

Abstract
Acute Myeloid Leukemia with FLT3 ITD mutations are associated with a poor prognosis characterized by a higher relapse rate, shorter relapse free survival, and decreased likelihood of response to therapy at relapse. FLT3 ITD signaling drives cell proliferation and survival. FLT3 ITD AML disease progression is associated with cytogenetic evolution and acquired tyrosine kinase inhibitor (TKI) resistance suggesting a potential role of genomic instability. There is growing evidence demonstrating a relationship between FLT3 signaling and increased DNA damage, specifically through increased reactive oxygen species (ROS) resulting in double strand breaks (DSB), as well as impaired DNA repair, involving deficiencies in the non-homologous end joining (NHEJ), alternative non-homologous end joining (ALT NHEJ) and homologous recombination (HR) pathways. The role of genomic instability in the pathogenesis of FLT3 ITD AML warrants further examination as it offers potential therapeutic targets.
Keywords: FLT3, AML, DNA repair, Genomic instability
Introduction
Activating mutations of FMS-like tyrosine kinase 3 (FLT3) are one of the most common molecular abnormalities in Acute Myeloid Leukemia (AML) and are present in about 30% of newly diagnosed patients [1]. Internal tandem duplications of FLT3 (FLT3 ITD mutations), which are found in about 23% of de novo AML, are localized to the juxtamembrane domain of the receptor represent the most common activating mutation and confer a poor prognosis [2]. FLT3 tyrosine kinase domain (TKD) mutations are found in about 7% of de novo AML with a less certain prognostic significance [2]. For the last several years efforts have been underway to develop targeted therapy for this subtype of AML, as FLT3 ITD AML is rarely cured with chemotherapy alone [3]. Current approaches incorporate allogeneic hematopoietic stem cell transplant in first remission for those patients who have a suitable donor and are medically qualified for transplantation [4]. The first hurdle to overcome for potential cure is the achievement of remission with induction therapy. The addition of the pan-kinase inhibitor midostaurin [5] or FLT3 inhibitor sorafenib [6] to conventional cytarabine and anthracycline induction have been shown in a randomized trials to improve overall survival and relapse free survival respectively. Hematopoietic stem cell transplant (HSCT) holds the most potential of a cure for patients with high-risk leukemias. Specific targeting of oncogenic FLT3 ITD in the context of high intensity induction followed by HSCT represents one avenue yet to be thoroughly evaluated to improve this poor prognosis leukemia subset [7].
The remission rate for FLT3 ITD mutated patients is reported to be similar to AML patients without FLT3 ITD mutations, although relapse rates are high and remission durations are often short, even after HSCT. Conventional multi-agent salvage therapy is less effective in FLT3 ITD AML than other relapsed AML, suggesting the rapid development of a chemotherapy resistant phenotype [8]. One potential explanation for the high relapse rate and early chemorefractoriness would be rapid clonal evolution through genomic instability. Evidence of cytogenetic evolution at the time of relapse supports this hypothesis. This review will explore the published data exploring mechanisms for genomic instability as manifest through distinct features of DNA damage and DNA damage response described in FLT3 ITD AML.
Competent DNA replication and repair are critical to genomic stability
Familial cancer syndromes provide a model for understanding the mechanisms leading to disruption in genomic integrity and oncogenesis. Familial cancer syndromes are genetic disorders in which an inherited genetic mutation predisposes the affected individuals to the development of cancer. Multiple genes and pathways participate in maintaining genomic stability, including those involved in the detection of DNA damage, and the activation of cell cycle checkpoints and DNA repair mechanisms. Several mutations associated with familial cancer syndromes occur in genes responsible for maintaining genomic stability, including p53 (Li-Fraumeni Syndrome), MSH2 & MLH1 (Lynch syndrome (HNPCC)), BRCA1 & BRCA2 (Familial breast cancer), FANCA-G (Fanconi anemia). Inherited defects in DNA damage response and repair can lead to a higher rate of the accumulation of DNA damage when the unaffected copy is mutated or lost. The mechanisms associated with the tolerance of DNA damage are often the same that confer the resistance to chemotherapies, which rely on generating DNA damage to kill the cancer cells. There are extensive protein signaling cascades involved in the DNA damage response and partial redundancy of repair pathways which allow for repair of DNA damage through multiple pathways (Figure 1). Some of these pathways have higher degrees of repair fidelity than others. Somatic mutations acquired during oncogenesis often overlap those described in predisposition syndromes highlighting the importance of these processes in malignant transformation. Understanding the overlapping biology across tumor types may allow for expansion of novel therapies beyond those developed for specific germline lesions.
Figure 1.
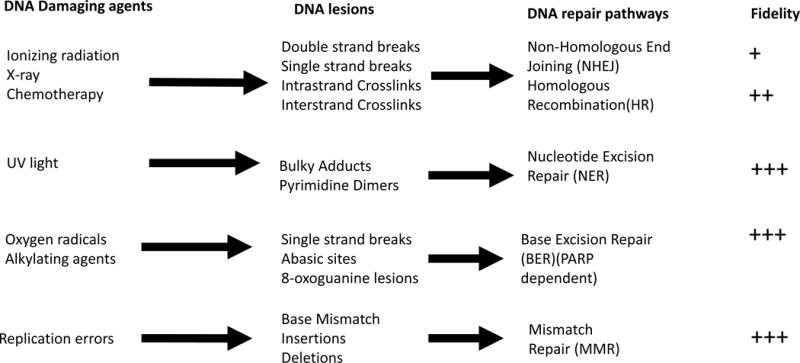
DNA Repair pathways. Adapted from Blanpain et al. [100]
Genomic instability in leukemia
Genomic instability is the acquisition of genomic abnormalities during cell division. Genomic instability drives tumorigenesis by activating oncogenes or deactivating tumor suppressor genes, making genomic instability a hallmark of cancer [9]. Despite leukemias having lower numbers of baseline mutations than almost all other cancers [10], high rates of genomic instability have been recognized specifically in myeloid malignancies containing activated tyrosine kinase (TK) pathways, such as BCR/ABL in chronic myeloid leukemia (CML), FLT3 ITD and c-KIT in AML, JAK2 in MPNs, and Ras mutations in myelodysplastic syndromes (MDS) [11].
