

Abstract
Peptidoglycan is an essential crosslinked polymer that surrounds bacteria and protects them from osmotic lysis. Beta-lactam antibiotics target the final stages of peptidoglycan biosynthesis by inhibiting the transpeptidases that crosslink glycan strands to complete cell wall assembly. Characterization of transpeptidases and their inhibition by beta-lactams has been hampered by lack of access to substrate. We describe a general approach to accumulate Lipid II in bacteria and to obtain large quantities of this cell wall precursor. We demonstrate utility by isolating Staphylococcus aureus Lipid II and reconstituting the synthesis of crosslinked peptidoglycan by the essential penicillin-binding protein 2, PBP2, which catalyzes both glycan polymerization and transpeptidation. We also show that we can compare the potencies of different beta-lactams by directly monitoring transpeptidase inhibition. The methods reported here will enable a better understanding of cell wall biosynthesis and facilitate studies of next-generation transpeptidase inhibitors.
Beta-lactams are an important family of antibiotics. The founding member of the beta-lactam family, penicillin, was acclaimed as a ‘miracle drug’ for its effectiveness in treating wound infections during World War II. More than seventy years have passed since penicillin entered the clinic and resistance to it is widespread. Several generations of beta-lactam antibiotics have been developed to counteract resistance as it has emerged, and beta-lactams remain a first-line therapy for treating many Gram-positive and Gram-negative infections.1 Nevertheless, methicillin-resistant Staphylococcus aureus (MRSA) strains are resistant to nearly all beta-lactams and MRSA infections are responsible for more than half of all lethal antibiotic-resistant infections in U.S. hospitals.2 Given the clinical importance of beta-lactams, it is remarkable that assays to directly monitor inhibition of their lethal targets, the transpeptidases, do not exist.
The transpeptidases are enzymes that catalyze the final stage of peptidoglycan biosynthesis. Peptidoglycan consists of a meshwork of crosslinked glycan strands and forms an essential structure surrounding the bacterial cytoplasmic membrane. Peptidoglycan biosynthesis is a highly conserved process that can be divided into three stages (Figure 1a). The first stage occurs in the cytoplasm and involves synthesis of a soluble precursor, the Park nucleotide, which contains a stem pentapeptide attached to UDP-N-acetyl muramic acid (UDP-MurNAc). In the second stage, the enzyme MraY catalyzes the coupling between the Park nucleotide and undecaprenyl phosphate in the cytoplasmic membrane to produce a lipid-linked monosaccharide peptide called Lipid I (Figure 1a).3 The glycosyltransferase MurG then transfers N-acetyl glucosamine (GlcNAc) from UDP-GlcNAc to the C4 position of Lipid I, forming a lipid-linked disaccharide peptide called Lipid II.4,5 While these steps are conserved in all bacteria, a series of subsequent modifications to the stem peptide occur in some bacteria. In S. aureus, for example, the stem pentapeptide of Lipid II contains a carboxamide at the second position,6 resulting from amidation of isoglutamate by the glutaminase GatD and the amidotransferase MurT.7 It also contains a pentaglycine branch at the third position, which is assembled by sequential transfer of glycine from a glycyl-tRNA donor to the lysine side chain in the stem peptide.8,9 Three enzymes, FemX, FemA, and FemB, are involved in synthesis of the pentaglycine branch (Figure 1a).10 Once Lipid II assembly is complete, it is translocated across the cytoplasmic membrane by a flippase.11,12 The final stage of peptidoglycan biosynthesis involves polymerization of Lipid II by peptidoglycan glycosyltransferases (PGTs) and crosslinking of the resulting glycan strands by transpeptidases.13,14 In many organisms, PGT and transpeptidase are parts of a bifunctional penicillin-binidng protein (PBP), and the two activities are directly coupled to one another.15 Therefore, in order to study transpeptidation in a given organism, one must have access to the native Lipid II from that organism because the stem peptide plays an important role, while analogues containing merely the stem peptide portion are not suitable as substrates.
Figure 1. Biosynthetic pathway for peptidoglycan assembly in Staphylococcus arueus.
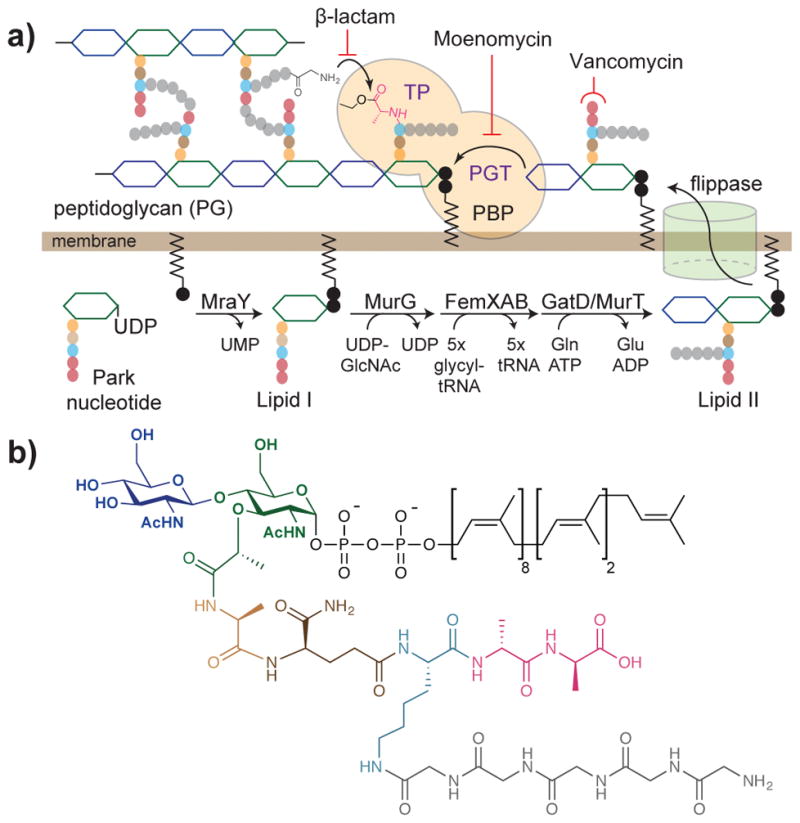
(a) The monomeric precursor of peptidoglycan, Lipid II, is synthesized and modified on the inner leaflet of the cytoplasmic membrane. It is then translocated by a flippase, and polymerized and crosslinked by peptidoglycan glycosyltransferase (PGT) and transpeptidase (TP) domains of penicillin-binding proteins (PBPs) respectively. The cell wall synthesis pathway is a target of multiple antibiotics. Moenomycin inhibits the PGT domain, beta-lactams inhibit the TP domain, and vancomycin binds to Lipid II. The acyl-enzyme intermediate formed on the catalytic serine during the transpeptidation reaction is depicted. The order of the Fem enzyme and GatD/MurT has not been experimentally established.7 (b) Structure of native S. aureus Lipid II.
Peptidoglycan crosslinking occurs via a two-step reaction in which the active-site serine of transpeptidases first attacks the terminal D-Ala-D-Ala amide bond in a stem pentapeptide to form a covalent acyl-enzyme intermediate, which then reacts with the nucleophilic amine from an adjacent strand to form a new peptide bond (Figure 1a).16 In S. aureus, the terminal amine on the pentaglycine branch acts as the nucleophile, resulting in a pentaglycine bridge between stem peptides. Beta-lactams, which are structural mimics of the D-Ala-D-Ala terminus that is conserved in Lipid II stem peptides, react with the active-site serine of transpeptidases to inactivate them.17 The crosslinking activity of transpeptidases in S. aureus and most other organisms has not been studied because it has not been possible to obtain sufficient quantities of their native Lipid II substrates.
Chemical, chemoenzymatic, and biosynthetic routes to Lipid II variants have been developed,13,14,18–27 but all are laborious. Moreover, each approach was developed for a specific Lipid II variant and considerable reengineering of the routes is required to obtain other Lipid II variants. S. aureus Lipid II is the most complex Lipid II variant in any organism (Figure 1b), and although S. aureus Lipid II analogs have been produced in small amounts,7,10 native S. aureus Lipid II has never been prepared.
In principle, the best way to obtain Lipid II is by direct isolation from bacterial cultures. Previously, this approach was found to yield only minute quantities of Lipid II.28 However, we recently developed a strategy to detect cellular Lipid II in S. aureus, and we found that a substantial amount of Lipid II accumulated when S. aureus was treated with either moenomycin, a natural product antibiotic that inhibits PGT activity and prevents Lipid II polymerization, or vancomycin, a glycopeptide antibiotic that binds and sequesters Lipid II (Figure 2a).29,30 We wondered whether it would be possible to accumulate enough Lipid II in S. aureus for biochemical studies.
