those from the other techniques (p-values < 0.05, paired t-test for ligand binding region Ca RMSD and ligand interacting residues heavy atom RMSD. Supplementary Table 2). The structural discrepancy calculated over all ligand contacting residues from all targets was only 1.5 Å using the integrated approach, further demonstrating the high accuracy of our predictions. Improvements were particularly noticeable when the sequential approach performed poorly (RMSD > 2 Å) and when target and template were bound to ligands with distinct pharmacology leading to substantial structural differences (e.g. ergotamine agonist bound serotonin receptors modeled from the antagonist bound dopamine D3 receptor). Remarkably, as shown in Fig. 3 and Supplementary Table 1, the level of prediction accuracy did not depend on the size, chemical property, conformational complexity or location of the binding site of the ligands. Unlike the alternative approaches, IPHoLD was able to predict correctly the conformation of ligands buried between TMHs at location where these helices are tightly packed in the homolog structures (e.g. fatty acid binding GPR40 and class B GPCR CRF1, Fig. 3f). These two challenging modeling cases highlight the considerable ligand induced fit effects on receptor structures recapitulated by IPHoLD. These results strongly suggest that the method should be applicable to a large diversity of GPCR-ligand complexes.
Figure 3. Near-atomic accuracy prediction of GPCR-bound ligand poses.

a. Beta 2 adrenergic receptor bound to antagonist timolol (3D4S) (Ligand heavy atom RMSD = 1.6 Å; Binding site Ca RMSD = 1.7 Å). b. M2 muscarinic acetylcholine receptor bound to antagonist 3-quinuclidinyl-benzilate (3UON) (Ligand heavy atom RMSD = 1.0 Å; Binding site Ca RMSD = 1.3 Å). c. Histamine H1 receptor bound to doxepin (3RZE) (Ligand heavy atom RMSD = 1.1 Å; Binding site Ca RMSD = 0.9 Å). d. Serotonin receptor 5HT2B bound to partial agonist ergotamine (4IB4) (Ligand heavy atom RMSD = 1.9 Å; Binding site Ca RMSD = 1.1 Å). e. CCR5 Chemokine Receptor bound to antagonist maraviroc (4MBS) (Ligand heavy atom RMSD = 2.1 Å; Binding site Ca RMSD = 1.6 Å). f-g. Class B CRF1R bound to CP-376395 small-molecule antagonist. The ligand is buried in the transmembrane core of the receptor. f. IPHoLD prediction (Ligand heavy atom RMSD = 2.2 Å; Binding site Ca RMSD = 1.1 Å). g. Rosetta prediction (Ligand heavy atom RMSD = 9.0 Å; Binding site Ca RMSD = 1.5 Å). blue: X-ray structure; yellow: predicted structure.
Quantitative prediction of ligand binding selectivity
Since our ultimate goal is to apply the technique to design GPCRs with novel ligand binding properties, we next tested whether ligand binding selectivity could be accurately predicted from homology models only and generated by our integrated technique. Because the approach does not directly calculate free energy of binding or ligand free energies, we restricted our predictions to changes in binding affinity upon receptor amino-acid variations for the same ligand (Method). We selected a dataset of 39 experimentally measured GPCR-ligand interactions encompassing a large diversity of ligand structures, chemistry, affinity ranges and pharmacology (Supplementary Table 3). All receptor targets (most were structurally-uncharacterized) were modeled from homologs in distinct (i.e. inactive or active) functional states sharing as low as 28% sequence identity with their homologs in the extracellular binding site (Supplementary Table 4). The selected receptor targets were either native receptor or point mutants described in the literature (e.g. L126A and T134A for the serotonin 5HT1B receptor, S193G for the dopamine D2 receptor34–36). We approximated the changes in binding affinity between variants by the difference in receptor/ligand interaction energies calculated from the GPCR/ligand bound models for each variant. Remarkably, except for glemanserin binding to 5HT2B and 5HT2A, all differences in ligand binding affinity between GPCR variants were qualitatively correctly predicted (Fig. 4a, Supplementary Table 3). Additionally, with a correlation coefficient r of 0.76 and standard error of around 1.4 kcal/mol only, our calculated interaction energy differences correlated quantitatively with the experimentally determined binding energy differences. These results indicate that our modeled ligand-bound GPCR structures are accurate enough to predict significant changes in ligand binding interaction energies and corresponding affinity differences.
Figure 4. Prediction and design of ligand binding selectivity to structurally-uncharacterized GPCRs.

a. Correlation between predicted and experimentally measured ligand binding energy differences of a ligand for two receptors (blue: benchmark predictions with published experimental binding affinities; green: blind predictions). R: Pearson correlation coefficient (two-tailed P value < 0.0001). b. Schematic rationale for the structure-based design of the dopamine D2 receptor ligand binding site to reprogram the receptor’s ligand binding selectivity as follows: by simultaneously decreasing the binding to the high-affinity ligand spiperone, increasing the binding to the low-affinity ligand SCH23390 without affecting the affinity to the high-affinity ligand raclopride. c. Low-energy ligand poses bound to one conformation of the D2 receptor highlighting the positions of the designed specificity switch mutations (magenta: spiperone; cyan: raclopride; green: SCH23390). d. Comparison between blind predicted and experimentally-measured changes in binding energy of the 3 ligands for 6 designed D2 variants. Experimental values correspond to the mean of at least 3 independent experiments (see Methods, Supplementary Table 5). Each designed mutation has a significant effect on spiperone and SCH binding (all unpaired two-sided t-test P values < 0.05) but not on raclopride binding (all unpaired two-sided t-test P values > 0.1).
Designed GPCRs with novel ligand binding selectivity
If important ligand receptor interactions can be recapitulated using predicted ligand bound receptor conformations, the models should prove accurate enough to guide the design of receptors with novel ligand binding selectivity profiles. To test this hypothesis, we selected the structurally-uncharacterized dopamine D2 receptor and identified 2 high (nanomolar) affinity (spiperone, raclopride) and 1 low (micromolar) affinity (SCH) ligands. As a proof of concept, we designed the D2 ligand binding site to simultaneously increase the affinity to SCH, while decreasing the affinity to spiperone without perturbing the affinity to raclopride (Fig. 4b, c).
Since these ligands are of different size and bind the receptor with distinct affinities, we modeled the receptor bound to each ligand separately. Because the design of ligand-receptor interactions is very sensitive to the atomic details of the models, we performed two rounds of ligand docking (cycle 2) where the second round was biased toward sampling dominant ligand poses identified in the first round (Method). This process enabled a finer-grained sampling of ligand poses and a refinement of the structural models after the first round of experimental validation (see below). While the high affinity ligands clustered mainly in one large family of low-energy ligand-bound receptor conformations, the SCH-bound receptor conformations were more structurally diverse, consistent with the low binding affinity of the ligand. Consequently, we performed the design calculations on one spiperone and one raclopride bound D2 structure, but with 20 representative SCH bound D2 conformations (Method, Supplementary Fig. 4).