The mechanisms of genomic instability in leukemia are best described and understood in BCR/ABL CML. As a manifestation of disease progression, CML cells accumulate karyotype abnormalities, including additional chromosomes, isochromosomes, translocations, and inversions, as well as mutations in p53 and Ras [12]. Increased DNA damage occurs in the form of DNA oxidation and DSBs caused by increased ROS [13]. BCR/ABL signaling influences multiple DNA repair pathways, including the homologous recombination (HR), non-homologous end joining (NHEJ) and nucleoside excision repair (NER) pathways [14]. The increased DNA repair allows for survival, but at the cost of fidelity, since many of these DNA repair mechanisms are error prone [15, 16]. Further analysis traced this genomic instability to several changes, including enhanced tyrosine phosphorylation of RAD51, leading to its aberrant function [17]; downregulation of BRCA1 [18]; stimulation of single-strand annealing, an error-prone DNA repair pathway [19]; and other changes in the Fanconi anemia/BRCA pathway that can be reverted by ectopic BRCA1 expression [20]. Cytogenetic evolution in BCR/ABL CML leads to disease progression and acquired resistance to small molecule inhibitors [21]. These mutations are thought to occur either due to DNA oxidation from ROS or from point mutations resulting from imprecise DNA repair due to dysfunctional DNA repair pathways [22].
Genomic instability in a different subset of kinase activated leukemias, myeloproliferative neoplasms (MPNs), is due to aberrant homologous recombination which likely contributes to malignant transformation to AML [23]. One common mutation in MPNs, an activating V617F point mutation in the tyrosine kinase JAK2, is found in greater than 80% of cases of polycythemia vera [24], 40% of cases of essential thrombocythemia, and 30% of cases of myelofibrosis [25]. JAK2 mutation and overexpression are associated with increased double strand breaks which require repair through DNA repair mechanisms. One primary repair mechanism is increased homologous recombination (HR) which can lead to loss of heterozygosity (LOH) states [26, 27, 28] as has been observed in other alterations conferring an MPN-like phenotype, such as FIP1L1-PDGFR rearrangements in eosinophilic leukemias, and FLT3 mutations in acute myeloid leukemia [28, 29]. MPN leukemia often has functional defects in HR pathways as evidenced by abnormal RAD51 foci formation upon exposure to ionizing radiation [23]. Epigenetic changes in BRCA promotor leading to “BRCAness” in AML represents one possible route for these functional deficits [30]. In these tumors, single strand DNA breaks would be repaired by base excision repair requiring Poly (ADP Ribose) Polymerase (PARP) pathway enzymes. Inhibition of PARP in this context can lead to synthetic lethality as demonstrated in BRCA mutated breast and ovarian cancers [23, 30, 31]. Consistent with this preclinical finding, MPN associated leukemia had a significantly higher response rate than other leukemias in the phase I study of the PARP inhibitor veliparib in combination with topotecan and carboplatin [32].
Genomic instability of FLT3 ITD AML suggests inefficient DNA repair
FLT3 ITD mutated AML is characterized by not only a high relapse rate and short relapse free survival, but a decreased likelihood of responding to therapy after relapse [33], resulting in shorter overall survival. Genomic instability, as evidenced by cytogenetic evolution at relapse, likely contributes to the chemoresistant phenotype and is described in a number of case series. A review of 15 patients treated between 2004 and 2013 with both a FLT3 ITD mutation and a normal karyotype examined diagnostic and relapse cytogenetic data. Twelve patients had persistent FLT3 ITD mutations at relapse, suggesting that FLT3 mutations, despite often occurring in less than all clones at diagnosis, play a role in chemoresistance leading to disease relapse [34]. Ten of the 12 patients displayed new karyotype abnormalities at relapse: Eight patients acquired only structural changes, 1 patient acquired only numerical changes, and 1 acquired both structural and numerical changes. Structural changes (translocations) often occur through the NHEJ pathway after DSB and are a feature of dysregulated FLT3 ITD related DNA damage repair (see below). McCormick et al. published a similar retrospective chart review and found that cytogenetic evolution was more frequent among FLT3 ITD AML patients. Ten out of 14 FLT3 ITD-AML patients acquired new cytogenetic abnormalities, compared to 7 out of 21 FLT3 wild type AML patients [35]. All 10 FLT3 ITD-AML patients with cytogenetic evolution acquired structural chromosomal abnormalities. A high frequency of cytogenetic progression, especially in the form of rare structural chromosomal abnormalities at relapse, suggests that FLT3 ITD mutations might increase the risk of relapse by the induction of genomic instability [34].
Review of FLT3 ITD AML genomic instability at Johns Hopkins Hospital revealed 50% of FLT3 ITD patients had new cytogenetic abnormalities at the time of relapse [36]. Of the 27 patients with new cytogenetic findings, 96%acquired at least one structural abnormality, 33%acquired at least one numerical abnormality, and 30% acquired both. Interestingly, patients with abnormal cytogenetics at diagnosis were not statistically more likely to acquire new cytogenetic abnormalities at relapse than patients with normal cytogenetics at diagnosis that relapsed (40% versus 54%, p=NS). Patients who acquired cytogenetic abnormalities had a higher median allelic burden at diagnosis (67.5% versus 24%). Patients with NPM1 mutations at diagnosis appeared to acquire new cytogenetic abnormalities at relapse less often (5/15) than NPM1 negative patients (7/11) (33% versus 64%, p=NS), although this observation did not reach statistical significance (Fishers exact test). NPM1 mutations have been shown to abrogate genomic instability, potentially explaining the more favorable outcomes of those with combined FLT3 ITD/NPM1 mutations [37].
Biologic mechanisms for FLT3 ITD genomic instability
Increased DNA Damage
DNA damage can occur through several well characterized mechanisms including DNA adduct formations with heavy metals, error prone DNA replication, and single strand and double strand breaks through ROS (Figure 1). Increased ROS levels have been observed in many cancers, including leukemia [38, 39]. Leukemia related oncogenes generate ROS, including BCR/ABL [40, 41, 42], as described above, and FLT3 ITD [43], with this ROS contributing to genomic instability [11].
FLT3 ITD AML cell lines, cells transfected with FLT3 ITD, and FLT3 ITD primary leukemia cells show increased ROS associated with STAT5 activation of RAC1, a critical member of the NADPH oxidase pathway [43]. When treated with CEP-701, a FLT3 inhibitor, a decrease in ROS production was observed in a dose-dependent manner in FLT3 ITD cells. In addition, a decrease in ROS production was observed when RAC1 protein was directly inhibited in FLT3 ITD cell lines. RAC1 activity plays an important role in the production of ROS. RAC1 activity decreased in FLT3 ITD cell lines when treated with the FLT3 ITD inhibitor CEP-701, suggesting FLT3 signaling directly influences RAC1 activity. ROS production also decreased when STAT5 expression was inhibited through siRNA. Binding of phosphorylated STAT5 to RAC1-GTP was increased in FLT3 ITD cell lines, suggesting that FLT3 signaling increases the production of ROS through both STAT5 and RAC1 [43].