Figure 2. Lipid II can be accumulated in bacteria using chemical probes that block Lipid II export or polymerization.
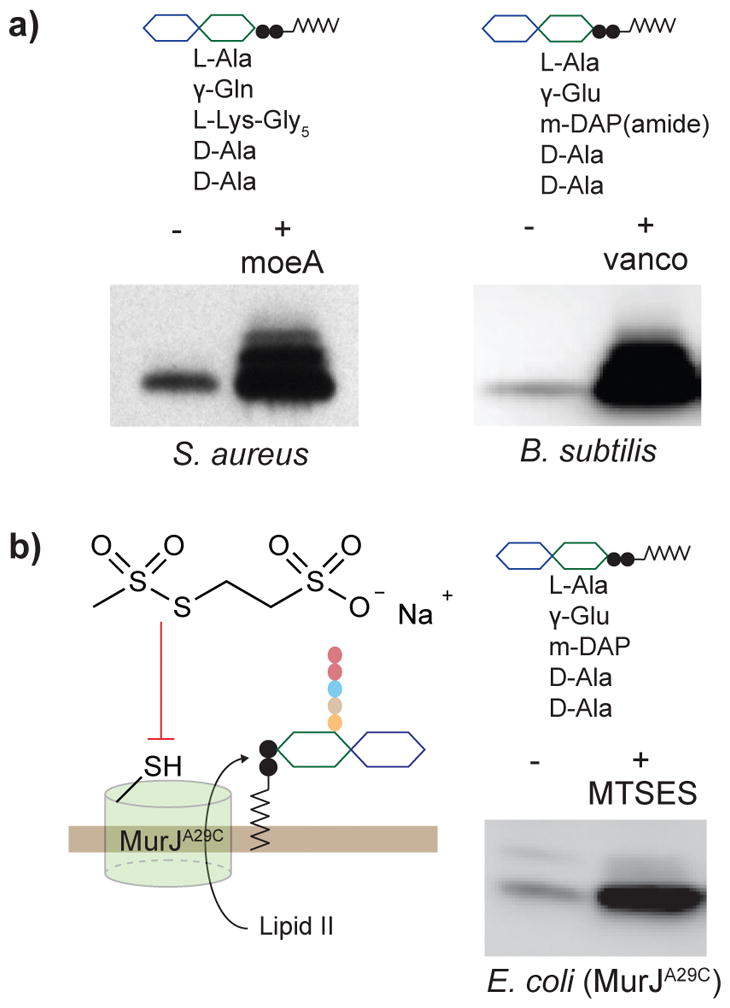
(a) Western blot (cropped) showing accumulation of Lipid II from S. aureus and B. subtilis in the presence of moenomycin and vancomycin, respectively. Lipid II was chemoenzymatically biotinylated to enable detection. Multiple bands are present due to Lipid II crosslinking during chemoenzymatic labeling.30 (b) Gram-negative Lipid II can be accumulated in an E. coli strain containing a mutant variant of the Lipid II flippase MurJ (A29C) that can be blocked with MTSES. (The full gels of the blots are reported in Supplementary Figures 8–10.)
Here, we show that large quantities of S. aureus Lipid II can be obtained easily. Using native S. aureus Lipid II, we have reconstituted the synthesis of crosslinked peptidoglycan by the essential penicillin-binding protein (PBP), PBP2 and have developed a transpeptidase activity assay to directly monitor beta-lactam inhibition.
Results
Chemical probes can be used to accumulate Lipid II
To determine how much Lipid II can be accumulated in S. aureus, we treated a small S. aureus culture (2 mL) with moenomycin (0.6 μg mL−1, 2x MIC) for varying amounts of time. We then extracted the cellular lipids with chloroform/methanol (CHCl3/MeOH) and selectively labeled Lipid II with a biotinylated probe (biotin-D-Lys, BDL) using purified S. aureus PBP4.29 BDL-labeled Lipid II was visualized by western blotting with streptavidin-HRP. We found that Lipid II levels in S. aureus increased by approximately 10-fold after 15 min of moenomycin treatment, and longer treatment did not further increase Lipid II levels (Figure 2a, Supplementary Results, Supplementary Figure 1a, d).
We next explored strategies to accumulate Lipid II in Bacillus subtilis, the model Gram-positive organism. We found the baseline Lipid II levels in B. subtilis to be very low, and we needed to increase the culture volume to 10 mL to detect any Lipid II (Figure 2a). Since B. subtilis is intrinsically resistant to moenomycin,31 we used vancomycin (8 μg mL−1, 8x MIC) to accumulate Lipid II. Vancomycin is useful because it dissociates readily from Lipid II, which facilitates downstream work-up.30 The optical density remained constant up to 20 min after addition of vancomycin and then it began to decrease due to cell lysis (Supplementary Figure 1b). At 20 min we extracted total lipids and, after PBP4-mediated biotinylation, we detected a 30-fold increase in B. subtilis Lipid II (Figure 2a, Supplementary Fig. 1e).
We also explored strategies to accumulate Escherichia coli Lipid II. E. coli has an outer membrane that prevents moenomycin and vancomycin from reaching their cellular targets.32 Instead, we accumulated Lipid II by blocking the activity of MurJ, the flippase that translocates Lipid II from the cytoplasm to the cell surface.11,12 To do so, we used a MurJ variant (MurJA29C) that is fully functional but is inactivated upon exposure to the thiol-reactive agent MTSES (2-sulfonatoethyl methanethiosulfonate),33 resulting in a build-up of Lipid II in the inner leaflet of the cytoplasmic membrane (Figure 2b).12 We treated the E. coli MurJA29C mutant (6 mL) with MTSES (1 mM, 8x MIC), extracted the lipids, and then visualized E. coli Lipid II after biotinylation. Lipid II increased 16-fold in the MTSES-treated MurJA29C mutant (Figure 2b, Supplementary Figure 1c, f).
Two-step extraction yields large quantities of Lipid II
To isolate accumulated S. aureus Lipid II, we extracted the lipids from six liters of moenomycin-treated culture using CHCl3/MeOH. We observed a thick, white interface layer between the aqueous and organic layers (Figure 3a). We collected the interface and organic layers separately, removed solvent, and re-dissolved the material in DMSO for analysis. Lipid II from each fraction was detected after chemoenzymatic biotinylation.29 We found that the interface layer contained much more Lipid II than the organic layer (Figure 3b). Conversely, thin layer chromatography (TLC) showed that cellular phospholipids such as phosphatidylglycerol were found mainly in the organic layer (Figure 3c, and Supplementary Figure 2a).
Figure 3. Large quantities of native S. aureus Lipid II can be isolated with good purity by extraction.

(a) Schematic showing the two-step extraction protocol. The first extraction (CHCl3/MeOH) produced three phases with a thick, white interface enriched in Lipid II. The second extraction (pyridinium acetate/butanol) separated Lipid II from water-soluble peptidoglycan precursors. (scale bar = 1cm) (b) Western blot (cropped) of biotinylated Lipid II in the interface and organic layers after the first extraction showed large amounts of Lipid II in the interface layer. (The full gel of the blot is reported in Supplementary Figure 11.) (c) TLC of the interface and organic layers showed that cellular phospholipids had partitioned into the organic layer. CL: cardiolipin; PG: phosphatidylglycerol; LPG: lysyl-phosphatidylglycerol. (d–e) Extracted ion chromatograms (EICs) of the interface layer after the first extraction (d) and the organic layer after the second extraction (e). The two-step extraction effectively separated the Park nucleotide from Lipid II. The samples were treated to remove the lipid on Lipid II prior to LC/MS analysis. (f) Structures of the Park nucleotide and delipidated Lipid II species.