Our first round of design calculations identified two sites on TMH2 (V912.61) and TMH3 (V1113.29) where selectivity switching mutations could be designed. Since the large diversity of SCH bound D2 receptor structures may reflect an intrinsic conformational heterogeneity, we selected mutations that displayed consensus effects on most of the models (Supplementary Fig. 4). Consistent with the predictions, all designed point mutations decreased the binding affinity to spiperone by up to 23-fold (Fig. 4d, Supplementary Fig. 5, Supplementary Table 5). The largest effect on SCH was observed for V91W, which increased the binding affinity to the ligand by close to 10-fold (Fig. 4d, Supplementary Fig. 5, Supplementary Table 5). Altogether, with a 130-fold change in ligand binding selectivity, the designed mutation V91W was the largest designed specificity switch in the first round of calculations. While the relative effect of the V111L mutation compared to that of V111I on the binding affinity for SCH was predicted correctly, both mutations decreased the affinity for SCH compared to WT in disagreement with the predictions (Fig. 4d, Supplementary Table 5).
This discrepancy prompted us to refine the selection of SCH-bound D2 conformations and identify ligand-bound models with slightly different ligand conformation that best fit the first round of measured affinities (Method, Supplementary Fig. 6). From this new set of experimentally-guided models, we performed a second round of design calculations. We selected two additional sites on TMH3 (C1183.36) and TMH7 (T4127.39) where mutations were predicted to increase the binding affinity to SCH while not affecting that of spiperone. Unexpectedly, the selected C118I mutation abolished significant binding to any ligand antagonists. The D2 variant displayed partial degradation suggesting that the mutation may destabilize the receptor, leading to partial unfolding (Supplementary Fig. 7a). The T412L designed specificity switch displayed 14-fold increase and only 2-fold increase in affinity for SCH and spiperone, respectively, in agreement with the predictions (Fig. 4d, Supplementary Table 5). Remarkably, while the designed mutations had significant effects on spiperone and SCH binding affinity, we observed no significant effects on the affinity for raclopride, consistent with our predictions (Fig. 4d, Supplementary Fig. 5, Supplementary Table 5). We then combined the V91W and T412L mutations to create a double mutant variant predicted to further alter the ligand binding selectivity. In good agreement with the predictions, the double mutant displayed a considerable 1410-fold change in ligand binding selectivity and bound SCH with slightly higher affinity than spiperone. By contrast, the affinity for raclopride remained almost identical to that of WT as intended by the calculations. Lastly, our designed D2 variants remained functional, bound dopamine and displayed Gi signaling responses to dopamine binding very similar to D2 WT in cell-based assays (Supplementary Fig. 7b,c). These results indicate that our designed mutations had very selective effects on the binding of antagonists.
With a 76% success rate on 21 designed receptor-ligand pairs, these results indicate that our integrated receptor homology modeling/ligand docking approach can provide structural templates accurate enough to design mutations altering ligand binding selectivity. The success rate even increased to 86% after the second round of design calculations guided by the sparse ligand binding data obtained during the initial experimental validation. Importantly, the ligands selected in this proof of concept study spanned a large range of binding affinity, chemistry, structures and receptor bound conformations. The ligands also selected distinct receptor conformations as assessed by the substantial difference in ligand binding site structures between the different ligand-bound receptors (Rmsd ranging between 1 Å to 1.3 Å, Supplementary Table 6). Overall, these results highlight the ability of IPHoLD to recapitulate non-trivial structure-energy relationships in receptor-ligand interactions. Importantly, most mutations selected in the first cycle of design calculations would have been identified using models from our first round of unbiased ligand docking simulations (Method, Supplementary Fig. 8). However, the second round of ligand docking generated a finer-grained ligand conformational ensemble including two models that matched best the experimental results. Therefore, our results indicate that ligand docking resampling approaches to enrich dominant ligand poses is an effective strategy for experimentally refining the computational design of ligand-protein interactions.
Limitations of the approach
Since homolog structural templates with as low as 25% sequence identity were sufficient to generate accurate models, we estimate that around 30% of all GPCRs (close to 250 receptors) and receptors from many other families could now be targeted using IPHoLD. Modeling receptors from more distant homologs is more challenging because it usually requires also de novo reconstructing part of the TMH core regions. Despite de novo remodeling a fraction of TMH6, we were able to correctly predict the class B GPCR CRF1-bound ligand conformation starting from a template sharing only 20% sequence identity (Fig. 3f). However, whether the near-atomic level of accuracy obtained in this study can be consistently achieved starting from distant homologs (< 20% sequence identity) that require the de novo rebuilding of multiple TMHs remains unclear.
IPHoLD requires additional software to generate ligand conformers. Our results suggest that near-native ligand poses can often be selected using the ligand rotamers generated from the software Marvin. However, in a few instances, no ligand poses closely matching the conformation observed in GPCR X-ray structures were selected by Marvin. These observations suggest that generating ensemble of ligand conformations using multiple softwares based on distinct energy functions may be necessary in the future.
Our approach relies also on an implicit solvent model which does not represent solvent molecules explicitly13,37. As such, IPHoLD failed at consistently generating native-like structures for ligand-receptor interactions that are largely mediated by water molecules such as in the ZM241385-bound A2A receptor. Due to the associated computational cost and complex physical properties of protein bound water molecules, the explicit modeling of solvent remains a challenge even in simple ligand docking simulations on experimentally determined protein structures38. Combining explicit solvent treatment with homology modeling of protein receptor and ligand docking techniques will likely require major advances in both conformational sampling algorithms and computer hardware.
Lastly, several families of GPCRs bind hormone peptides or even proteins (e.g. chemokines) but modeling these complexes remains beyond the scope of the present study and will necessitate further computational developments.
Discussion
Accurately predicting protein-ligand interactions in absence of experimental protein structures remains a major challenge especially for conformationally-flexible membrane receptors such as GPCRs which bind and respond to several ligands. To address this problem, we developed and validated IPHoLD, an Integrated Protein Homology modeling, Ligand Docking and protein design approach to accurately model and design ligand-GPCR complexes even without experimental structure on the molecules (Fig. 1). By simultaneously reconstructing ligand binding sites and docking ligands, we considerably improved ligand-bound pose predictions as compared to traditional modeling approaches, reaching near-atomic accuracy in most cases (Fig. 2, 3). IPHoLD enabled quantitative prediction of the binding selectivity of a wide diversity of ligands to multiple structurally-uncharacterized GPCRs (Fig. 4a). Lastly, by combining IPHoLD with experimental validations, we redesigned the ligand binding site of the structurally-uncharacterized dopamine D2 receptor and rationally engineered receptor variants with novel binding selectivity profiles for 3 distinct ligands (Fig. 4b–d).