As noted above, FLT3 ITD associated leukemia is associated with high levels of ROS which results in both DNA oxidation and DSBs. Specifically, FLT3 ITD causes an increase in both NOX protein through the rate limiting substrate NADPH, as well as p22phox protein, a membrane bound NOX complex subunit [44]. The increase in these protein levels results in the production of H2O2, which diffuses from the nuclear membrane to the nucleus where it causes oxidative damage to the DNA [44]. DNA oxidation contributes to carcinogenesis by two mechanisms: epigenetic modifications [45] and mutagenesis [46]. DNA oxidation correlates with promoter methylation leading to decreased transcription of tumor suppressor genes. Importantly, it was discovered that FLT3 ITD activated ROS caused oxidation of the tumor suppressor gene, and negative FLT3 regulator, DEP-1 phosphatase [47, 48]. Furthermore, DNA oxidation can directly lead to mutagenesis if not repaired correctly. In addition to oxidative DNA damage, the increase in ROS results in DNA double strand breaks, as measured through quantification of gamma H2AX, a histone family member that contributes to nucleosome formation and the structure of DNA, which is phosphorylated by PI3 kinase, a signal transduction enzyme located downstream of the FLT3 receptor, in response to DSBs [49]. FLT3 ITD cells show a rapid loss of H2AX phosphorylation after etoposide treatment, which is not seen in FLT3 wild type cells. Loss of H2AX phosphorylation has been shown to correlate to DSB repair. In FLT3 ITD cells, treatment with a FLT3 inhibitor, PKC412, reversed the loss of H2AX phosphorylation previously seen. Therefore, inhibiting FLT3 ITD activation resulted in decreased DNA repair in FLT3 ITD samples, representing a mechanism for FLT3 ITD cells to persist under genotoxic stress of chemotherapy [50].
FLT3 Mouse Model
In mice, FLT3 expression is important for B cell development. Mice with disrupted FLT3 signaling due to loss of either FLT3 ligand or FLT3 receptor show deficiencies in B cell progenitors [51, 52]. FLT3 ITD knock-in mouse models also show decreases in B cells due to a block in B cell development at the early pro B cell stage as a result of impaired VDJ recombination [53, 54]. VDJ requires the RAG1/RAG2 endonuclease initiated DSB at recombination signal sequences and a rejoining of the DNA strands by the NHEJ pathway. In lymphocytes lacking proteins required for the NHEJ pathway, VDJ recombination errors occur resulting in chromosomal abnormalities [55]. In the FLT3 ITD mouse model, RAG1 and RAG2 expression are reduced in early pro B cells, which would suggest a reduction in DNA cleavage. Importantly, in FLT3 ITD knock in mice, early pro B cells also showed an accumulation of DSBs at Jh recombination signals, and reduced D-Jh rearrangements, suggesting an impaired ligase activity at DNA breaks. FLT3 ITD cells were repaired using microhomology sequences, and larger microhomology sequences, suggesting the use of the ALT-NHEJ DNA repair pathway. This is also supported by evidence of decreased expression of Ku86, which is important for the NHEJ pathway, and increased expression of PARP1, which is important for the ALT NHEJ pathway [53]. Restoring Ku86 expression (and the NHEJ pathway) led to greater B cell development. Lastly, treatment of B cells from FLT3 ITD mice with CEP-701 led to increased Ku86, restoring the NHEJ pathway and B cell differentiation [53].
Aberrant DNA Repair
In addition to increasing DNA damage, there is evidence to suggest that FLT3 ITD mutations influence DNA repair mechanisms. Several mechanisms exist to repair DSBs which require either the use of a homologous template or nonhomologous end-joining, both of which are critical to maintaining genomic integrity [56]. Upregulated DNA repair mechanisms may contribute to chemoresistance and loss of repair fidelity may contribute to mutagenesis and disease progression.
Double strand DNA breaks are joined without the use of a homologous template through the NHEJ pathway. If the DNA ends are modified prior to ligation, this pathway could lead to mutations. In FLT3 ITD cells, there is inefficient repair of DSBs by the NHEJ pathway, leading to an increased frequency of errors [29, 43]. In particular, NHEJ repaired products from FLT3 ITD cells showed increases in deletion sizes [29]. Treatment with CEP-701, a FLT3 inhibitor, resulted in more efficient NHEJ repair and a decreased frequency of misrepair [29, 43]. It is possible that the deficiency in NHEJ repair leads to compensatory repair in the form of upregulation of other pathways [57].
It has been demonstrated that FLT3 ITD expression leads to increased activity of an alternate nonhomologous end-joining (ALT NHEJ) pathway [29], which is more error-prone than the classical NHEJ pathway, and can result in the formation of translocations [58]. In FLT3 mutated cells, levels of Ku proteins, which are important for the NHEJ pathway, are decreased, and levels of DNA Ligase III-alpha protein, which is important for the ALT NHEJ pathway, are increased [29]. Treatment with CEP-701 resulted in a decrease in levels of Ligase III-alpha protein, and an increase in levels of Ku proteins. It was also noted that the majority of DNA repair occurred at sequences of microhomology, also indicative of ALT NHEJ repair [29].
In addition to the abnormalities seen in the DNA repair pathways involving nonhomologous end-joining, the pathways involving the use of a homologous template also appear to be abnormal. Pathways involving the use of homologous template include homologous recombination and single-strand annealing (SSA). HR, the most precise DNA repair pathway, is dependent upon the protein RAD51. RAD51 transcription significantly correlates with FLT3 ITD transcription, suggesting that FLT3 mutations lead to increased RAD51 expression. Importantly, treatment of FLT3 ITD cells with the FLT3 inhibitor, PKC412, resulted in reduced RAD51 production. Additionally, RAD51 gene expression was associated with downstream STAT5 phosphorylation in FLT3 ITD cell lines [50]. FLT3 ITD AML cells and FLT3 transfected cells showed increased sister chromatid exchange, a measure of HR mediated crossing over and mechanism for loss of heterozygosity.