To better characterize the interface fraction, we hydrolyzed the pyrophosphate linkage in Lipid II to remove the undecaprenyl chain, which interferes with mass spectrometry (MS) analysis.34 LC/MS analysis showed a small peak corresponding to hydrolyzed Lipid II and a large peak identified as the Park nucleotide (Figure 3d). The Park nucleotide has been shown to accumulate in cells when treated with certain cell wall inhibitors, including moenomycin.35,36 To remove the Park nucleotide, we extracted the interface layer with a mixture of pyridinium acetate/butanol/H2O and collected the organic phase for analysis (Figure 3a).37 After pyrophosphate cleavage, we observed only hydrolyzed Lipid II (Figure 3e–f). MS/MS analysis confirmed the presence of the pentaglycine branch and isoglutamine on the stem peptide (Supplementary Figure 3a, b). We searched for related Lipid II precursors, e.g., those lacking any of the five glycines or the carboxamide modification, but did not detect them. TLC analysis of the organic phase from the second extraction showed one major spot that migrated similarly to a synthetic Lipid II analog (Supplementary Figure 2b). This simple two-step extraction procedure thus provides Lipid II largely free of cellular lipids and the Park nucleotide. We quantified isolated S. aureus Lipid II by two different methods and found the yield to be approximately 500 μg/L (Supplementary Figure 4a–c), which is sufficient for many thousands of biochemical assays. In larger scale isolations, we can obtain milligram quantities of Lipid II for structural studies.
The fact that accumulated S. aureus Lipid II partitions into the interface of a three-phase extraction facilitates easy purification, but it was not clear whether Lipid II variants with different stem peptides would display similar partitioning behavior. B. subtilis and E. coli Lipid II differ substantially from S. aureus Lipid II by the lack of a branching peptide; 38 moreover, the presence of a γ–Glu rather than a γ–Gln could affect solubility (Figure 2a–b).39 After the first CHCl3/MeOH extraction of lipids from both organisms, we observed an interface layer similar to that observed in S. aureus. We collected the interface layer and subjected it to the second extraction. MS/MS analysis of the delipidated samples confirmed the presence of Lipid II (Supplementary Figure 3d–e). For E. coli, only the canonical structure was observed, but for B. subtilis we observed a minor amount (~10%) of a Lipid II variant containing a tripeptide lacking the D-Ala-D-Ala terminus (Supplementary Figure 3c). Because amidation on the m-DAP residue in B. subtilis Lipid II occurs after completion of the stem pentapeptide, the tripeptide likely resulted from hydrolysis of the pentapeptide by carboxypeptidases.40 The yield of E. coli Lipid II was a respectable 100 μg/L, but the yield of B. subtilis Lipid II was lower (20 μg/L, Supplementary Figure 4d–e). Even so, the amount is sufficient for a large number of biochemical assays.
Reconstitution of peptidoglycan crosslinking by PBP2
With native S. aureus Lipid II in hand, we set out to synthesize peptidoglycan using purified components. The major penicillin-binding protein (PBP) in S. aureus is PBP2, a bifunctional enzyme that carries out both Lipid II polymerization and glycan strand crosslinking (transpeptidation).41 We incubated S. aureus Lipid II with purified PBP2, then analyzed the products after mutanolysin digestion by LC/MS (Figure 4a, Supplementary Figure 5a).14 We detected monomeric fragments (muropeptides) and crosslinked fragments (dimeric and trimeric muropeptides); the composition of the peptidoglycan produced by PBP2 in vitro closely resembled that of the isolated S. aureus sacculus (Figure 4b). A time course of the PBP2 reaction revealed that monomeric muropeptides (resulting from uncrosslinked peptidoglycan) stopped accumulating within 10 min, a time point that coincided with the appearance of substantial amounts of crosslinked muropeptides (Figure 4c). After 1 h, the monomeric and dimeric species began to decrease as the trimeric species increased (Supplementary Figure 6). Reaction with a PBP2 variant containing an inactive transpeptidase domain (PBP2S398G) resulted in only monomeric muropeptides, identical to the products observed when the monofunctional PGT SgtB was used for reaction (Supplementary Figure 5b–c). Therefore, we have reconstituted the synthesis of crosslinked S. aureus peptidoglycan using native Lipid II.
Figure 4. The synthesis of crosslinked peptidoglycan was reconstituted using native S. aureus Lipid II.

(a) Schematic for analysis of peptidoglycan formed by S. aureus PBP2. (b) LC/MS extracted ion chromatogram of S. aureus PBP2 and Lipid II (left) produces peak A, the monomeric muropeptide, and peak B and C, the crosslinked dimeric and trimeric muropeptides; composition of in vitro peptidoglycan formed by PBP2 closely resembles that of the enzyme-digested S. aureus sacculus. The following ions were extracted from each chromatogram: A: 1253.5856 ([M+H]); B: 1029.0617 ([M+2H]2+); and C: 1194.2204 ([M+3H]3+). (c) Time-course analysis of PBP2 reaction shows that its transpeptidase activity follows glycan polymerization. The y-axis (% maximum) is calculated from the integrated EIC intensity of the monomeric muropeptide (polymerization) or the sum of dimeric and trimeric muropeptides (crosslinking), with each normalized to the highest intensity detected in the 90-minute period. Data represent averaged values of two experimental results.
A transpeptidase assay to characterize beta-lactams
We sought to develop a transpeptidase assay to characterize inhibition by beta-lactams. Previously, other bifunctional PBPs have been shown to incorporate D-amino acids during in vitro peptidoglycan synthesis.42,43 We wondered whether S. aureus PBP2 would incorporate BDL into peptidoglycan in vitro, and if so, whether BDL labeling would enable detection of product. We incubated purified PBP2 with native Lipid II and BDL for 1 h, then separated products by polyacrylamide gel electrophoresis (PAGE), and visualized them after western blotting via chemiluminscence (Figure 5a). We observed a band at the top of the well (Figure 5b), indicating that the products were highly crosslinked and unable to enter the gel matrix.44 In order to better analyze the product, we added lysostaphin, an endopeptidase that specifically cleaves S. aureus crosslinks,45 to the reaction mixture before quenching. The lysostaphin-treated sample showed strong chemiluminescent signals across a wide range of molecular weights, corresponding to BDL-labeled linear peptidoglycan polymers of different lengths (Figure 5b). The extent of crosslinking shows that PBP2 strongly prefers its native pentaglycine substrate to BDL for crosslinking. When the PBP2S398G mutant was used in the reaction, we observed no signal in the presence or absence of lysostaphin (Supplementary Figure 7a). Therefore, BDL incorporation into peptidoglycan provides a simple assay for monitoring PBP2 transpeptidase activity in vitro.
Figure 5. Direct transpeptidase activity assay enables comparison of beta-lactam inhibition of S. aureus PBP2.
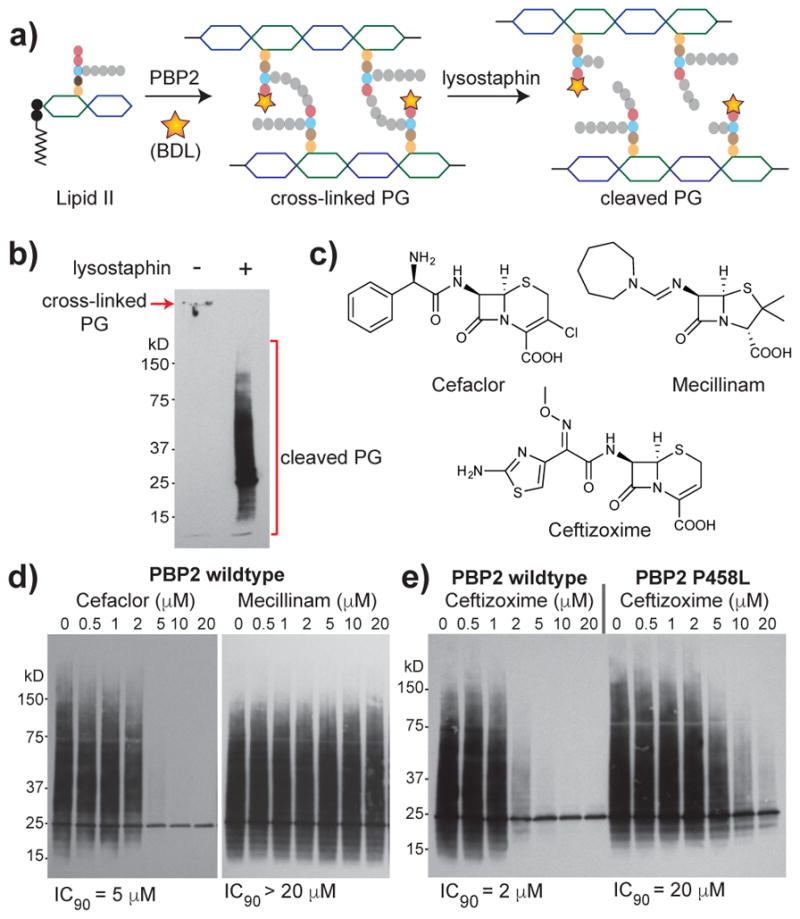
(a) Schematic depicting lysostaphin digestion of polymerized, crosslinked and biotinylated PG.(b) Western blot of crosslinked peptidoglycan produced by PBP2 with (right) and without (left) post-reaction lysostaphin treatment. Product detection was enabled by BDL incorporation during PBP2 reaction. (c) Structures of beta-lactams examined in d–e. (d) Cefaclor inhibits PBP2 activity but mecillinam does not up to the highest concentration tested. (e) Ceftizoxime inhibits wild-type PBP2 at lower concentrations than the PBP2P458L variant identified in a cefitzoxime-resistant S. aureus mutant. For all experiments, 1 μM of enzyme was used.