Our results indicate that modeling ligand-bound receptor conformations by mimicking induced fit and conformational selection mechanisms of ligand binding considerably improve the structural and energetic accuracy of receptor-ligand interactions in homology modeling. By contrast, traditional sequential approaches which first model ligand-free GPCR structures and then dock ligands often suffer from inaccurate de novo modeled loop conformations and cannot fully capture ligand induced fit effects. These sequential techniques rarely generate optimal ligand bound structures on their own (Fig. 2, Supplementary Fig. 1) and often required experimental information on ligand-receptor interactions and human expert guidance to select native-like GPCR-ligand complexes in blind contests21–23. Sequential techniques will likely remain useful for modeling receptors from well-characterized classes or with canonical ligand binding properties. However, there is growing interest in applying rational structure modeling techniques to poorly characterized receptors and for uncovering novel classes of ligand compounds. To our knowledge, efforts in this direction have remained very sparse20. In our blind and benchmarking predictions, only objective physical criteria (based on model energy and convergence of the simulations without experimental information) were used to select models. Additionally, the high level of conformational relaxation of ligand-bound GPCR complexes enabled to substantially improve the modeling of activating ligand agonist bound binding site structures even when starting from inactivating ligand antagonist bound GPCR homolog structures. More generally, our successful predictions encompassed ligands with a wide diversity of chemical, pharmacological, structural and signaling activating properties. Unlike alternative techniques, we were also able to correctly model ligands bound in the non canonical binding sites of the class B GPCR corticotropin-releasing factor receptor 1 and of the free fatty acid receptor FFAR1 (Table 1, Fig. 4, Supplementary Table 3, 4). Therefore, our technique is general and should be applicable even to classes of poorly characterized receptors (e.g. orphan) with no available experimental structural information. In principle, the only information necessary to run IPHoLD is a starting protein structural template with sufficient homology to the target receptor (i.e. at least 25% sequence identity) and the chemical structure of a binding ligand.
A wide range of synthetic, cell biology applications and cell-based therapies would benefit from the ability to reprogram the ligand binding selectivity and signaling responses of membrane receptors that are often promiscuous and difficult to study or utilize in their native forms. While empirical approaches have provided a few examples of engineered receptors31,32, these techniques lack the rational physical basis to predictably reprogram ligand binding affinities and selectivity. The ability to considerably modulate the binding affinities of two ligands without perturbing the binding to other ligands in our proof-of-concept study demonstrates the power of rational structure-based approaches. Our results also highlight the need to explicitly model ligand-bound receptor conformational diversity, which was critical to recapitulate precise, selective ligand-receptor contacts and guide the design of novel ligand binding specificity determinants.
Our method should prove particularly useful in ligand/drug discovery approaches and in the design of receptors with reprogrammed ligand binding and biosensing responses for the numerous classes of membrane protein targets that remain poorly characterized and difficult to crystallize.
Online Methods
Selection of homolog structure templates
For each target, homolog template structures were searched using a sequence-structure alignment approach by HHpred39. Specifically, HHpred was run with the local alignment option and three iterations of BLAST followed by global MAC realignment. The top-ranked homolog templates were selected.
Integrated Protein Homology Modeling-Ligand Docking (IPHoLD) protocol
IPHoLD addresses the combinatorial explosion problem of sampling simultaneously ligand positions and loop conformations in 4 consecutive steps gathered in 2 cycles which provide efficiency while not substantially sacrificing the sampling of receptor and ligand conformational diversity. IPHoLD was developed by integrating and adapting the RosettaMembrane homology modeling and Rosetta Ligand protocols as follows.
Cycle 1: modeling an ensemble of low-energy ligand-bound receptor conformations
In cycle 1, alignments from HHpred were used to thread the target sequence onto the backbone of the selected template. The alignments were systematically checked and eventually manually corrected to match those of the GPCR database (GPCRdb40,41) in the extracellular regions. This step became important after we identified that HHpred had not correctly aligned the conserved prolines in TMH4 and extracellular loop cysteines of the target serotonin 5HT1B receptor to that of the template Dopamine D3 receptor (3PBL) in the blind GPCR-DOCK 2013 prediction contest23. Regions of the target receptor displaying alignment gaps with the homolog template were selected for de novo rebuilding. These regions included extracellular loops for all targets (except for the close homolog B2AR, B1AR pair) as well as one transmembrane helix for a few targets.
Ligand conformers were generated as described below in the ligand conformer generation section.
Step 1. Coarse-grained ligand docking on starting receptor templates
In each of 10000 independent simulations, coarse-grained ligand conformers are first docked onto a coarse-grained model of the starting receptor template guided by anchor points initially placing the center of mass of the ligand at distinct positions spanning the entire ligand binding site. At this stage, extracellular loops that will later be rebuilt in presence of the ligand (in step 2) are removed from the receptor template structures. In our benchmark, the regions reconstructed de novo contributed at most 40% of the ligand-receptor contacts even for receptor targets modeled from relatively distant homologs. Therefore, a majority of the binding site is composed of residues already built in the homolog template structure, which justifies the first step of ligand pose selection prior to loop rebuilding. Each ligand conformer orientation is randomized a thousand times and the positions displaying perfect surface complementarity with the receptor coarse-grained template structure are gathered in a set of “optimal” ligand-bound conformers. From 10 to 100 thousands initial ligand conformers and positions, only a hundred to a few hundreds are selected that display perfect shape complementarity and enough structural diversity (i.e. redundant conformation and position are discarded).
Step 2. Coarse-grained loop rebuilding of selected ligand-bound receptor templates
In each of 10000 independent simulations, one “optimal” ligand pose is randomly selected and the ligand-bound receptor template is subjected to coarse-grained de novo loop (and TMH in a few targets) rebuilding. This step constructs and selects loop conformations making favorable contacts with low-energy docked ligands. The models are scored using the coarse-grained energy function of RosettaMembrane42 for the receptor and a simplified potential for the ligand approximating Van der Waals contacts and penalizing atom overlaps.
Step 3. Ligand-bound loop rebuilt model all-atom relaxation and selection
Each of the 10000 loop rebuilt and ligand bound coarse-grained models is then fully relaxed at all-atom to allow the non-rebuilt parts of the template to accommodate the target sequence, relax in the presence of the bound ligand with an all-atom representation and generate a large diversity of low-energy receptor conformations compatible with multiple distinct ligand conformers. At this stage, the ligand is not redocked but kept in its starting conformer. Typically, five repeats of structure relaxation involving several cycles of side-chain repacking and structure minimization with increasing weights for the repulsive term of the Lennard-jones potential (starting from a heavily damped and reaching the regular high-resolution weight) are performed and the lowest energy structure is selected. At this stage, the models are scored using the all-atom energy function of RosettaMembrane37.
The 10% lowest energy models are clustered by receptor structural similarity and the centers of the top 20 most populated clusters are used as starting receptor conformations for cycle2.