The upregulated HR in response to increased DSB provides a potential explanation for the acquired copy neutral loss of heterozygosity (LOH) seen commonly in FLT3 ITD leukemia [59]. Demonstration of this LOH is seen routinely on chromosome 13, which contains the FLT3 gene and in the mouse model of FLT3 ITD pathogenesis LOH is associated with increased disease aggressiveness [60]. In diagnostic samples from 85 patients with either FLT3 ITD mutations, FLT3 TKD mutations or FLT3 wild type (WT), LOH on chromosome 13 was observed in 12 patient samples, all of which had FLT3 ITD mutations. The LOH was determined to not be due to deletion, and was therefore copy neutral loss of heterozygosity (CN-LOH) at 13q, which always involved the FLT3 gene. The FLT3 ITD allelic ratio (AR) was significantly higher (median 2.5) in these samples than in samples without CN-LOH (median 0.65). Paired diagnostic and relapse samples revealed that the homozygous clone becomes the dominant clone at relapse and therefore CN-LOH at 13q appears to be a late event in AML pathogenesis where the FLT3 ITD evolves from a heterozygous clone. FLT3 ITD with high AR is associated with significant gene expression differences in 642 genes when compared to WT and normal CD34 bone marrow samples through microarray studies. The three most represented gene expression differences included genes involved in cell cycle, and DNA replication, DNA recombination and DNA repair. The DNA replication and repair genes included genes involved in double-strand DNA break repair, specifically FEN1, KPNA2, POLA1, SOD1, SOD2, and XRCC5. These gene expression differences likely play a role in the development of CN-LOH [61].
Alternatively, the SSA pathway is inhibited by RAD51, and DNA repair through SSA pathway is always mutagenic [62]. Interestingly, the SSA is also upregulated in FLT3 mutated cells. Treatment with PKC412 reduced the frequency of, but did not sufficiently suppress SSA in FLT3 mutated cells. SSA is dependent upon PI3k and Ras signaling. In BCR/ABL cells, inhibition of PI3K, Akt, and MEK1 through small molecule inhibitors led to a dose-dependent reduction of SSA [63].
Single strand breaks (SSB), when not repaired, become double strand breaks during replication. If FLT3 ITD signaling were to impair SSB repair mechanisms, such as nucleotide excision repair and mismatch repair, additional DSB may accumulate and contribute to the genomic instability seen in this subset of patients. BCR/ABL affects the efficiency of both nucleotide excision repair [64] and mismatch repair [65]. Due to the similarities observed in BCR/ABL and FLT3 ITD influence on aberrant DNA repair, it is reasonable to consider the possibility that FLT3 ITD also affects the efficiency of SSB repair, although to our knowledge this has not yet been investigated.
Other mechanisms of genomic instability
In addition to changes seen at the karyotype level, activating FLT3 mutations have been associated with other factors reflective of genomic instability, including, microsatellite instability, shorter telomeres, and a high frequency of somatic mutations.
Microsatellite instability is seen in both MDS and AML, and has been found to occur more frequently in specific subsets of patients [66, 67]. Microsatellite instability (MSI) correlated with chromosomal instability in myeloid malignancies [68]. In a series of de novo AML patients analyzed to assess whether MSI was associated with a specific molecular subgroup, both FLT3 ITD mutations and mixed-lineage leukemia (MLL) rearrangements were found to be associated with MSI [66]. MLL is associated with overexpression of FLT3 WT [69]. Microsatellite instability might be a reflection of the mutator phenotype seen in this patient population. In particular, in colorectal cancers with high MSI from defective mismatch repair, a higher frequency of mutations in tyrosine kinase genes (ERBB2, KIT, FGFR3, EGFR, PDGFRA, ERBB4, and FGRR1) has been described [70]. Due to coactivated pathways downstream of these TKs, mechanisms for development of MSI and genomic instability are likely similar amongst tumors with activating TK mutations [70].
Telomere related genome instability in cancer is well described [71]. AML cells harboring FLT3 ITD mutations are associated with shorter telomeres than those with other mutations [72, 73]. AML patients with abnormal cytogenetics have shorter telomeres than patients with normal cytogenetics, suggesting short telomeres may be caused by the same mechanisms that lead to genomic instability [74, 75]. Short telomere length has influence on DNA damage response through proteins such as telomere-associated protein telomeric repeat-binding factor 2 (TRF2) which disassociates from the telomere complex, and upregulate DNA damage response and repair mechanisms [73, 76]. These mechanisms may also influence chemotherapeutic resistance in clones with short telomeres.
Recent work exploring the changes at the genomic level in FLT3 ITD AML which occur in matched diagnostic and relapsed samples suggest a preferential acquisition of DNA (C>A & C>G) transversions at the time of relapse suggesting potential role for chemotherapy induced mutation acquisitions [77]. These transversions are also a common event in carcinogenesis of tobacco induced lung cancer [78]. Non-FLT3 mutated AML also demonstrates a propensity for DNA transversions at the time of relapse [79] but it is unclear if the rate of these tranversions are more frequent in FLT3 mutated cases.
Therapeutic interventions
Although FLT3 inhibitors show promising results in preclinical studies, when used as monotherapy in clinical trials, only partial and transient responses are achieved in most patients [80]. Second generation inhibitors are under development in combination therapies, but acquired resistance remains a major obstacle to achieving successful long term control of FLT3 ITD disease [81]. Recently more attention and effort have been turned towards understanding the underlying mechanisms of acquired resistance.
One widely recognized mechanism of resistance of FLT3 ITD leukemias to oral tyrosine kinase inhibitor (TKI) therapy involves the emergence of new FLT3 TKD mutations [81, 82]. Newer inhibitors are being investigated that have shown to target both ITD and TKD mutations, including crenolanib [83, 84], and gilteritinib (ASP2215) but large studies of outcomes of patients, and resistance in these patients, are yet to be described [85, 86].
Another mechanism of resistance involves the upregulation of parallel and downstream signal transduction pathways [82]. The dysregulation of three main signaling pathways associated with FLT3 mutations includes: PI3K/Akt/mTOR, Ras/Raf/MEK/ERK, and JAK/STAT, all of which are though to also confer a survival advantage. Consideration of multitargeted therapy may be beneficial in resistant disease due to codependency between these pathways [81]. Several preclinical studies have demonstrated that combining inhibitors leads to antileukemic synergy in TKI resistant cells. In sorafenib resistant leukemia cells with acquired TKD mutations, the combination of sorafenib and crenolanib resulted in enhanced FLT3 signaling suppression, including suppression of downstream targets, ERK and Akt, and greater apoptotic effects [87]. In the same sorafenib resistant leukemia cells with acquired TKD mutations, inhibiting both FLT3 and ERK/Akt signaling through the combinations of sorafenib and a MEK inhibitor, and sorafenib and an mTOR inhibitor, both also showed synergistic apoptotic effects [87]. Clinical studies in other TK mutated cancers have also demonstrated success in inhibiting multiple signaling pathways, including dual inhibition of the PI3k/Akt/mTOR and Raf/MEK/ERK pathway in KRAS and BRAF mutated cancers [88, 89].