We next investigated inhibition of transpeptidase activity by beta-lactams using this assay. We briefly incubated PBP2 with various concentrations of beta-lactams before adding native Lipid II and BDL (Figure 5c). Cefaclor, a second-generation cephalosporin, was originally reported to bind poorly to PBP2;46 subsequent studies showed that it has a high affinity for PBP2 but undergoes rapid deacylation.47 We found that 5 μM cefaclor completely inhibited PBP2 transpeptidase activity under the reaction condition (Figure 5d). In contrast, mecillinam showed no detectable inhibition of PBP2 up to the highest concentration tested (Figure 5d), which this is consistent with reports that mecillinam has a poor affinity for S. aureus PBP2.46 We also characterized other beta-lactams including penicillin G, methicillin, and imipenem and found their inhibitory activities to be consistent with reported data from competition binding assays (Supplementary Figure 7b–c).46 Since our transpeptidase activity assay accurately reflected relative inhibitory potencies of different beta-lactams, we wondered if it would be useful for characterizing the effects of beta-lactam resistance mutations. A single amino acid substitution in PBP2 (PBP2P458L) was previously identified in a ceftizoxime-resistant S. aureus strain and the mutant protein was reported to show reduced binding to ceftizoxime.48 We found that higher concentrations of ceftizoxime were required to inhibit the transpeptidase activity of PBP2P458L compared with the wild-type protein (Figure 5e). On the other hand, we detected no increase in the inhibitory concentration of oxacillin against PBP2P458L (Supplementary Figure 7d), consistent with previous report that the ceftizoxime-resistant strain was still susceptible to oxacillin.48
Discussion
Obtaining useful amounts of Lipid II for biochemical studies of peptidoglycan biosynthetic enzymes has been a formidable challenge. Access to even simple Lipid II variants has been limited to a few laboratories with specialized expertise. Here we have reported a strategy to obtain practical quantities of native Lipid II from bacteria. This strategy uses chemical probes to accumulate Lipid II, and we have shown that different organisms require different treatments. While Lipid II can be accumulated in Gram-positive organisms using antibiotics, we used a specially engineered E. coli strain to accumulate canonical Gram-negative Lipid II. In this strain, Lipid II flippase activity can be inhibited using a thiol-reactive probe, preventing Lipid II export to the periplasm.12 Since most Gram-negative organisms produce the same Lipid II structure as E. coli,38 we expect that Lipid II isolated from this E. coli strain will be useful for studying peptidoglycan biosynthetic enzymes from other Gram-negative organisms. We have also developed a simple extraction procedure to isolate the accumulated Lipid II that exploits a three-phase extraction system to remove cellular phospholipids. The procedure is straightforward and it should be possible for anyone to obtain Lipid II within a day, enabling a wide range of applications.
Although we were surprised that so much Lipid II could be obtained from cells, our isolated yields are generally consistent with a recent study quantifying undecaprenyl species in E. coli and S. aureus.49 Under the assumption that all undecaprenyl species are converted to Lipid II when cells are treated with antibiotics or chemical probes, we would expect to obtain about 0.5 milligram of Lipid II per liter of culture, which is approximately what we obtained for S. aureus. Whereas milligram quantities of Lipid II can be readily isolated from a few liters of S. aureus culture, less can be obtained from E. coli and B. subtilis. Since the pool level of undecaprenyl species are similar in E. coli and S. aureus, the difference in isolated yield of Lipid II suggests that accumulation was not as effective and/or that more Lipid II is lost during the isolation. Structural differences in Lipid II could alter the partitioning in the extractions. In order to improve isolated yield of Lipid II from E. coli and B. subtilis, the extraction procedure can be modified or cells can be further engineered to better block Lipid II consumption. Nevertheless, the amount of Lipid II that can already be obtained from one-liter culture of B. subtilis is enough for at least 100 enzymatic reactions.
A major impetus for developing a method to obtain native Lipid II from different organisms was to enable studies on transpeptidases, the lethal targets of beta-lactams. Although beta-lactams have been studied for decades, it has not been possible to directly study inhibition of transpeptidase activity by beta-lactams. Instead, inhibitory potencies of different beta-lactams have been inferred from competition binding assays.16 Typically, bacterial membranes or purified PBPs are incubated with increasing concentrations of a test beta-lactam for a period of time and then a large excess of a radiolabeled or fluorescent beta-lactam is added. The concentration of the test beta-lactam that blocks labeling by the reporter beta-lactam is used as a measure of potency. While competition assays are useful, they can produce erroneous results. For example, while cefaclor was originally reported to be inactive against S. aureus PBP2,46 later studies showed that its inactivity was due to rapid deacylation rather than poor binding.47 Another limitation of these competition assays is that they do not work well for non-covalent inhibitors because the covalent reporter, which binds irreversibly, outcompetes the non-covalent inhibitors. Methods to isolate useful quantities of native Lipid II from different organisms make it possible to study transpeptidase activity and inhibition directly. We have developed a non-radiometric activity assay that exploits transpeptidase-mediated BDL incorporation during in vitro peptidoglycan assembly to enable product detection, and have shown that this assay is useful for evaluating beta-lactam inhibition of S. aureus PBP2. The results reproduce what is known about the relative potencies of beta-lactams for this enzyme from competition binding assays. While it is possible that BDL incorporation affects the extent of crosslinking, it does not affect the assay result as the products in either case are due to transpeptidase activity and TP inhibitors would affect the formation of both equally. The BDL incorporation assay cannot, however, differentiate inhibitors of polymerization from inhibitors of transpeptidation because transpeptidation by PBP2 requires peptidoglycan polymer. If there is a question about which step is being inhibited, the mass spectrometry assay can provide the answer because it can monitor both polymerization and crosslinking.
We expect direct transpeptidase assays will enable identification of other classes of transpeptidase inhibitors, including non-covalent inhibitors. Other classes of transpeptidase inhibitors may be particularly useful for overcoming methicillin-resistant S. aureus infections as MRSA strains have acquired an intrinsically beta-lactam resistant transpeptidase, PBP2a.50 The activity assay reported here can be readily adapted to study the mechanism of PBP2a – which has so far not been reconstituted, and its inhibition.
Online Method Section
Materials and general methods
Biotin-D-lysine (BDL) and synthetic Lipid II analog were prepared as previously described.29,42 Moenomycin A was isolated from Flavomycin stock. Vancomycin hydrochloride was purchased from Sigma-Aldrich. 2-sulfonatoethyl methanethiosulfonate (MTSES) was purchased from Toronto Research Chemicals. Beta-lactam drugs were purchased from the indicated vendors: piperacillin sodium salt (VWR), imipenem monohydrate (Toronto Research Chemicals), methicillin sodium salt, mecillinam vetranal, cefaclor, oxacillin sodium salt and cephradine (Sigma-Aldrich). S. aureus lipids, 16:0 phosphatidylglycerol (abbreviated as PG), 14:0 cardiolipin (abbreviated as CL), and 16:0 lysyl-phosphatidylglycerol (abbreviated as LPG), were purchased from Avanti Polar Lipids. Nalgene Oak Ridge High-Speed Centrifuge Tubes used for lipid extractions were purchased from Thermo Scientific. Streptavidin-HRP antibody was purchased from Pierce (Catalog #21130). Amersham ECL Prime Western Blotting Detection Reagent was purchased from GE Healthcare. Primers were purchased from Integrated DNA Technologies. Restriction endonucleases were purchased from New England Biolabs. Vectors and expression hosts were obtained from Novagen. Non-stick conical vials and pipette tips used for enzymatic reactions were from VWR. Tryptic Soy Broth and Luria Broth were purchased from Becton Dickinson.