Cycle 2: Fine-grained ligand docking on the receptor conformational ensemble
In cycle 2, ligands are re-docked onto each ligand-free receptor conformation selected from cycle 1 to ensure that, unlike in cycle 1, ligand conformers can fully sample the receptor conformational ensemble and select the optimal receptor conformation.
Step 4. Ligand docking refinement
In this cycle, coarse-grained followed by full-atom ligand docking is performed as described previously15,27. In this protocol, the receptor structure is kept mostly rigid with the exception of side-chain repacking and constrained minimization of the residues in direct contact with the ligand. 3000 ligand-docked models are generated for each input receptor model and the final structures are scored using the Rosetta ligand all-atom energy function15,27.
From the 10% lowest total score models, the 1,000 models displaying the lowest receptor-ligand interface (i.e. interaction) score (Rosetta function term interface_delta) are selected and clustered. After aligning the protein coordinates of the selected models, all the ligands are clustered into families of ligand poses displaying similar location and conformation using a two-step clustering method described below in the ligand conformer clustering section. In the structure prediction benchmark, the top 10 clusters by size were further ranked using specific energy terms defined below. Within each of the top 5 reranked clusters, the optimal ligand conformer according to the specific energy term is selected as a final ligand model. Following the GPCR-DOCK blind prediction selection criteria, among the 5 final models, the ligand bound model with the lowest ligand heavy atom RMSD to the native X-ray structure is reported in our study.
The energy term selection criteria are defined as follows: for small size ligands (molecular weight less than 350 Dalton), the hydrogen bond term (Rosetta function term if_hbond_sc) is used. For larger ligands (molecular weight higher than 350 Dalton), because Van der Waals interactions and surface complementarity often play a major contribution in receptor-ligand interactions, the entire interface energy (Rosetta function term interface_delta) is used.
Selection of the effective number of independent IPHoLD simulations
IPHoLD modeling simulations are performed using multiple independent trajectories to sample relevant combinations of ligand and receptor conformations. As explained below, IPHoLD was designed to considerably reduce the conformational space to be sampled. The number of trajectories was selected to provide an effective tradeoff between accuracy and speed of the calculations.
The first most important step leading to a considerable reduction in the ligand conformational space to be sampled is the coarse-grained ligand docking step. Typically, this selection step reduced the total possible number of ligand positions from 10 thousands or 100 thousands to one hundred or a few hundreds ligand poses. In principle, 10000 simulations enable each “optimal” ligand pose to be selected multiple times and to generate multiple loop rebuilt receptor structures. As shown in Supplementary Fig. 2, generating up to 100000 loop rebuilt structures from the same set of ligand poses did not improve the sampling of receptor and ligand conformational diversity and the selection of near-native ligand-bound extracellular loop and binding site conformations.
During the development of our approach, we also assessed whether an additional cycle 1 of coarse-grained ligand docking and loop rebuilding starting from loop rebuilt receptor structures would improve the final relaxed ligand-bound models. We found no significantly more accurate ligand-bound models after a second cycle of coarse-grained ligand and loop modeling.
The ligand docking refinement step (cycle 2, step 4) is performed using the center models of the 20 largest clusters of low-energy ligand-bound receptor relaxed structures from step 3. The number of selected models was optimized during the blind prediction of ergotamine-bound 5HT1B and 5HT2B structures and on the three benchmark test cases described in Supplementary Fig. 2. We found that 20 starting receptor models enabled enough receptor conformational diversity and performing ligand docking on up to 100 starting models did not enrich for near-native ligand poses. 3000 ligand docking trajectories for each starting receptor model is sufficient to guarantee convergence and is similar to the number of trajectories reported for the same simulations performed by RosettaLigand starting from a single receptor structure15.
To test the effects of cycle 1 simulation and model number on the structure prediction accuracy (Supplementary Fig. 2), we selected the ligand-bound GPCR targets Adenosine receptor A2A (3UZA), β1 adrenergic receptor (2VT4), serotonin receptor 5HT1B (4IAR) and serotonin receptor 5HT2B (4IB4) because they displayed different ligand size and number of ligand conformers and all involved substantial loop reconstruction. Specifically, 4, 200 and 84 ligand conformations were generated by MarvinSketch (http://www.chemaxon.com) for 4UZA, 2VT4 and 5HT2B, respectively. Each ligand conformer is placed at 3 distinct locations in the binding site and its position randomized a 1000 times. Therefore, the total number of initial ligand positions is 12000, 600000 and 252000, for 4UZA, 2VT4 and 5HT2B, respectively. 51, 35, 36 residues were rebuilt on the extracellular regions of 4UZA, 2VT4 and 5HT2B, respectively.
Supplementary Fig. 2 compares the results of IPHoLD simulations performed with 10000 versus 50000 and 100000 independent trajectories for steps 1–3. The results are compared using the following 3 metrics:
The ligand binding site RMSD to native of the selected receptor models in step 3. This metric reflects the conformational diversity and accuracy of the ligand binding structure resulting from the loop modeling and ligand-induced fit effects. Undersampling would result in narrower binding site rmsd distribution and less accurate receptor models (i.e. higher rmsd to native).
The diversity of bound ligand conformation in the selected receptor models in step 3. The diversity is calculated from the rmsd distribution of the model ligand conformations after alignment to the native ligand conformation. This metric reflects the diversity and accuracy of ligand conformations selected to rebuild loops and refine receptor structure for induced fit effects during steps 1–3. Undersampling would result in narrower ligand rmsd distribution (i.e. less diverse ligand conformer selected) and less accurate ligand poses (i.e. higher rmsd to native).
The accuracy of the selected ligand-bound pose at the end of the simulation in step 4. This metric reflects the diversity and accuracy of the final docked ligand conformations after superposition of the model and native receptor structures. Undersampling the receptor conformations and ligand conformations would result in narrower rmsd distribution and less accurate ligand poses (i.e. higher rmsd to native).
As shown in Supplementary Fig. 2, the 3 above described rmsd metrics remained very similar whether 10K, 50K or 100K independent simulations were performed. These results suggest that 10K simulations do not lead to substantial undersampling of ligand and receptor conformations. 10K simulations are sufficient to generate enough receptor and ligand conformational diversity for selecting near-native ligand poses.
Ligand conformer clustering
A two-step clustering method was used to identify the dominant ligand binding modes. The program first clusters ligands by their geometric centers into families with similar docking sites and then clusters the ligand poses belonging to the same docking site into families of similar conformations. For both clustering steps, the average linkage hierarchy-clustering method was used. Instead of using a pre-defined distance cut-off, the optimal cut-offs were identified using the elbow strategy43 for each ligand within a 5–8 Å range for ligand docking site and a 2–3 Å range for ligand conformation.