Interestingly, several of the studies described above show additional suppression of DNA damage and DNA repair from the use of inhibitors of downstream signal transduction pathways. For example, inhibiting STAT5 reduced the production of the ROS responsible for causing DSBs [43], and inhibitors of PI3K, Akt, and MEK1 reduced SSA [63]. Furthermore, all of these pathways have been implicated in DNA repair independent of FLT3 signaling [90, 91, 92, 93, 94].
While many of the studies above note FLT3 inhibitors reduce DNA DSBs and reduce DNA repair efficiency and errors, they do so insufficiently. Targeting DNA repair mechanisms offers another potential therapeutic strategy, and one that has proven to be successful in other malignancies with DNA repair deficits [95]. One strategy to potentially decrease the rates of chemoresistance is to suppress the activated kinase pathways at the time of minimal residual clonal disease. This strategy has been successful in the use of TKI therapy for BCR/ABL acute lymphoblastic leukemia (ALL) in its use in the post HSCT periods. Likewise FLT3 inhibitor therapy in the post-transplant period is showing promise in improvement of outcomes in the post-transplant periods [7], and one could argue this success is at least partially due to suppression of ongoing genomic instability and clonal evolution by suppression of the activated kinase pathways.
Alternative therapeutic approaches could include strategies to induce a synthetic lethality in persistent clonal disease with aberrant DNA repair pathways. This can be accomplished through blockade of the PARP dependent pathways relied upon by many activated kinase tumors or those deficient in BRCA protein members which both have been found to have function HR deficits. The strategy that has shown the most promise is in the minimal reisidual disease state for ovarian cancers where the PARP1/2 inhibitor olaparib has been approved by the FDA for maintenance therapy for BRCA mutation associated ovarian cancers. HR defects in our preclinical models and clinical studies of leukemias with activated kinases (non-FLT3 disease) suggest this form of maintenance may be applicable to MPN associated leukemias [23, 32, 96]. Due to mechanistic similarities in DNA repair, and in particular HR deficiencies, PARP inhibition may warrant evaluation in FLT3 ITD clonal disease. The utility of PARP inhibitors in the prevention of acquisition of chromosomal translocations has been hypothesized [97], which may be advantageous at prevention of disease after therapy as we describe above.
Lastly, recent promising approaches in solid tumor oncology involve the use of immune checkpoint inhibitors to increase immune recognition of malignant cells. Several lines of evidence point to increased efficacy of these strategies in malignant clones with high levels of genomic instability [98]. These genetically unstable malignant clones create higher levels of neo-antigens which can stimulate immune recognition of the malignant clone [99]. Immune checkpoint inhibitor therapy is now being introduced into clinical trials in acute leukemias and one might hypothesize that leukemia with high level genomic instability such as FLT3 ITD and BCR/ABL associated leukemias would have the greatest biologic rationale for potential therapeutic benefit.
Conclusions
Genomic instability represents a mechanism many cancers use to evade chemotherapeutics and immune recognition. FLT3 ITD associated genomic instability may represent an opportunity for development of therapy which is selective for clonal disease rather than conventional therapeutics which target cells with normal DNA damage response. The potential utility of including strategies that target aberrant DNA repair warrants consideration in light of their success in BRCA associated solid tumors. Finally, novel applications of immune checkpoint inhibitor therapy in this specific leukemic subtype holds promise based on observations of increased activity in solid tumors associated with genomic instability.
Acknowledgments
Financial support: KW Pratz’s effort on these studies was supported as a co-investigator for translational science team at Johns Hopkins (UM1 CA186691) and cancer center support grant (P30 CA006973)
Footnotes
Conflict of Interest: No conflicts of interest exist for any authors of this manuscript.
References
- 1.Moreno I, Martin G, Bolufer P, et al. Incidence and prognostic value of FLT3 internal tandem duplication and D835 mutations in acute myeloid leukemia. Haematologica. 2003;88:19–24. [PubMed] [Google Scholar]
- 2.Yanada M, Matsuo K, Suzuki T, et al. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations for acute myeloid leukemia: a meta-analysis. Leukemia. 2005;19:1345–9. doi: 10.1038/sj.leu.2403838. [DOI] [PubMed] [Google Scholar]
- 3.Pratz KW, Luger SM. Will FLT3 inhibitors fulfill their promise in acute meyloid leukemia? Curr Opin Hematol. 2014;21:72–8. doi: 10.1097/MOH.0000000000000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pratz KW, Levis M. How I treat FLT3-mutated AML. Blood. 2016 doi: 10.1182/blood-2016-09-693648. [Epub Nov 21, 2016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stone RM, Mandrekar S, Sanford BL, et al. The Multi-Kinase Inhibitor Midostaurin (M) Prolongs Survival Compared with Placebo (P) in Combination with Daunorubicin (D)/Cytarabine (C) Induction, High-Dose C Consolidation, and As Maintenance (maint) Therapy in Newly Diagnosed Acute Myeloid Leukemia (AML) Patients (pts) Age 18–60 with FLT3 Mutations: An International Prospective Randomized P-Controlled Double-Blind Trial (CALGB 10603/RATIFY [Alliance]) Blood (Annual Meeting Abstract) 2015;126:6. [Google Scholar]
- 6.Röllig C, Serve H, Hüttmann A, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. The Lancet Oncology. 2015;16:1691–9. doi: 10.1016/S1470-2045(15)00362-9. [DOI] [PubMed] [Google Scholar]
- 7.Pratz KW, Gojo I, Karp JE, et al. Prospective Study of Peri-Transplant Use of Sorafenib As Remission Maintenance for FLT3-ITD Patients Undergoing Allogeneic Transplantation. Blood (Annual Meeting Abstract) 2015;126:3164. [Google Scholar]