S. aureus strain was grown at 37 °C in Tryptic Soy Broth (TSB) media under aeration with shaking. B. subtilis and E. coli MurJA29C strain were grown at 37 °C in Luria Broth (LB) media under aeration with shaking. LC/MS chromatograms were obtained on an Agilent Technologies 1100 series LC-MSD instrument using electrospray ionization (ESI). HRMS data was obtained on a Bruker Maxis Impact LC-q-TOF Mass Spectrometer using ESI. Western blots were developed using Biomax Light Film (Kodak) or imaged using an Azure C400 imaging system. ImageJ was used for densitometric analysis of western blots.
Small-scale Lipid II accumulation in bacteria
The small-scale lipid extraction from bacteria was modified from a previously published protocol.29 For S. aureus sample, an overnight culture of S. aureus RN4220 was diluted to OD600= 0.1, and allowed to grow to mid-exponential phase (OD600= 0.4–0.5) in TSB at 37 °C. The culture was divided into two (10 mL each). One culture was treated with moenomycin (0.6 μg/mL, 2x minimum inhibitory concentration (MIC)), whereas the other was not. At the indicated time, a 1-mL aliquot was taken out from each culture to measure O.D. and then transferred to a glass tube containing 3.5 mL of CHCl3: MeOH (1: 2). The mixture was vortexed for 10 min at 25 °C, following which any cell debris was removed with centrifugation for 10 min at 4,000× g. The supernatant was collected and transferred to a new glass tube with 2 mL CHCl3 and 1.5 mL PBS (pH 7.4). The mixture was vortexed for 10 min, and centrifuged for 10 min at 4,000 × g to achieve phase separation. The bottom organic layer was collected and dried under N2. The dried fractions were resuspended in 20 μL DMSO.
For B. subtilis sample, a similar protocol was performed with the following modifications: a culture of B. subtilis py79 (10 mL) in LB at OD600= 0.4–0.5 was treated with vancomycin (8 μg/mL, 8x MIC) for 20 min. The culture was centrifuged to collect cell pellet, which was then resuspended in 1 mL PBS (pH 7.4) for lipid extraction as described above. During phase separation, the top aqueous phase appeared cloudy, and was acidified with 20% H3PO4 (25 μL) to pH 3 to limit Lipid II solubility in water. The bottom organic layer was collected and dissolved in DMSO (20 μL).
For E. coli Lipid II isolation, a 6-mL culture of E. coli MurJA29C strain NR2186 (ref. 12) grown in LB at OD600= 0.3 was treated with MTSES (1 mM, 8x MIC) for 10 min. The culture was pelleted at 4,000 × g and resuspended in 1 mL of PBS (pH 7.4) and added with 2.7 mL CHCl3 and 1.3 mL MeOH. The mixture was pelleted 4000 × g for 2 min. The supernatant was collected and transferred to a new glass tube containing 2 mL of CHCl3 and 1.3 mL of PBS (pH 7.4). 3 mL of 100 mM HCl was added to adjust the aqueous phase to pH 1. The mixture was vortexed for 5 min and centrifuged at 4,000 × g for two minutes. The aqueous layer was removed. The organic layer was washed with 3 mL of H2O. The organic layer was collected, dried and resuspended in 320 μL of DMSO.
Western blot analysis of biotinylated Lipid II
The protocol for Lipid II biotinylation and detection was previously reported.29 Briefly, the lipid extract dissolved in DMSO (2 μL) was added to a mixture containing S. aureus PBP4 (4 μM), BDL (3 mM) in a reaction buffer (12.5 mM HEPES (pH 7.5), 2 mM MnCl2, and 250 μM Tween-80) to reach a total volume of 10 μL with a final DMSO concentration of 20%. The reaction was incubated at room temperature for 1 h, and quenched with 10 μL of 2x SDS loading buffer. 3 μL of the final mixture was loaded onto a 4–20% gradient polyacrylamide gel and let run at 200 V for 40 min. The products were transferred onto Immuno-Blot PVDF membrane (BioRad). BDL-Lipid II was detected by blotting with streptavidin-HRP (1:10000 dilution). In contrast, BDL-labeled Park nucleotide did not transfer onto the membrane and gave no signals after blotting.
Large-scale Lipid II accumulation and its two-step extraction from bacteria
Large-scale lipid extraction was performed on 6 L (4× 1.5L) of S. aureus RN4220 cultures. An overnight culture of S. aureus RN4220 in TSB media (15 mL) was used to inoculate each 1.5-L culture. The cultures were grown at 37 °C with shaking. Moenomycin was added at a final concentration of 0.6 μg/mL to each 1.5 L culture when OD600= 0.5–0.6 to accumulate of Lipid II. The moenomycin-treated cultures were grown for another 20 min before harvesting cell pellets. The pellets from 6 L cultures were resuspeneded in 60 mL PBS (pH 7.4), and divided equally into 4 × 125-mL Erlenmeyer flaks, each of which contains a mixture of 52.5 mL CHCl3: MeOH (1:2). The mixture was stirred for 1 h at room temperature to ensure cell lysis. The mixture (about 70 mL) from each Erlenmeyer flask was poured into two Teflon tubes, which were centrifuged at 4,000 × g for 10 min at 4 °C. The cell debris was visible as a pellet at the bottom of the tube, while the supernatant that contains the solubilized cellular contents was collected. For each two tubes, the supernatants were combined and poured into a clean 125-mL Erlenmeyer flask containing 30 mL CHCl3 and 22.5 mL PBS. The mixture was stirred at high speed for 1 h for thorough mixing of the layers. The homogenized mixture was quickly poured into three clean Teflon tubes and centrifuged at 4,000 × g for 10 min at 4 °C. It is important to quickly transfer the heterogeneous mixture into three tubes so that the composition of the mixture in each tube is roughly the same. In each Teflon tube, white materials were observed in between the top aqueous and bottom organic layer, resulting in an interface fraction. The top aqueous layer may appear hazy at first, but the haziness settled into the interface fraction upon the Teflon tube warmed up to room temperature, giving a clear aqueous layer. The interface fraction was collected by first removing the aqueous layer slowly using a Pasteur pipette. It was unavoidable to take up some volume of the aqueous layer while transferring the interface. The combined interface was dried in vacuo. We note that the interface fraction was not observed in the small-scale lipid extraction from S. aureus, since the volume of organic solvent used per cell mass was greater in small-scale extraction.
For the second extraction to remove the Park nucleotide in the interface, the combined dried interface was dissolved in a 15 mL organic mixture of 6M pyridinium acetate: n-butanol (1:2) (note: 6M pyridinium acetate was prepared by mixing 51.5 mL glacial acetic acid with 48.5 mL pyridine51,52), and washed with 15 mL of aqueous solvent (n-butanol saturated water) in a separatory funnel. The aqueous layer was extracted again with 10 mL organic solvent (1: 2/ 6M pyridinium acetate: n-butanol) to maximize Lipid II extraction. The organic layers were combined and washed with aqueous solvent (n-butanol saturated water) for three times (10 mL × 3) to remove the water-soluble Park nucleotides. The clean organic layer was concentrated in vacuo, and re-dissolved in MeOH.
In large-scale Lipid II extraction, a 1.5-L culture of B. subtilis py79 grown in LB at 37 °C was treated with vancomycin (8 μg/mL, 8x MIC) at OD600= 0.5–0.6 for 20 min, and a 1.5-L culture of E. coli MurJA29C (NR2186) in LB at 37 °C was treated with MTSES (1 mM, 8x MIC) at OD600= 0.5–0.6 for 20 min. The identical protocol of the two-step extraction of Lipid II described above was performed for both cultures.
Thin-layer chromatography (TLC) analyses of phospholipids and Lipid II
For phospholipid analysis, the dried interface and organic layers from the first extraction were each dissolved in 200 μL MeOH, and 10 μL was spotted for TLC. Authentic phospholipids dissolved in MeOH were used as standards. The TLC plate was eluted in solvent CHCl3: MeOH: CH3COOH of 60: 30: 10 (by volume), and detected by spraying with a solution of CuSO4 (100 mg/mL) in 8% phosphoric acid, followed by heating at 180 °C for 10 min until brown spots appeared.53
For Lipid II analysis, isolated Lipid II or synthetic Lipid II analog in MeOH was spotted on a TLC plate. The plate was eluted in solvent CH2Cl2: MeOH: 1% NH4OH of 6: 3.5: 1, stained with cerium ammonium molybdate reagent, and heated on a heating block until blue spots appeared.22
LC/MS analysis of delipidated Lipid II
For removal of the lipid tail, a Lipid II sample in DMSO (10 μL) was incubated with H2O (80 μL) and 100 mM ammonium acetate at pH 4.2 (10 μL). The mixture was boiled at 100 °C on a heating block for 30 min. Under this condition, Lipid II is cleaved at the phosphodiester linkage. The reaction was then lyophilized.