Ligand conformer generation
Ligand conformers were generated prior to docking using MarvinSketch (http://www.chemaxon.com) or Openeye program (Omega)44. In the blind prediction of the ergotamine-bound serotonin receptors, Openeye’s default parameters were used for ligand conformer generation. In all remaining stages of the study, ligand protonation states were first calculated by the MarvinSketch program at pH=7.4 before generating ligand conformers. To allow ligands with different sizes to sample the same range of internal energies, conformers for each ligand were generated until they covered an energy window equal to 10 kcal/mol.
Sequential Rosetta homology modeling and ligand docking
The sequential protocol was run as described previously21,22,27 except for a few parameters (i.e. number of models, selection of ligand poses) which were adapted to match those of the integrated IPHoLD approach, allowing a direct comparison of the 2 techniques. 10,000 GPCR models were generated and the lowest 10% by total energy were clustered by receptor structural similarity. The centers of the top 20 most populated clusters were used as starting receptor conformations for ligand docking. 3000 ligand bound models were generated for each starting structures using the same protocol and starting ligand conformers than cycle2 in the integrated IPHoLD approach. The lowest 10% ligand-bound GPCR models by total energy were selected and re-ranked by ligand-receptor interaction (i.e. interface) energy. The lowest 1000 ligand poses by interface energy were clustered as described above prior to model selection.
Sequential approach using Modeller and Glide
The online server of Modeller11 (http://toolkit.tuebingen.mpg.de/modeller) was used to generate homology models for each target protein. Modeller was given the same target-template sequence alignment than Rosetta and IPHoLD techniques. Modeller models were retrieved and used for ligand docking using the software Glide16 in Maestro (version 9.6.012, Schrodinger). Before generating ligand-docked complex models, both receptor structure and ligand were prepared using ‘Protein preparation’ and ‘ligand Preparation’ protocols in Maestro. Standard precision Glide ligand docking protocol was used as described in the manual to output up to 5 selected models by the program. The most accurate model (i.e. lowest ligand rmsd to the target X-ray structure) was used for comparison in the benchmark.
Structural accuracy of receptor models
The accuracy of the models was assessed by calculating Ca RMSD and all-atom RMSD over two sets of protein residues: 1. A large “ligand binding region” comprising the extracellular half of all TMHs and the extracellular loop 2 between the conserved Cys residue (Cys45.50, 45 indicates between TM helix 4 and 5) and the N-terminal residue of TM helix 5; For CRF1R, the ligand binding region is defined by the intracellular half of transmembrane helices 3, 5 and 6 that include the binding pocket for the ligand. 2. A smaller “interacting residues” region comprising only the residues interacting with the ligands (i.e. with at least one heavy atom within 4 Å of a ligand atom).
Sequence identity between templates and targets was calculated over the full length receptor and the ligand binding region.
Ligand binding site design
Structure models of different ligand-bound dopamine receptor 2 (Spiperone, Raclopride, SCH23390) were generated and selected as described above, except that the final ligand docking refinement step was performed twice as follows.
A first cycle of ligand docking was performed as described above in the blind prediction and benchmark without any bias. Analysis of the largest families of low-energy ligand poses indicated that the interaction between the amine moiety of each of the 3 ligands with Asp3.32 114 of D2 (which is a conserved interaction in all class A GPCRs45) was the most frequent polar contact observed in the blindly selected models (Supplementary Fig. 8 A). Since several selected models did not display that contact and because the design of protein-ligand interactions is very sensitive to the atomic details of the models, we decided to enrich the population of models for those displaying the consensus polar contact.
We therefore performed a second ligand docking refinement with a distance constraint between the amine moiety of the ligand and Asp 114. While a large fraction of the low-energy selected poses remained similar to those from the unbiased simulations (Supplementary Fig. 8 B), all the ligand-bound models now displayed the constrained polar interaction. The simulations with the high-affinity spiperone and raclopride ligands led to one dominant family of models (by energy and size) and the representative center model of that family was selected for the design calculations. Concerning the weak-affinity ligand SCH, since no dominant family of poses was identified (i.e. multiple ligand conformations clustered in families of similar sizes and energies), we performed the design calculations using 20 center models sampling distinct ligand conformations accommodating the consensus contact (Supplementary Fig. 4 A).
Mutations were designed at residues belonging to the ligand binding pocket of the receptor using a set of RosettaScripts designed and adapted from previous studies46. The example of XML protocols used in this study is described at the end of the method section. 200 structures were generated for each mutation and the model with the lowest interface energy (i.e. ligand-receptor interaction energy) was selected. For the SCH23390-D2DR complex occupying multiple ligand-bound conformations, mutations were first selected that display consensus effects on a majority of the representative center models of the 20 selected clusters (Supplementary Fig. 4).
To improve the quality of the SCH23390-D2DR models after the first round of experimental validation, all models (i.e. around 500) from the 20 clusters were threaded and repacked, energy minimized with the designed mutations to identify those consistent with the experimental results of the first set of designed mutations. This automated energy-based selection process enabled the identification of 2 ligand bound models that fit best the experimental results for performing the second round of design calculations.
Classification of the designed mutational effects calculated from the D2-ligand models generated by the unbiased docking simulations (Supplementary Fig. 8C)
The effects of the designed mutations were calculated using the selected ligand-bound D2 models. The absolute mean effect of the mutations for each ligand was calculated after removing the outliers (i.e. models generating very large effects) using the ROUT method implemented in the software prism (Graphpad) with Q (chance to remove a false outlier) of only 5%. Half of the absolute mean (0.5*|mean|) was defined as the threshold for a significant mutational effect. Three categories of effects on ligand binding affinity were then derived from the calculated ligand-receptor interaction energy difference from WT, ΔE: “increase” for ΔE ≤ −0.5*|mean|; “decrease” for ΔE ≥ 0.5*|mean|; minor effect or “negligible” for 0.5*|mean| > ΔE > −0.5*|mean|. For each ligand and each D2 receptor mutant variant, the number of models within each category of effects is reported in Supplementary Fig. 8C.
Benchmark target selection
For the structure prediction benchmark, we selected a representative sample of 23 GPCR pairs with sequence identity between 20% and 50% and bound to a large diversity of ligands. X-ray structures of target and template ligand-bound GPCRs were extracted from the Protein Data Bank. We first performed ligand docking control simulations using the X-ray structure of our selected targets and an ensemble of ligand conformations generated using the software Marvin to ensure that the ligand conformations were accurate and diverse enough for identifying near-native ligand bound conformations (i.e. within 2 Å heavy atom RMSD of the native ligand conformation). If not, the targets were removed from the dataset. These ligand docking control simulations were performed using the RosettaLigand protocol15 using the same input parameters than the ligand docking refinement protocol (cycle 2) of IPHoLD.