- 8.Levis M, Ravandi F, Wang ES, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011;117:3294–301. doi: 10.1182/blood-2010-08-301796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pikor L, Thu K, Vucic E, et al. The detection and implication of genome instability in cancer. Cancer Metastasis Reviews. 2013;32:341–52. doi: 10.1007/s10555-013-9429-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer genes. Nature. 2013;499:214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer letters. 2008;270:1–9. doi: 10.1016/j.canlet.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 12.Perrotti D, Jamieson C, Goldman J, et al. Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest. 120:2254–64. doi: 10.1172/JCI41246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skorski T. Chronic myeloid leukemia cells refractory/resistant to tyrosine kinase inhibitors are genetically unstable and may cause relapse and malignant progression to the terminal disease state. Leuk & Lymphoma. 2011;52:23–9. doi: 10.3109/10428194.2010.546912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skorski T. Genomic instability: The cause and effect of BCR/ABL tyrosine kinase. Curr Hematol Malig Rep. 2007;2:69–74. doi: 10.1007/s11899-007-0010-6. [DOI] [PubMed] [Google Scholar]
- 15.Slupianek A, Nowicki MO, Koptyra M, et al. BCR/ABL modifies the kinetics and fidelity of DNA double-strand breaks repair in hematopoietic cells. DNA repair. 2006;5:243–50. doi: 10.1016/j.dnarep.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nowicki MO, Falinski R, Koptyra M, et al. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood. 2004;104:3746–53. doi: 10.1182/blood-2004-05-1941. [DOI] [PubMed] [Google Scholar]
- 17.Slupianek A, Dasgupta Y, Ren SY, et al. Targeting RAD51 phosphotyrosine-315 to prevent unfaithful recombination repair in BCR-ABL1 leukemia. Blood. 2011;118:1062–8. doi: 10.1182/blood-2010-09-307256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deutsch E, Jarrousse S, Buet D, et al. Down-regulation of BRCA1 in BCR-ABL-expressing hematopoietic cells. Blood. 2003;101:4583–8. doi: 10.1182/blood-2002-10-3011. [DOI] [PubMed] [Google Scholar]
- 19.Cramer K, Nieborowska-Skorska M, Koptyra M, et al. BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair. Cancer Res. 2008;68:6884–8. doi: 10.1158/0008-5472.CAN-08-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valeri A, Alonso-Ferrero ME, Rio P, et al. Bcr/Abl interferes with the Fanconi anemia/BRCA pathway: implications in the chromosomal instability of chronic myeloid leukemia cells. PLoS One. 2010;5:e15525. doi: 10.1371/journal.pone.0015525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bixby D, Talpaz M. Mechanisms of resistance to tyrosine kinase inhibitors in chronic myeloid leukemia and recent therapeutic strategies to overcome resistance. ASH Education Program Book. 2009;2009:461–76. doi: 10.1182/asheducation-2009.1.461. [DOI] [PubMed] [Google Scholar]
- 22.Koptyra M, Falinski R, Nowicki MO, et al. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 2006;108:319–27. doi: 10.1182/blood-2005-07-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pratz KW, Koh BD, Patel AG, et al. Poly (ADP-Ribose) Polymerase Inhibitor Hypersensitivity in Aggressive Myeloproliferative Neoplasms. Clin Cancer Res. 2016;22:3894–902. doi: 10.1158/1078-0432.CCR-15-2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 25.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–97. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 26.Plo I, Nakatake M, Malivert L, et al. JAK2 stimulates homologous recombination and genetic instability: potential implication in the heterogeneity of myeloproliferative disorders. Blood. 2008;112:1402–12. doi: 10.1182/blood-2008-01-134114. [DOI] [PubMed] [Google Scholar]
- 27.Nakatake M, Monte-Mor B, Debili N, et al. JAK2(V617F) negatively regulates p53 stabilization by enhancing MDM2 via La expression in myeloproliferative neoplasms. Oncogene. 2012;31:1323–33. doi: 10.1038/onc.2011.313. [DOI] [PubMed] [Google Scholar]
- 28.Slupianek A, Hoser G, Majsterek I, et al. Fusion tyrosine kinases induce drug resistance by stimulation of homology-dependent recombination repair, prolongation of G(2)/M phase, and protection from apoptosis. Molecular and cellular biology. 2002;22:4189–201. doi: 10.1128/MCB.22.12.4189-4201.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan J, Li L, Small D, et al. Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: implications for genomic instability and therapy. Blood. 2010;116:5298–305. doi: 10.1182/blood-2010-03-272591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cramer-Morales K, Nieborowska-Skorska M, Scheibner K, et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood. 2013;122:1293–304. doi: 10.1182/blood-2013-05-501072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tobin LA, Robert C, Rapoport AP, et al. Targeting abnormal DNA double-strand break repair in tyrosine kinase inhibitor-resistant chronic myeloid leukemias. Oncogene. 2013;32:1784–93. doi: 10.1038/onc.2012.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pratz KW, Rudek MA, Gojo I, et al. A Phase I study of topotecan, carboplatin and the PARP inhibitor veliparib in acute leukemias, aggressive myeloproliferative neoplasms and chronic myelomonocytic leukemia. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levis M, Ravandi F, Wang E, et al. Results From a Randomized Trial of Salvage Chemotherapy Followed by Lestaurtinib for FLT3 Mutant AML Patients in First Relapse. Blood (Annual Meeting Abstracts) 2009:114. doi: 10.1182/blood-2010-08-301796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gourdin TS, Zou Y, Ning Y, et al. High frequency of rare structural chromosome abnormalities at relapse of cytogenetically normal acute myeloid leukemia with FLT3 internal tandem duplication. Cancer genetics. 2014;207:467–73. doi: 10.1016/j.cancergen.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCormick SR, McCormick MJ, Grutkoski PS, et al. FLT3 mutations at diagnosis and relapse in acute myeloid leukemia: cytogenetic and pathologic correlations, including cuplike blast morphology. Arch Pathol Lab Med. 2010;134:1143–51. doi: 10.5858/2009-0292-OA.1. [DOI] [PubMed] [Google Scholar]
- 36.Rebechi M, Hand W, Smith BD, et al. Structural Chromosomal Changes Are Common Manifestation of FLT3 ITD Relapse and Presence of Chromosomal Progression Is Independent of Normal Karyotype at Diagnosis. Blood(Annual Meeting Abstract) 2016;128:2868. [Google Scholar]
- 37.Poletto M, Lirussi L, Wilson DM, 3rd, et al. Nucleophosmin modulates stability, activity, and nucleolar accumulation of base excision repair proteins. Mol Biol Cell. 2014;25:1641–52. doi: 10.1091/mbc.E13-12-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou F, Shen Q, Claret FX. Novel roles of reactive oxygen species in the pathogenesis of acute myeloid leukemia. Journal of leukocyte biology. 2013;94:423–9. doi: 10.1189/jlb.0113006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hole PS, Darley RL, Tonks A. Do reactive oxygen species play a role in myeloid leukemias? Blood. 2011;117:5816–26. doi: 10.1182/blood-2011-01-326025. [DOI] [PubMed] [Google Scholar]