For LC/MS analysis, the lyophilized sample was resuspended in H2O, centrifuged at 16,000 × g for 10 min to remove precipitates. The supernatant was subjected to LC/MS analysis conducted with ESI-MS operating in negative mode. The instrument was equipped with a Waters Symmetry Shield RP18 column (5 μM, 3.9 × 150 mm) with a matching column guard. The fragments were separated using the following method: 0.5 mL/min H2O (0.1% formic acid) for 5 min followed by a gradient of 0% acetonitrile (ACN) (0.1% formic acid)/H2O (0.1% formic acid) to 20% ACN (0.1% formic acid)/H2O (0.1% formic acid) over 40 min. The molecular ion corresponding to the hydrolyzed Lipid II was extracted. MS/MS fragmentations of the species were also obtained.
Estimations of Lipid II quantities isolated from bacteria
A sample of native bacterial Lipid II in DMSO (4 μL) was mixed with varying known concentrations of synthetic Lipid II analog in DMSO (4 μL of 200 μM, 100 μM or 50 μM). The mixture was subjected to hydrolysis of the lipid tail prior to LC/MS analysis as described above. In the extracted ion chromatograms (EICs), the integrated area corresponding to each species was tabulated and plotted. A linear standard curve was obtained for the synthetic analog, and was used to estimate the concentration of native Lipid II.
In addition, an orthogonal quantification approach was used to estimate the amount of S. aureus Lipid II. A serial dilution of S. aureus Lipid II or the synthetic analog was prepared separately, and subjected to chemeozymatic biotinylation by PBP4.29 BDL-Lipid II signals on western blot were quantified using ImageJ, and a standard curve was obtained for the synthetic analog. The linear region of the curve was used to estimate the amount of S. aureus Lipid II.
Cloning of S. aureus PBP2[M59-S716] and its point mutants
The S. aureus pbp2[M59-S716] construct was cloned into pET42a(+) plasmid using Gibson assembly protocol. Briefly, the nucleotide region of S. aureus pbp2 gene encoding amino acid 59 to 716 was amplified using primer pair F’pET42a_PBP2 and R’pET42a_PBP2 (Supplementary Table 1). The PCR product was analyzed and purified using agrose gel electrophoresis. The pET42a(+) plasmid was amplified using the primer pair F’pET42a and R’pET42b (Supplementary Table 1). The purified PCR product and the linearized vector were assembled using Gibson assembly protocol. The inserted pbp2[M59-S716] gene was confirmed by sequencing (Beckman Coulter Sequencing Facility). E. coli NovaBlue strain was used for cloning.
The point mutants of pbp2[M59-S716] were made with the appropriate primers (Supplementary Table 1) using QuickChange site-directed mutagenesis kit (Stratagene). The construct was confirmed by sequencing.
Overexpression and purification of S. aureus PBP2[M59-S716] and its point mutants
The plasmid encoding S. aureus PBP2[M59-S716] collected from the NovaBlue cloning strain was transformed into E. coli. BL21(DE3) culture for overexpression and purification. A 15 mL culture of E. coli BL21 (DE3) harboring the plasmid encoding for S. aureus PBP2[M59-S716] grown in LB supplemented with 50 μg/ mL kanamycin was grown overnight, which was then used to inoculate 1.5 L LB medium (1:100) supplemented with 50 μg/mL kanamycin. The culture was grown at 37 °C with shaking until OD600 reached 0.4–0.5, and then cooled down to 17 °C before induction with 0.5 mM IPTG for 17 h with shaking. Cells were harvested by centrifugation (5250 × g, 20 min, 4 °C) and pellets were resuspended on ice with 30 mL of lysis buffer (10 mM Tris pH 8.0, 1 M NaCl, 10 mM MgCl2, 10% v/v glycerol and 40 mM CHAPs) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and 100 μg/mL DNase from bovine pancreas (Sigma Aldrich). The resuspended cells were passed through a cell disrupter (3 × 10,000 psi, 4 °C) for three times. The cell lysate was then pelleted by ultracentrifugation (100,000 × g, 30 min, 4 °C). The resulting supernatant containing PBP2[M59-S716] protein was applied to 1.5 mL pre-washed Ni-NTA resin (Qiagen) at 4 °C (the Ni-NTA resin was washed with dH2O and equilibrated with lysis buffer). After collecting flow through (FT), the resin was washed with buffer A (10 mM Tris pH 8.0, 0.2 M NaCl, and 0.28 mM LDAO) to facilitate detergent exchange (20 mL, W1). The resin was then washed with buffer A containing 60 mM imidazole (20 mL, W2). The protein was eluted with buffer A containing increasing imidazole concentrations (10 mL of 100 mM, 200 mM and 500 mM each, E1–E3). The fractions were analyzed on using SDS-PAGE electrophoresis. The fractions containing PBP2 protein (E1– E3) were combined and concentrated to ~10 mg/mL using a 50 kD MWCO Amicon Ultra Centrifuge Filter Device (Millipore). The concentrated PBP2 sample was further purified using size-exclusion chromatography with a Superdex S200 column equilibrated in buffer A. The fractions indicating monomeric protein were combined and concentrated using the 50 kD MWCO Amicon Ultra Centrifuge Filter Device, while the concentration was measured on nanodrop using the calculated extinction coefficient of PBP2 [M59-S716]. The final yield was approximately 10 mg per 1.5 L culture. S. aureus PBP2 [M59-S716] is referred to PBP2 subsequently. PBP2S398G and PBP2P458L were purified using the same protocol.
LC/MS assays for evaluations of PBP2 activities in vitro
S. aureus Lipid II in DMSO (40 μM) was incubated with purified PBP2 (1 μM) in 1x reaction buffer (50 mM HEPES, pH 7.5, 10 mM CaCl2) in a total of 10 μL reaction volume for 1 h at 25 °C. The reaction was quenched at 95 °C for 5 min, and then treated with mutanolysin (from Streptomyces globisporus, Sigma, 1 U) for 1.5 h at 37 °C followed by another 1 U aliquot for 1.5 h. The resulting disaccharides were reduced with sodium borohydride (10 μL of 10 mg/mL solution, 30 min). Phosphoric acid (20%, 1.2 μL) was then added to adjust the pH to ~4. Then reaction mixture was the lyophilized, redissolved in 12 μL H2O and subjected to LC/HRMS analysis, conducted with ESI-MS operating in positive mode on a Bruker qTOF mass spectrometer. The instrument was equipped with a Waters Symmetry Shield RP18 column (5 μM, 3.9 × 150 mm) with a matching column guard. The fragments were separated using the following method: 0.5 mL/min H2O (0.1% formic acid) for 5 min followed by a gradient of 0% acetonitrile (ACN) (0.1% formic acid)/H2O (0.1% formic acid) to 40% ACN (0.1% formic acid)/H2O (0.1% formic acid) over 25 min. Molecular ions corresponding to expected muropeptides were extracted. The reactions with PBP2S398G or SgtB were carried out using identical conditions.
The time-course analysis was performed by quenching the PBP2 reaction at various time points and subjected to mutanolysin digestion. The integrated areas of the peaks corresponding to monomeric muropeptides and dimeric muropeptides based on ion counts were measured and plotted; chromatograms showing the peak corresponding to the trimeric muropeptide were shown.
Isolation and enzyme digestion of S. aureus sacculus
The protocol was modified from a previous report.54 Briefly, an overnight culture of S. aureus (2 mL) was centrifuged at 10,000 rpm for 5 min. The pellet was resuspended in 1 mL 0.25% SDS in 0.1 M Tris/HCl, pH ~7.0, and boiled at 100 °C for 20 min. The suspension was centrifuged at 10,000 rpm for 5 min, and the pellet was washed with 1.5 mL H2O for at least three times to remove SDS. The washed pellet was resuspended in 1 mL H2O and sonicated in a water bath for 30 min, after which, 500 μL of a solution containing 15 μg/mL DNase from bovine, 60 μg/mL RNase in 0.1 M Tris-HCl, pH 6.8 was added. After shaking at 37 °C for 2 h, the mixture was boiled at 100 °C for 3 min to inactivate enzymes, centrifuged (5 min, 10,000 rpm) and washed with water once (1 mL). To release wall teichoic acid, the pellet was suspended in 500 μL of 1M HCl and incubated with shaking at 37 °C for 4 h. The pellet was centrifuged (5 min, 10,000 rpm) and washed with water until pH is 5 ~6. The pellet was resuspended in a 100 μL digestion buffer of 12.5 mM NaH2PO4, pH 5.5, and treated with 10 μL of mutanolysin (5 U/mL in H2O), and was incubated with shaking at 37 °C for 16 h. After digestion, the sample was boiled at 100 °C for 3 min to inactivate enzymes. The sample was centrifuged (5 min, 10,000 rpm), and added 50 μL NaBH4 (10 mg/mL) at room temperature for 30 min. The pH of the sample was adjusted to 4 with 20% phosphoric acid, and lyophilized. The lyophilized materials were resuspended in 500 μL H2O, and 20 μL was used for LC/MS analysis. The LC/MS condition is the same as described above for PBP2 reactions.