The selected GPCR targets were: Adenosine receptor A2A (3EML, 3UZA), β1 adrenergic receptor (2VT4), β2 adrenergic receptor (2RH1, 3D4S), C-X-C chemokine receptor type 4 (3ODU), C-C chemokine receptor type 5 (4MBS), Dopamine receptor type 3 (3PBL), Serotonin receptor 1B (4IAR), Serotonin receptor 2B (4IB4), Histamine receptor (3RZE), M2 Muscarinic acetylcholine receptor (3UON), Sphingosine-1-phosphate receptor 1 (3V2W), Opiate receptor-like 1 (4EA3), κ-opioid receptor (4DJH), δ-opioid receptor (4N6H), Angiotensin type I receptor (4YAY), M3 Muscarinic acetylcholine receptor (4DAJ), OX1 orexin receptor (4ZJ8), GPR40 free fatty-acid receptor (FFAR1) (4PHU), Class B GPCR corticotropin-releasing factor receptor 1 (4K5Y). All the targets were modeled as described above except for Class B GPCR CRF1 and Class A GPCR GPR40. Both receptors bind ligands at binding sites located outside of the common extracellular ligand binding pocket. Therefore, we assigned starting ligand anchor locations in the initial coarse-grained ligand docking step to the core of the TMH regions at the native binding sites of the ligand. These modifications were also applied to the classical Rosetta and the Modeller/Glide protocols.
Ligand heavy atom RMSD and native atomic contact recovery (ligand-protein contact defined by ligand-protein heavy atom distance within 4Å) were used to compare the model to X-ray structures.
For the ligand binding energy prediction benchmark, ligands with different affinity to receptor subtypes in the serotonin and dopamine receptor families were selected. A detailed list of the ligands and their affinity can be found in the supplementary table 3. The dopamine and serotonin-bound dopamine D2 receptor models were generated starting from the B2AR active state structure. The binding affinities to different ligands were extracted from published binding database34–36.
IPhoLD performance
A single trajectory for the first loop modeling-ligand docking cycle (i.e. step 2 in Fig. 1) takes around 10 minutes on a standard CPU (e.g. Intel Xeon E5-2640v3, 2.6 GHz). A single trajectory for the second ligand docking and refinement cycle (i.e. step 3 in Fig. 1) takes around 3 minutes on the same hardware. Since a typical job involves 10000 cycle 1 and 60000 cycle 2 trajectories, the total computational cost for modeling a ligand-bound GPCR is: 1667 + 3000 = 4667 CPU hours.
Statistical Analysis
The results from different methods were statistically compared. All normality tests, t test and correlation calculations were carried out using GraphPad Prism 3.0 (GraphPad Software, San Diego).
Except for the comparison of the Cα or all atom RMSD of residues interacting with ligands (Supplementary Table 2), all data sets passed the D’Agostino & Pearson omnibus normality test (P value > 0.05) and the results from different methods were analyzed using paired two-sided t-tests. To compare the RMSD of residues interacting with ligands, a Wilcoxon matched-pairs signed rank test was used instead.
Figure 2 (Supplementary Table 1)
In comparing ligand heavy atom RMSD and native atomic contact recovery between IPHoLD and the other two methods, IPHoLD significantly improves the results (in all case paired t-test P value < 0.001). Specifically:
Ligand rmsd (Fig. 2A):
IPHoLD outperforms Rosetta: paired two-sided t-test P value < 0.0001
IPHoLD outperforms Modeller/Glide: paired two-sided t-test P value < 0.0001
Native Contact recovery (Fig. 2B):
IPHoLD outperforms Rosetta: paired two-sided t-test P value < 0.0001
IPHoLD outperforms Modeller/Glide: paired two-sided t-test P value < 0.0001
Supplementary Table 2
Ligand binding region (Ca RMSD): IPHoLD outperforms Rosetta and Modeller/Glide: paired two-sided t-test P values = 0.0002 and 0.0491, respectively.
Ligand interacting residues (Ca RMSD): IPHoLD outperforms Rosetta and Modeller/Glide: two-sided Wilcoxon matched-pairs signed rank test P values = 0.0002 and 0.0317 (on 82% of the dataset; P value = 0.1860 on the entire dataset), respectively.
Ligand interacting residues (all atom RMSD): IPHoLD outperforms Rosetta and Modeller/Glide: two-sided Wilcoxon matched-pairs signed rank test P values = 0.0004 and 0.0079, respectively.
Figure 4A
Pearson correlation coefficient (r) between binding energy prediction and experimental results was statistically calculated (two-tailed P value < 0.0001). Both datasets pass D’Agostino & Pearson omnibus normality test (P value >0.1).
Figure 4D and Supplementary Figure 4
Nonlinear regression to calculate Kd or Ki. See Supplementary method section “Ligand saturation assay” and “Ligand competition assay”
Supplementary Table 5
Spiperone: each designed mutation has a significant effect on spiperone binding: all unpaired two-sided t-test P values < 0.05.
SCH: each designed mutation has a significant effect on SCH binding: all unpaired two-sided t-test P values < 0.03.
Raclopride: no designed mutation has a significant effect on raclopride binding: all unpaired two-sided t-test P values > 0.1.
Mutagenesis and Cell Transfection
The cDNA clone for human dopamine D2DR (long isoform) receptor was a generous gift from the Wensel lab. D2DR mutants were generated using the QuikChange mutagenesis kit (Stratagene) and confirmed by DNA sequencing (Eurofins Genomics). All mutants were generated in the human D2DR tagged in the N-terminal with the HA epitope. HEK293T cells were transfected with D2DR cDNA using Genejuice (Invitrogen) as described by the manufacturer. Cells were maintained in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum supplemented with 1% Glutamax. Cells were grown in a 37 °C humidified environment with 5% CO2.
Cell membrane sample preparation
Membranes were prepared from transiently transfected HEK293T cells as described previously34,47. After 48hrs transfection, cells were scrapped and centrifuged at 300 × g for 15 min at 4 °C. The pellets were resuspended in 5 ml of hypotonic buffer (5 mM Tris H-Cl, 10 mM EDTA, pH 6.8) supplemented with protease inhibitors. The cells were lysed through a 25 gauge needle. The homogenates were centrifuged at 2000 × g for 15 min at 4 °C. The supernatant was further centrifuged at 200,000 × g for 40min at 4 °C. The final membrane pellets were resuspended by homogenization in 1 ml of binding buffer (20 mM Tris, 150 mM NaCl, 1mM MgCl2, 10mM EDTA, pH 7.4).