- 40.Sattler M, Verma S, Shrikhande G, et al. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J Biol Chem. 2000;275:24273–8. doi: 10.1074/jbc.M002094200. [DOI] [PubMed] [Google Scholar]
- 41.Landry WD, Woolley JF, Cotter TG. Imatinib and Nilotinib inhibit Bcr-Abl-induced ROS through targeted degradation of the NADPH oxidase subunit p22phox. Leuk Res. 2013;37:183–9. doi: 10.1016/j.leukres.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 42.Kim JH, Chu SC, Gramlich JL, et al. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood. 2005;105:1717–23. doi: 10.1182/blood-2004-03-0849. [DOI] [PubMed] [Google Scholar]
- 43.Sallmyr A, Fan J, Datta K, et al. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood. 2008;111:3173–82. doi: 10.1182/blood-2007-05-092510. [DOI] [PubMed] [Google Scholar]
- 44.Stanicka J, Russell EG, Woolley JF, et al. NADPH oxidase-generated hydrogen peroxide induces DNA damage in mutant FLT3-expressing leukemia cells. J Biol Chem. 2015;290:9348–61. doi: 10.1074/jbc.M113.510495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Hagan HM, Wang W, Sen S, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell. 2011;20:606–19. doi: 10.1016/j.ccr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valavanidis A, Vlachogianni T, Fiotakis K, et al. Pulmonary Oxidative Stress, Inflammation and Cancer: Respirable Particulate Matter, Fibrous Dusts and Ozone as Major Causes of Lung Carcinogenesis through Reactive Oxygen Species Mechanisms. Int J Environ Res Public Health. 2013;10:3886–907. doi: 10.3390/ijerph10093886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Godfrey R, Arora D, Bauer R, et al. Cell transformation by FLT3 ITD in acute myeloid leukemia involves oxidative inactivation of the tumor suppressor protein-tyrosine phosphatase DEP-1/PTPRJ. Blood. 2012;119:4499–511. doi: 10.1182/blood-2011-02-336446. [DOI] [PubMed] [Google Scholar]
- 48.Arora D, Stopp S, Bohmer SA, et al. Protein-tyrosine phosphatase DEP-1 controls receptor tyrosine kinase FLT3 signaling. J Biol Chem. 2011;286:10918–29. doi: 10.1074/jbc.M110.205021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burma S, Chen BP, Murphy M, et al. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 50.Seedhouse CH, Hunter HM, Lloyd-Lewis B, et al. DNA repair contributes to the drug-resistant phenotype of primary acute myeloid leukaemia cells with FLT3 internal tandem duplications and is reversed by the FLT3 inhibitor PKC412. Leukemia. 2006;20:2130–6. doi: 10.1038/sj.leu.2404439. [DOI] [PubMed] [Google Scholar]
- 51.Mackarehtschian K, Hardin JD, Moore KA, et al. Targeted disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic progenitors. Immunity. 1995;3:147–61. doi: 10.1016/1074-7613(95)90167-1. [DOI] [PubMed] [Google Scholar]
- 52.McKenna HJ, Stocking KL, Miller RE, et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood. 2000;95:3489–97. [PubMed] [Google Scholar]
- 53.Li L, Piloto O, Nguyen HB, et al. Knock-in of an internal tandem duplication mutation into murine FLT3 confers myeloproliferative disease in a mouse model. Blood. 2008;111:3849–58. doi: 10.1182/blood-2007-08-109942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li L, Zhang L, Fan J, et al. Defective nonhomologous end joining blocks B-cell development in FLT3/ITD mice. Blood. 2011;117:3131–9. doi: 10.1182/blood-2010-05-286070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jankovic M, Nussenzweig A, Nussenzweig MC. Antigen receptor diversification and chromosome translocations. Nat Immunol. 2007;8:801–8. doi: 10.1038/ni1498. [DOI] [PubMed] [Google Scholar]
- 56.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Allen C, Halbrook J, Nickoloff JA. Interactive competition between homologous recombination and non-homologous end joining. Mol Cancer Res. 2003;1:913–20. [PubMed] [Google Scholar]
- 58.Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4/ligase IV during chromosomal translocation formation. Nat Struct Mol Bio. 2010;17:410–6. doi: 10.1038/nsmb.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Darling DC, Mufti GJ. Internal Tandem Duplication Mutation Of FLT3 (FLT3/ITD) Induces Increased Homologous Recombination DNA Repair Activity, Drug Resistance and Sister Chromatid Exchanges In Acute Myeloid Leukaemia (AML) Blood. 2013;122:1244. [Google Scholar]
- 60.Li L, Bailey E, Greenblatt S, et al. Loss of the wild-type allele contributes to myeloid expansion and disease aggressiveness in FLT3/ITD knockin mice. Blood. 2011;118:4935–45. doi: 10.1182/blood-2011-01-328096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stirewalt DL, Pogosova-Agadjanyan EL, Tsuchiya K, et al. Copy-neutral loss of heterozygosity is prevalent and a late event in the pathogenesis of FLT3/ITD AML. Blood Cancer J. 2014;4:e208. doi: 10.1038/bcj.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stark JM, Pierce AJ, Oh J, et al. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Molecular and cellular biology. 2004;24:9305–16. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fernandes MS, Reddy MM, Gonneville JR, et al. BCR-ABL promotes the frequency of mutagenic single-strand annealing DNA repair. Blood. 2009;114:1813–9. doi: 10.1182/blood-2008-07-172148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Canitrot Y, Falinski R, Louat T, et al. p210 BCR/ABL kinase regulates nucleotide excision repair (NER) and resistance to UV radiation. Blood. 2003;102:2632–7. doi: 10.1182/blood-2002-10-3207. [DOI] [PubMed] [Google Scholar]
- 65.Stoklosa T, Poplawski T, Koptyra M, et al. BCR/ABL inhibits mismatch repair to protect from apoptosis and induce point mutations. Cancer Res. 2008;68:2576–80. doi: 10.1158/0008-5472.CAN-07-6858. [DOI] [PubMed] [Google Scholar]
- 66.Nomdedeu JF, Perea G, Estivill C, et al. Microsatellite instability is not an uncommon finding in adult de novo acute myeloid leukemia. Ann Hematol. 2005;84:368–75. doi: 10.1007/s00277-005-1035-3. [DOI] [PubMed] [Google Scholar]
- 67.Das-Gupta EP, Seedhouse CH, Russell NH. Microsatellite instability occurs in defined subsets of patients with acute myeloblastic leukaemia. Br J Haematol. 2001;114:307–12. doi: 10.1046/j.1365-2141.2001.02920.x. [DOI] [PubMed] [Google Scholar]
