

Abstract
Experimental in vivo tumor models are essential for comprehending the dynamic process of human cancer progression, identifying therapeutic targets, and evaluating antitumor drugs. However, current rodent models are limited by high costs, long experimental duration, variability, restricted accessibility to the tumor, and major ethical concerns. To avoid these shortcomings, we investigated whether tumor growth on the chick chorio-allantoic membrane after human glioblastoma cell grafting would replicate characteristics of the human disease. Avascular tumors consistently formed within 2 days, then progressed through vascular endothelial growth factor receptor 2-dependent angiogenesis, associated with hemorrhage, necrosis, and peritumoral edema. Blocking of vascular endothelial growth factor receptor 2 and platelet-derived growth factor receptor signaling pathways by using small-molecule receptor tyrosine kinase inhibitors abrogated tumor development. Gene regulation during the angiogenic switch was analyzed by oligonucleotide microarrays. Defined sample selection for gene profiling permitted identification of regulated genes whose functions are associated mainly with tumor vascularization and growth. Furthermore, expression of known tumor progression genes identified in the screen (IL-6 and cysteine-rich angiogenic inducer 61) as well as potential regulators (lumican and F-box-only 6) follow similar patterns in patient glioma. The model reliably simulates key features of human glioma growth in a few days and thus could considerably increase the speed and efficacy of research on human tumor progression and preclinical drug screening.
Keywords: angiogenesis, animal model alternatives, glioblastoma
Glioblastoma multiforme (GBM) brain tumors are among the most lethal human cancers with a mean survival time of only several months (1). Existing in vivo GBM models are based on inoculation of glioma cells into rodent brains (2) or the use of transgenic mice with modified expression of tumor suppressors or oncogenes in glial cell lineages (3, 4). However, these models suffer from generally poor penetrance or variable growth rate, a major obstacle for collecting clearly graded (prevascular/vascularized) tumor samples exploitable in gene profiling experiments. Rodent models are further limited by the difficulty of obtaining morphological data during tumor progression, resulting in a high number of animals required to obtain conclusive results, paralleled by an elevated level of animal suffering. The latter fact has been increasingly discussed and severely questioned by public opinion (5). A reproducible and fast in vivo GBM model, in which both morphological and gene regulation steps of human GBM can be studied, would greatly facilitate discovery and validation of new therapeutic targets. Ideally, such a model would incorporate the widely accepted guidelines aimed at reduction of animal number and suffering, as well as the refinement and replacement of models (rule of the 3Rs) (6). For decades, the avian embryo has been a model organism of choice in developmental biology, whereas the field of basic cancer research has focused mainly on mouse models because of the potential given by genetic engineering. The complete chick genome has now been sequenced (www.nhgri.nih.gov/11510730), new techniques allowing gene function studies in chick embryos are emerging (7, 8), and a vascular marker for normal and angiogenic chick capillaries has been identified (9). Anticipating the impact of these advances, we established a robust and highly reproducible GBM progression model on the chorio-allantoic membrane (CAM), a densely vascularized, extraembryonic tissue. This model allows fast and precise analysis of the principal steps of human GBM progression and response to comprehensive treatment with new anticancer drugs. Furthermore, we provide evidence that gene regulation in the course of this short-term assay predicts gene expression in patient GBMs.
Materials and Methods
Cells and Embryos. U87 human glioma cells (American Type Culture Collection) were maintained in DMEM with 10% FBS, antibiotics, and l-glutamine. Fertilized chicken eggs (Gallus gallus) (EARL Morizeau, Dangers, France) were handled as described (10).
Experimental Glioma Assay. On embryonic day 10, a plastic ring was placed on the CAM (10), and 3 million to 5 million U87 cells in 20 μl of medium were deposited after gentle laceration of surface. Digital photos were taken under a stereomicroscope (Nikon SMZ800). When required, tumor size-matching was done based on tumor volume calculation: V = 4/3πr3, with 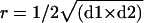 . For edema quantification, experimental glioma at day 2 (D2) or control CAMs, which received cell-free medium, were fixed in vivo with 4% paraformaldehyde and spread out on transparent plastic tubes. CAM surfaces were measured on cross sections, and statistical comparison between the two groups was performed by using the Student's t test.