Ligand saturation binding assay
Ligand saturation binding assays were carried out as previously described34,47. 300ug/ml membrane suspensions (measured by Bradford assay) were incubated in binding buffer (20 mM Tris, 150 mM NaCl, 1mM MgCl2, 10mM EDTA, pH 7.4) and titrated with different concentrations of labeled antagonist ([3H]Spiperone or [3H]-Raclopride; PerkinElmer, purity > 97%). Samples were incubated for 60 min on ice (120min for [3H] Raclopride) prior to filtration through Whatman GF/C filters using filter holder manifold (Millipore) and washed three times using ice-cold TBS buffer. The filters were counted using a Beckman LS1701 scintillation counter after 12hrs. Specific antagonist binding was defined as the total binding subtracted by that measured for same amount of cell membrane samples from pcDNA transfected HEK293T cells. Saturation binding data were analyzed by fitting to a one-site saturation binding curve using GraphPad Prism 3.0 (GraphPad Software). The function describing the one-site saturation binding curve: Specific binding =Bmax*[X]/([X]+Kd) ([X] is the concentration of labeled ligands)
Ligand competition assay
300ug/ml membrane suspensions were incubated with increasing concentrations of antagonist SCH23390 (SCH23390 Hydrochloride: Calbiochem, purity 100.00%) in the presence of saturating labeled antagonist. Samples were incubated for 3hrs on ice prior to filtration through Whatman GF/C filters and washed three times using ice-cold TBS buffer using filter holder manifold (Millipore). Competition assay data were analyzed by fitting to a one-site competition binding curve using GraphPad Prism 3.0 (GraphPad Software). The function describing the one-site competition-binding curve: logIC50 = log(10^logKi*(1+[labeled-antagonist]/Kd(labeled-antagonist))
Cellular signaling of the designed D2 variants
HEK-293 cells stably expressing a transient receptor potential cation channel (TRPC4B) were generously provided by Dr. Michael Zhu (University of Texas Houston Health and Science). TRPC4B channels are a family of ion channels that are activated specifically by Gi/o proteins, and therefore constitute an appropriate reporter system of the functionality of D2 receptors. These cells were cultured in DMEM supplemented with 10% FBS and 1% G418 (selection antibiotic) and designed for use with FLIPR Membrane Potential (FMP) assay kits (Molecular Devices). The fluorescent signal from the FMP dye is modulated by shifts in cellular membrane potential upon ion channel activation. Therefore, changes in fluorescence are directly linked to Gi activation by D2 receptors48,49.
96-well flat-bottom clear plates (Falcon) were pre-coated with poly-D-lysine (Sigma). Next, 150,000 cells were transferred into each well of the treated plates and transiently co-transfected using Lipofectamine 2000 (Thermo Fisher) with pcdna plasmids encoding the appropriate D2 variant. Cell count was optimized using Life technologies’ recommended settings for reverse transfection (3 to 4 times standard). The quantity of DNA used for each D2DR mutant variant was optimized for maximal D2DR WT fluorescent signal (10ng).
Following a 24-hour transfection, assays were performed by incubating cells in FMP dye for half an hour, before placing them in a preheated (32 C) Flexstation 3 plate reader. After a 20 second reading to establish the baseline fluorescence, a saturating level of dopamine (20uM, Sigma) was added to induce activation of the D2DR receptors, reaching a maximal signal at around a recording time of 2 minutes. The plate was then rinsed multiple times with PBS to remove the dye for performing an ELISA to verify proper expression of D2DR variants. In order to determine surface expression of D2DR variants, cells were fixed with paraformaldehyde (PFA; 4% in PBS) and incubated with primary anti-HA antibodies (1/300 dilution), followed with secondary anti-IgG (1/2000 dilution) (Cell Signaling).
All fluorescence traces reported for the D2 variants were subtracted from fluorescent signals measured for Mock samples (cells transfected with empty pcdna vectors) under identical conditions.
Code availability
The methods developed in this study will be made available and released through a Baylor College of Medicine server.
Data availability
The data and protocols generated in this study will be made available and released through a Baylor College of Medicine server.
XML protocol example
<dock_design>
<SCOREFXNS>
<myscore weights=ligand.wts/>
</SCOREFXNS>
<TASKOPERATIONS>
<ReadResfile name=res_po/>
<DetectProteinLigandInterface name=edto design=1 cut1=6.0 cut2=8.0
cut3=10.0 cut4=12.0/>
<ProteinLigandInterfaceUpweighter name=up interface_weight=1.5/>
<InitializeFromCommandline name=init/>
</TASKOPERATIONS>
<FILTERS>
<LigInterfaceEnergy name="interfE" scorefxn=myscore energy_cutoff=-5.5/>
</FILTERS>
<MOVERS>
<AddOrRemoveMatchCsts name=cstadd cst_instruction=add_new/>
<EnzRepackMinimize name=desmin design=1 repack_only=0
scorefxn_minimize=myscore scorefxn_repack=myscore minimize_rb=1 minimize_sc=1
minimize_bb=0 cycles=5 minimize_lig=1 min_in_stages=0 backrub=0
task_operations=res_po, up, init/>
<FavorNativeResidue name=fnr bonus=1.0/>
<InterfaceScoreCalculator name=interface_rescore chains=X
scorefxn=myscore/>
</MOVERS>
<PROTOCOLS>
<Add mover_name=cstadd/>
<Add mover_name=fnr/>
<Add mover_name=desmin/>
<Add filter_name=interfE/>
<Add mover_name=interface_rescore/>
</PROTOCOLS>
</dock_design>
Supplementary Material
Acknowledgments
We thank the members of the Barth lab for insightful discussions during this study and critical comments on the manuscript. This work was supported by a grant from the National Institute of Health (1R01GM097207) and by a supercomputer allocation from XSEDE (MCB120101) to P.B.
Footnotes
Competing Financial Interests statement
The authors declare no competing financial interests.
Author contributions
P.B. designed the study; J.A. and P.B. developed the integrated homology modeling and ligand docking protocol; X.F. optimized the parameters for the ligand docking simulations and ligand pose selection; X.F. and K-Y.C. performed the blind and benchmark structure predictions; X.F. performed the ligand binding selectivity prediction benchmark, the design calculations; X.F. and M.Y. performed the experimental validations; P.B. and X.F. analyzed and discussed the results; P.B. wrote the manuscript.