Western blot assay to study PBP2 transpeptidase activity
Briefly, Lipid II (1 μL of 100 μM stock in DMSO) was incubated with PBP2 (1 μL of 10 μM), BDL (1.5 μL of 20 mM) and 10x reaction buffer (1 μL of 500 mM HEPES, pH 7.5, 100 mM CaCl2) to reach a total volume of 10 μL. The reaction was incubated at room temperature for 15 min, and heat quenched briefly at 100 °C for 1 min. Lysostaphin (0.5 μL of 1 mg/mL) was added to the reaction mixture to resolve the crosslinked product. The reaction was shaken at 37 °C for 3 h. To quench the reaction, 10 μL of 2x SDS loading buffer was added. The protocol for western blot analysis described in the earlier section was used. Reactions using PBP2S398G with or without lysostaphin treatment were performed.
Characterization of PBP2 enzymatic inhibition by beta-lactams
The aforementioned western blot assay for PBP2 transpeptidase activity was modified slightly to allow studies of beta-lactam inhibition. Briefly, PBP2 (1 μL of 10 μM) was pre-incubated for 10 minutes with varying concentrations of beta-lactams (0– 20 μM) in a reaction mixture containing BDL (1.5 μL of 20 mM) and 10x reaction buffer (1 μL of 500 mM HEPES, pH 7.5, 100 mM CaCl2) for 5 min. Lipid extract (1 μL of 100 μM stock in DMSO) was then added to the reaction (final reaction volume: 10 μL) and let to incubate at room temperature for 15 min, and heat quenched briefly at 100 °C for 1 min. Lysostaphin (0.5 μL of 1 mg/mL) was added to the reaction mixture to resolve the crosslinked product. The reaction was shaken at 37 °C for 3 h. To quench the reaction, 10 μL of 2x SDS loading buffer was added. Western blot analysis was the same as above. For studies on the resistance mutation, PBP2P458L was used in place of the wild-type protein.
Image processing tools
Photoshop and ImageJ were used to process and quantify blots.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments
The authors thank Jennifer X. Wang at Harvard Small Molecule Mass Spectrometry Facility for assistance in running LC/MS and LC/MS/MS and interpretation of MS/MS spectra. This work was funded by U.S. National Institutes of Health grants GM100951 to N.R., R01 GM076710 to S. W., R01 GM066174 to D.K, and a Singapore A*STAR NSS (PhD) scholarship to Y.Q.
Footnotes
Author contributions
Y.Q. and V.S. developed methods to isolate and quantify Lipid II from S. aureus, E. coli and B. subtilis based in part on studies conducted by F.R. and K.S. Y.Q. and V.S. purified S. aureus PBP2; Y.Q. performed studies on PBP2 transpeptidase activity and characterized beta-lactam inhibition; N.R. constructed E. coli MurJA29C strain. S.W. and D.K. designed and supervised the project. The manuscript and figures were prepared by Y.Q., V.S., S.W., and D.K. with input from all authors.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Walsh CT, Wecewiscz T. Antibiotics: Challenges, Mechanisms, Opportunities. Washington, DC: ASM press; 2016. [Google Scholar]
- 2.Centers for Disease Control and Prevention Office of Infectious Disease. [Accessed December 19, 2016];Antibiotic resistance threat in the United States. 2013 Available at: http://www.cdc.gov/drugresistance/threatreport-2013.
- 3.Chung BC, et al. Crystal structure of MraY, an essential membrane enzyme for bacterial cell wall synthesis. Science. 2013;341:1012–1016. doi: 10.1126/science.1236501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Men H, Park P, Ge M, Walker S. Substrate synthesis and activity assay for MurG. J Am Chem Soc. 1998;120:2484–2485. [Google Scholar]
- 5.Ha S, et al. The kinetic characterization of Escherichia coli MurG using synthetic substrate analogues. J Am Chem Soc. 1999;121:8415–8426. [Google Scholar]
- 6.Siewert G, Strominger JL. Biosynthesis of the peptidoglycan of bacterial cell walls XI. Formation of the isoglutamine amide group in the cell walls of Staphylococcus aureus. J Biol Chem. 1968;243:783–790. [PubMed] [Google Scholar]
- 7.Münch D, et al. Identification and in vitro analysis of the GatD/MurT enzyme-complex catalyzing Lipid II amidation in Staphylococcus aureus. PLoS Pathog. 2012;8:e1002509. doi: 10.1371/journal.ppat.1002509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsuhashi M, Dietrich CP, Strominger JL. Incorporation of glycine into the cell wall glycopeptide in Staphylococcus aureus: role of sRNA and lipid intermediates. Proc Natl Acad Sci U S A. 1965;54:587–594. doi: 10.1073/pnas.54.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bumsted RM, Dahl JL, Söll D, Strominger JL. Biosynthesis of the peptidoglycan of bacterial cell walls X. Future study of the glycyl transfer ribonucleic acids active in peptidoglycan synthesis in Staphylococcus arueus. J Biol Chem. 1968;243:779–782. [PubMed] [Google Scholar]
- 10.Schneider T, et al. In vitro assembly of a complete, pentaglycine interpeptide bridge containing cell wall precursor (lipid II-Gly5) of Staphylococcus aureus. Mol Microbiol. 2004;53:675–685. doi: 10.1111/j.1365-2958.2004.04149.x. [DOI] [PubMed] [Google Scholar]
- 11.Ruiz N. Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli. Proc Natl Acad Sci U S A. 2008;105:15553–15557. doi: 10.1073/pnas.0808352105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sham LT, et al. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science. 2014;345:220–222. doi: 10.1126/science.1254522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye XY, et al. Better substrates for bacterial transglycosylases. J Am Chem Soc. 2001;123:3155–3156. doi: 10.1021/ja010028q. [DOI] [PubMed] [Google Scholar]
- 14.Lebar MD, et al. Forming cross-linked peptidoglycan from synthetic Gram-negative Lipid II. J Am Chem Soc. 2013;135:4632–4635. doi: 10.1021/ja312510m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Born P, Breukink E, Vollmer W. In vitro synthesis of cross-linked murein and its attachment to sacculi by PBP1A from Escherichia coli. J Biol Chem. 2006;281:26985–26993. doi: 10.1074/jbc.M604083200. [DOI] [PubMed] [Google Scholar]
- 16.Waxman DJ, Strominger JL. Penicillin-binding proteins and the mechanism of action of beta-lactam antibiotics. Annu Rev Biochem. 1983;52:825–869. doi: 10.1146/annurev.bi.52.070183.004141. [DOI] [PubMed] [Google Scholar]
- 17.Tipper DJ, Strominger JL. Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-D-alanyl-D-alanine. Proc Natl Acad Sci U S A. 1965;54:1133–1141. doi: 10.1073/pnas.54.4.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lo MC, et al. A New Mechanism of Action Proposed for Ramoplanin. J Am Chem Soc. 2000;122:3540–3541. [Google Scholar]
- 19.Schwartz B, Markwalder JA, Wang Y. Lipid II: total synthesis of the bacterial cell wall precursor and utilization as a substrate for glycosyltransfer and tanspeptidation by penicillin binding protein (PBP) 1b of Eschericia coli. J Am Chem Soc. 2001;123:11638–11643. doi: 10.1021/ja0166848. [DOI] [PubMed] [Google Scholar]
- 20.VanNieuwenhze MS, et al. The First Total Synthesis of Lipid II: The Final Monomeric Intermediate in Bacterial Cell Wall Biosynthesis. J Am Chem Soc. 2002;124:3656–3660. doi: 10.1021/ja017386d. [DOI] [PubMed] [Google Scholar]
- 21.Breukink E, et al. Lipid II Is an intrinsic component of the pore induced by nisin in bacterial membranes. J Biol Chem. 2003;278:19898–19903. doi: 10.1074/jbc.M301463200. [DOI] [PubMed] [Google Scholar]