- 68.Gaymes TJ, Mohamedali AM, Patterson M, et al. Microsatellite instability induced mutations in DNA repair genes CtIP and MRE11 confer hypersensitivity to poly (ADP-ribose) polymerase inhibitors in myeloid malignancies. Haematologica. 2013;98:1397–406. doi: 10.3324/haematol.2012.079251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brown P, Levis M, Shurtleff S, et al. FLT3 inhibition selectively kills childhood acute lymphoblastic leukemia cells with high levels of FLT3 expression. Blood. 2005;105:812–20. doi: 10.1182/blood-2004-06-2498. [DOI] [PubMed] [Google Scholar]
- 70.Lin EI, Tseng LH, Gocke CD, et al. Mutational profiling of colorectal cancers with microsatellite instability. Oncotarget. 2015;6:42334–44. doi: 10.18632/oncotarget.5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.De Lange T. Telomere-related genome instability in cancer. Cold Spring Harbor symposia on quantitative biology. 2005;70:197–204. doi: 10.1101/sqb.2005.70.032. [DOI] [PubMed] [Google Scholar]
- 72.Aalbers AM, Calado RT, Young NS, et al. Telomere length and telomerase complex mutations in pediatric acute myeloid leukemia. Leukemia. 2013;27:1786–9. doi: 10.1038/leu.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Watts JM, Dumitriu B, Hilden P, et al. Telomere Length Is Associated with Specific Mutations and Mutation Classes in Patients with Acute Myeloid Leukemia. Blood. 2014;124:2280. [Google Scholar]
- 74.Swiggers SJ, Kuijpers MA, de Cort MJ, et al. Critically short telomeres in acute myeloid leukemia with loss or gain of parts of chromosomes. Genes, chromosomes & cancer. 2006;45:247–56. doi: 10.1002/gcc.20286. [DOI] [PubMed] [Google Scholar]
- 75.Hartmann U, Brummendorf TH, Balabanov S, et al. Telomere length and hTERT expression in patients with acute myeloid leukemia correlates with chromosomal abnormalities. Haematologica. 2005;90:307–16. [PubMed] [Google Scholar]
- 76.Okamoto K, Bartocci C, Ouzounov I, et al. A two-step mechanism for TRF2-mediated chromosome-end protection. Nature. 2013;494:502–5. doi: 10.1038/nature11873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Garg M, Nagata Y, Kanojia D, et al. Profiling of somatic mutations in acute myeloid leukemia with FLT3-ITD at diagnosis and relapse. Blood. 2015;126:2491–501. doi: 10.1182/blood-2015-05-646240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–10. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pratz K, Levis M. Incorporating FLT3 inhibitors into acute myeloid leukemia treatment regimens. Leuk Lymphoma. 2008;49:852–63. doi: 10.1080/10428190801895352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weisberg E, Barrett R, Liu Q, et al. FLT3 inhibition and mechanisms of drug resistance in mutant FLT3-positive AML. Drug Resist Updat. 2009;12:81–9. doi: 10.1016/j.drup.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wander SA, Levis MJ, Fathi AT. The evolving role of FLT3 inhibitors in acute myeloid leukemia: quizartinib and beyond. Ther Adv Hematol. 2014;5:65–77. doi: 10.1177/2040620714532123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Galanis A, Ma H, Rajkhowa T, et al. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood. 2014;123:94–100. doi: 10.1182/blood-2013-10-529313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zimmerman EI, Turner DC, Buaboonnam J, et al. Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood. 2013 doi: 10.1182/blood-2013-07-513044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mori M, Kaneko N, Ueno Y, et al. ASP2215, a novel FLT3/AXL inhibitor: Preclinical evaluation in acute myeloid leukemia (AML) ASCO Meeting Abstracts. 2014;32:7070. [Google Scholar]
- 86.Ueno Y, Kaneko N, Saito R, et al. ASP2215, a novel FLT3/AXL inhibitor: Preclinical evaluation in combination with cytarabine and anthracycline in acute myeloid leukemia (AML) ASCO Meeting Abstracts. 2014;32:7071. [Google Scholar]
- 87.Zhang W, Gao C, Konopleva M, et al. Reversal of acquired drug resistance in FLT3-mutated acute myeloid leukemia cells via distinct drug combination strategies. Clin Cancer Res. 2014;20:2363–74. doi: 10.1158/1078-0432.CCR-13-2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Naidoo J, Drilon A. KRAS-Mutant Lung Cancers in the Era of Targeted Therapy. Adv Exp Med Biol. 2016;893:155–78. doi: 10.1007/978-3-319-24223-1_8. [DOI] [PubMed] [Google Scholar]
- 89.Larkin J, Ascierto PA, Dreno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867–76. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 90.Meyn RE, Munshi A, Haymach JV, et al. Receptor Signaling as a Regulatory Mechanism of DNA Repair. Radiother Oncol. 2009;92:316–22. doi: 10.1016/j.radonc.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kurosu T, Nagao T, Wu N, et al. Inhibition of the PI3K/Akt/GSK3 pathway downstream of BCR/ABL, Jak2-V617F, or FLT3-ITD downregulates DNA damage-induced Chk1 activation as well as G2/M arrest and prominently enhances induction of apoptosis. PLoS One. 2013;8:e79478. doi: 10.1371/journal.pone.0079478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chang L, Graham PH, Hao J, et al. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014;5:e1437. doi: 10.1038/cddis.2014.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cardnell RJ, Feng Y, Diao L, et al. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin Cancer Res. 2013;19:6322–8. doi: 10.1158/1078-0432.CCR-13-1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kirkpatrick DS, Bustos DJ, Dogan T, et al. Phosphoproteomic characterization of DNA damage response in melanoma cells following MEK/PI3K dual inhibition. Proc Natl Acad Sci U S A. 2013;110:19426–31. doi: 10.1073/pnas.1309473110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rouleau M, Patel A, Hendzel MJ, et al. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gojo I, Beumer JH, Pratz KW, et al. A phase 1 study of the PARP inhibitor veliparib in combination with temozolomide in acute myeloid leukemia. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wray J, Williamson EA, Singh SB, et al. PARP1 is required for chromosomal translocations. Blood. 2013;121:4359–65. doi: 10.1182/blood-2012-10-460527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yaghmour G, Pandey M, Ireland C, et al. Role of Genomic Instability in Immunotherapy with Checkpoint Inhibitors. Anticancer Res. 2016;36:4033–8. [PubMed] [Google Scholar]
- 99.Lu YC, Robbins PF. Cancer immunotherapy targeting neoantigens. Semin Immunol. 2016;28:22–7. doi: 10.1016/j.smim.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Blanpain C, Mohrin M, Sotiropoulou PA, et al. DNA-Damage Response in Tissue-Specific and Cancer Stem Cells. Cell Stem Cell. 2011;8:16–29. doi: 10.1016/j.stem.2010.12.012. [DOI] [PubMed] [Google Scholar]