References
- 1.Kolb P, et al. Structure-based discovery of beta2-adrenergic receptor ligands. Proc Natl Acad Sci U S A. 2009;106:6843–8. doi: 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deupi X, Kobilka BK. Energy landscapes as a tool to integrate GPCR structure, dynamics, and function. Physiology (Bethesda) 2010;25:293–303. doi: 10.1152/physiol.00002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katritch V, Cherezov V, Stevens RC. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol Sci. 2012;33:17–27. doi: 10.1016/j.tips.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manglik A, et al. Structural Insights into the Dynamic Process of beta2-Adrenergic Receptor Signaling. Cell. 2015;161:1101–11. doi: 10.1016/j.cell.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nygaard R, et al. The dynamic process of beta(2)-adrenergic receptor activation. Cell. 2013;152:532–42. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kahsai AW, et al. Multiple ligand-specific conformations of the beta2-adrenergic receptor. Nat Chem Biol. 2011;7:692–700. doi: 10.1038/nchembio.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–56. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim TH, et al. The role of ligands on the equilibria between functional states of a G protein-coupled receptor. J Am Chem Soc. 2013;135:9465–74. doi: 10.1021/ja404305k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stevens RC, et al. The GPCR Network: a large-scale collaboration to determine human GPCR structure and function. Nat Rev Drug Discov. 2013;12:25–34. doi: 10.1038/nrd3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pieper U, et al. Coordinating the impact of structural genomics on the human alpha-helical transmembrane proteome. Nat Struct Mol Biol. 2013;20:135–8. doi: 10.1038/nsmb.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2008;426:145–59. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- 12.Kelm S, Shi J, Deane CM. MEDELLER: homology-based coordinate generation for membrane proteins. Bioinformatics. 2010;26:2833–40. doi: 10.1093/bioinformatics/btq554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen KY, Sun J, Salvo JS, Baker D, Barth P. High-resolution modeling of transmembrane helical protein structures from distant homologues. PLoS Comput Biol. 2014;10:e1003636. doi: 10.1371/journal.pcbi.1003636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang J, et al. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis IW, Baker D. RosettaLigand docking with full ligand and receptor flexibility. J Mol Biol. 2009;385:381–92. doi: 10.1016/j.jmb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 16.Repasky MP, Shelley M, Friesner RA. Flexible ligand docking with Glide. Curr Protoc Bioinformatics. 2007;Chapter 8(Unit 8):12. doi: 10.1002/0471250953.bi0812s18. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Z, Felts AK, Friesner RA, Levy RM. Comparative performance of several flexible docking programs and scoring functions: enrichment studies for a diverse set of pharmaceutically relevant targets. J Chem Inf Model. 2007;47:1599–608. doi: 10.1021/ci7000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moustakas DT, et al. Development and validation of a modular, extensible docking program: DOCK 5. J Comput Aided Mol Des. 2006;20:601–19. doi: 10.1007/s10822-006-9060-4. [DOI] [PubMed] [Google Scholar]
- 19.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–61. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang XP, et al. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature. 2015;527:477–83. doi: 10.1038/nature15699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michino M, et al. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat Rev Drug Discov. 2009;8:455–63. doi: 10.1038/nrd2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kufareva I, Rueda M, Katritch V, Stevens RC, Abagyan R. Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure. 2011;19:1108–26. doi: 10.1016/j.str.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kufareva I, Katritch V, Stevens RC, Abagyan R. Advances in GPCR modeling evaluated by the GPCR Dock 2013 assessment: meeting new challenges. Structure. 2014;22:1120–39. doi: 10.1016/j.str.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forrest LR, Tang CL, Honig B. On the accuracy of homology modeling and sequence alignment methods applied to membrane proteins. Biophys J. 2006;91:508–17. doi: 10.1529/biophysj.106.082313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stamm M, Forrest LR. Structure alignment of membrane proteins: Accuracy of available tools and a consensus strategy. Proteins. 2015;83:1720–32. doi: 10.1002/prot.24857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qian B, et al. High-resolution structure prediction and the crystallographic phase problem. Nature. 2007;450:259–64. doi: 10.1038/nature06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Combs SA, et al. Small-molecule ligand docking into comparative models with Rosetta. Nat Protoc. 2013;8:1277–98. doi: 10.1038/nprot.2013.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischer M, Coleman RG, Fraser JS, Shoichet BK. Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat Chem. 2014;6:575–83. doi: 10.1038/nchem.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spyrakis F, BidonChanal A, Barril X, Luque FJ. Protein flexibility and ligand recognition: challenges for molecular modeling. Curr Top Med Chem. 2011;11:192–210. doi: 10.2174/156802611794863571. [DOI] [PubMed] [Google Scholar]
- 30.Cavasotto CN, Abagyan RA. Protein flexibility in ligand docking and virtual screening to protein kinases. J Mol Biol. 2004;337:209–25. doi: 10.1016/j.jmb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Conklin BR, et al. Engineering GPCR signaling pathways with RASSLs. Nat Methods. 2008;5:673–8. doi: 10.1038/nmeth.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roth BL. DREADDs for Neuroscientists. Neuron. 2016;89:683–94. doi: 10.1016/j.neuron.2016.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen ED, Norn C, Frimurer TM, Meiler J. Assessment and challenges of ligand docking into comparative models of G-protein coupled receptors. PLoS One. 2013;8:e67302. doi: 10.1371/journal.pone.0067302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodriguez GJ, Yao R, Lichtarge O, Wensel TG. Evolution-guided discovery and recoding of allosteric pathway specificity determinants in psychoactive bioamine receptors. Proc Natl Acad Sci U S A. 2010;107:7787–92. doi: 10.1073/pnas.0914877107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007;35:D198–201. doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang C, et al. Structural basis for molecular recognition at serotonin receptors. Science. 2013;340:610–4. doi: 10.1126/science.1232807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barth P, Schonbrun J, Baker D. Toward high-resolution prediction and design of transmembrane helical protein structures. Proc Natl Acad Sci U S A. 2007;104:15682–7. doi: 10.1073/pnas.0702515104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lemmon G, Meiler J. Towards ligand docking including explicit interface water molecules. PLoS One. 2013;8:e67536. doi: 10.1371/journal.pone.0067536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–8. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Isberg V, et al. GPCRdb: an information system for G protein-coupled receptors. Nucleic Acids Res. 2016;44:D356–64. doi: 10.1093/nar/gkv1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Isberg V, et al. GPCRDB: an information system for G protein-coupled receptors. Nucleic Acids Res. 2014;42:D422–5. doi: 10.1093/nar/gkt1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yarov-Yarovoy V, Schonbrun J, Baker D. Multipass membrane protein structure prediction using Rosetta. Proteins. 2006;62:1010–25. doi: 10.1002/prot.20817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tibshirani R, Walther G, Hastie T. Estimating the number of data clusters via the Gap statistic. Journal of the Royal Statistical Society B. 2001;63:411–423. [Google Scholar]
- 44.Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. Journal of Chemical Information and Modeling. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Venkatakrishnan AJ, et al. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–94. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 46.Tinberg CE, et al. Computational design of ligand-binding proteins with high affinity and selectivity. Nature. 2013;501:212–6. doi: 10.1038/nature12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen KY, Zhou F, Fryszczyn BG, Barth P. Naturally evolved G protein-coupled receptors adopt metastable conformations. Proc Natl Acad Sci U S A. 2012;109:13284–9. doi: 10.1073/pnas.1205512109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo J, Zhu Y, Zhu MX, Hu H. Cell-based calcium assay for medium to high throughput screening of TRP channel functions using FlexStation 3. J Vis Exp. 2011 doi: 10.3791/3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sung YM, Wilkins AD, Rodriguez GJ, Wensel TG, Lichtarge O. Intramolecular allosteric communication in dopamine D2 receptor revealed by evolutionary amino acid covariation. Proc Natl Acad Sci U S A. 2016;113:3539–44. doi: 10.1073/pnas.1516579113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data and protocols generated in this study will be made available and released through a Baylor College of Medicine server.