- 22.Tsukamoto H, Kahne D. N-methylimidazolium chloride-catalyzed pyrophosphate formation: application to the synthesis of Lipid I and NDP-sugar donors. Bioorg Med Chem Lett. 2011;21:5050–5053. doi: 10.1016/j.bmcl.2011.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bugg TD, Braddick D, Dowson CG, Roper DI. Bacterial cell wall assembly: still an attractive antibacterial target. Trends in biotechnology. 2011;29:167–173. doi: 10.1016/j.tibtech.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 24.Patin D, et al. Colicin M hydrolyses branched lipids II from Gram-positive bacteria. Biochimie. 2012;94:985–990. doi: 10.1016/j.biochi.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 25.Zapun A, et al. In vitro reconstitution of peptidoglycan assembly from the Gram-positive pathogen Streptococcus pneumoniae. ACS Chem Biol. 2013;8:2688–2696. doi: 10.1021/cb400575t. [DOI] [PubMed] [Google Scholar]
- 26.Lebar MD, et al. Reconstitution of peptidoglycan cross-linking leads to improved fluorescent probes of cell wall synthesis. J Am Chem Soc. 2014;136:10874–10877. doi: 10.1021/ja505668f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang LY, et al. Enzymatic synthesis of lipid II and analogues. Angew Chem Int Ed. 2014;53:8060–8065. doi: 10.1002/anie.201402313. [DOI] [PubMed] [Google Scholar]
- 28.Guan Z, Breazeale SD, Raetz CRH. Extraction and identification by mass spectrometry of undecaprenyl diphosphate-MurNAc-pentapeptide-GlcNAc from Escherichia coli. Anal Biochem. 2005;345:336–339. doi: 10.1016/j.ab.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 29.Qiao Y, et al. Detection of lipid-linked peptidoglycan precursors by exploiting an unexpected transpeptidase reaction. J Am Chem Soc. 2014;136:14678–14681. doi: 10.1021/ja508147s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee W, et al. The Mechanism of Action of Lysobactin. J Am Chem Soc. 2016;138:100–103. doi: 10.1021/jacs.5b11807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McPherson DC, Popham DL. Peptidoglycan synthesis in the absence of class A penicillin-binding proteins in Bacillus subtilis. J Bacteriol. 2003;185:1423–1431. doi: 10.1128/JB.185.4.1423-1431.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silhavy TJ, Kahne D, Walker S. The bacterial cell envelope. Cold Spring Harb Perspect Biol. 2010;2 doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Butler EK, et al. Structure-function analysis of MurJ reveals a solvent-exposed cavity containing residues essential for peptidoglycan biogenesis in Escherichia coli. J Bacteriol. 2013;195:4639–4649. doi: 10.1128/JB.00731-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Higashi Y, Strominger JL, Sweeley CC. Biosynthesis of the peptidoglycan of bacterial cell walls XXI. Isolation of free C55-isoprenoid alcohol and of lipid intermediates in peptidoglycan synthesis from Staphylococcus arureus. J Biol Chem. 1970;245:3697–3702. [PubMed] [Google Scholar]
- 35.Park JT. Uridine-5′-pyrophosphate derivatives I. Isolation from Staphylococcus aureus. J Biol Chem. 1952;194:877–884. [PubMed] [Google Scholar]
- 36.Kohlrausch U, Höltje JV. Analysis of murein and murein precursors during antibiotic-induced lysis of Escherichia coli. J Bacteriol. 1991;173:3425–3431. doi: 10.1128/jb.173.11.3425-3431.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson JS, Matsuhashi M, Haskin MA, Strominger JL. Biosynthesis of the peptidoglycan of bacterial cell walls II. Phospholipid carriers in the reaction sequence. J Biol Chem. 1967;242:3180–3190. [PubMed] [Google Scholar]
- 38.Vollmer W, Blanot D, de Pedro MA. Peptidoglycan structure and architecture. FEMS Microb Rev. 2008;32:149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- 39.Sieber P, Riniker B. Protection of carboxamide functions by the trityl residue. Application to peptide synthesis. Tet Lett. 1991;32:739–742. [Google Scholar]
- 40.Hoyland CN, et al. Structure of the LdcB LD-carboxypeptidase reveals the molecular basis of peptidoglycan recognition. Structure. 2014;22:949–960. doi: 10.1016/j.str.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lovering AL, Safadi SS, Strynadka NCJ. Structural perspective of peptidoglycan biosynthesis and assembly. Annu Rev Biochem. 2012;81:451–478. doi: 10.1146/annurev-biochem-061809-112742. [DOI] [PubMed] [Google Scholar]
- 42.Lupoli TJ, et al. Transpeptidase-mediated incorporation of D-amino acids into bacterial peptidoglycan. J Am Chem Soc. 2011;133:10748–10751. doi: 10.1021/ja2040656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuru E, et al. In situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent D-amino acids. Angew Chem Int Ed. 2012;51:12519–12523. doi: 10.1002/anie.201206749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Helassa N, et al. The membrane anchor of penicillin-binding protein PBP2a from Streptococcus pneumoniae influences peptidoglycan chain length. FEBS J. 2012;279:2071–2081. doi: 10.1111/j.1742-4658.2012.08592.x. [DOI] [PubMed] [Google Scholar]
- 45.Thumm G, Gotz F. Studies on prolysostaphin processing and characterization of the lysostaphin immunity factor (Lif) of Staphylococcus simulans biovar staphylolyticus. Mol Microbiol. 1997;23:1251–1265. doi: 10.1046/j.1365-2958.1997.2911657.x. [DOI] [PubMed] [Google Scholar]
- 46.Georgopapadakou NH, Liu FY. Binding of beta-lactam antibiotics to penicillin-binding proteins of Staphylococcus aureus and Streptococcus faecalis: relation to antibacterial activity. Antimicrob Agents Chemother. 1980;18:834–836. doi: 10.1128/aac.18.5.834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chambers HF, Miick C. Characterization of penicillin-binding protein 2 of Staphylococcus aureus: deacylation reaction and identification of two penicillin-binding peptides. Antimicrob Agents Chemother. 1992;36:656–661. doi: 10.1128/aac.36.3.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Łęski TA, Tomasz A. Role of penicillin-binding protein 2 (PBP2) in the antibiotic susceptibility and cell wall cross-linking of Staphylococcus aureus: evidence for the cooperative functioning of PBP2, PBP4, and PBP2A. J Bacteriol. 2005;187:1815–1824. doi: 10.1128/JB.187.5.1815-1824.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barreteau H, et al. Quantitative high-performance liquid chromatography analysis of the pool levels of undecaprenyl phosphate and its derivatives in bacterial membranes. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2009;877:213–220. doi: 10.1016/j.jchromb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 50.Hartman B, Tomasz A. Altered penicillin-binding proteins in methicillin-resistant strains of Staphylococcus aureus. Antimicrob Agents Chemother. 1981;19:726–735. doi: 10.1128/aac.19.5.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heijenoort Yv, et al. Membrane intermediates in the peptidoglycan metabolism of Escherichia coli: possible roles of PBP 1b and PBP 3. J Bacteriol. 1992;174:3549–3557. doi: 10.1128/jb.174.11.3549-3557.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anderson JS, Meadow PM, Haskin MA, Strominger JL. Biosynthesis of the peptidoglycan of bacterial cell walls: I Utilization of uridine diphosphate acetylmuramyl pentapeptide and uridine diphosphate acetylglucosamine for peptidoglycan synthesis by particulate enzymes from Staphylococcus aureus and Micrococcus lysodeikticus. Archives of Biochemistry and Biophysics. 1966;116:487–515. doi: 10.1016/0003-9861(66)90056-7. [DOI] [PubMed] [Google Scholar]
- 53.Oku Y, Kurokawa K, Ichihashi N, Sekimizu K. Characterization of the Staphylococcus aureus mprF gene, involved in lysinylation of phosphatidylglycerol. Microbiology. 2004;150:45–51. doi: 10.1099/mic.0.26706-0. [DOI] [PubMed] [Google Scholar]
- 54.Kühner D, Stahl M, Demircioglu DD, Bertsche U. From cells to muropeptide structures in 24 h: Peptidoglycan mapping by UPLC-MS. Sci Rep. 2014;4 doi: 10.1038/srep07494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.