. For edema quantification, experimental glioma at day 2 (D2) or control CAMs, which received cell-free medium, were fixed in vivo with 4% paraformaldehyde and spread out on transparent plastic tubes. CAM surfaces were measured on cross sections, and statistical comparison between the two groups was performed by using the Student's t test.
In Situ Hybridization, Histology, and Immunohistochemistry. In situ hybridization with a vascular endothelial growth factor receptor 2 (VEGFR-2) riboprobe was performed as described (11). For histology, tumors were cut into 10- to 20-μm cryosections and stained with hematoxylin-erythrosin-safran. Primary antibodies used were anti-human vimentin (clone V9, NeoMarkers, Fremont, CA, 1:200 or AB1620, Chemicon, 1:40), rabbit anti-human desmin (clone D33, DAKO, 1:500), mouse anti-chick tenascin C (TN-C, clone M1B4, 1:800), mouse anti-human TN-C [clone B2–813, 1:800 (12)], and mouse anti cytochrome c (clone 6H2.B4, Pharmingen, 1:200). Corresponding secondary antibodies were from Molecular Probes (Alexa Fluor 488, 546, or 647; 1:2,000). Chick blood vessels were visualized by using SNA-1 lectin coupled to FITC (1:200, Vector Laboratories). Fluorescent labeling was viewed by confocal microscopy (Olympus FluoView 500).
Treatment of Experimental Glioma with Receptor Tyrosine Kinase Inhibitors. Gleevec (Imatinib mesylate) and PTK787/ZK 222584 were provided by Novartis (Basel). On D2, size-matched tumors were divided into control and treatment groups. Drugs were dissolved according to the manufacturers' recommendations and deposited locally at 50 μg per egg per day. Controls received solvent of the corresponding drug. Quantification of drug effects on pericyte, tumor cell, and vessel density were determined in five to seven representative tumors per group. The contrast of red-green-blue images of equal pixel size was adjusted to 100% in photoshop (Adobe Systems, San Jose, CA), and relative cell density was expressed as the percentage of pixels in the corresponding color channel per total number of pixels. Blood vessels were counted manually. Statistical analysis of control vs. Gleevec- or PTK787/ZK 222584-treated tumors was carried out by using the Mann–Whitney U test.
Gene Expression Profiling. RNA was extracted from pools of 10 size-matched tumors at D2 and D4 by using Qiagen (Valencia, CA) RNeasy columns. Five micrograms of RNA was reverse-transcribed (cDNA Synthesis System Kit, Roche Diagnostics). The purified cDNA was transcribed in the presence of Cy3-UTP or Cy5-UTP (Megascript D7 kit, Ambion, Austin, TX). cRNAs were purified (High Pure RNA Tissue Kit Columns, Roche Diagnostics), fragmented by heating, mixed (1:1), and hybridized in duplicates in 35-μl volume on array slides (9,850 genes, Human 30k Array A, MWG Biotech, Ebersberg, Germany). Scanning and spot analysis were done according to the manufacturer's guidelines (GMS 428 scanner and jaguar 2.0 software, Affymetrix, Santa Clara, CA). Only spots with reproducible >1.5-fold change were considered. A global mean normalization was applied to the data, and fold changes were expressed as a mean value ± SEM from three independent hybridizations.
Real-Time Quantitative PCR (qPCR). Experimental glioma and biopsies [astrocytoma (AS) II and GBMs] from patients who gave their informed consent (Centre Hospitalier Universitaire, Bordeaux) were snap-frozen. RNA was purified by using RNeasy columns (Qiagen) and reversed-transcribed with SuperScript II RNase H-Reverse Transcriptase (Invitrogen) by using oligo(dT)15 priming.
Human-specific primers for qPCR were designed and evaluated for amplification efficiency. Primer sequences were: α-tubulin (NM_006082), 5′-GAGTGCATCTCCATCCACGTT-3′, 5′-TAGAGCTCCCAGCAGGCATT-3′; IL-6, 5′-CCAGGAGCCCAGCTATGAAC-3′, 5′-CCCAGGGAGAAGGCAACTG-3′; cysteine-rich angiogenic inducer 61 (CYR61), 5′-TGAGGAGCATTAAGGTATTTCGAA-3′, 5′-CGTGGCTGCATTAGTGTCCAT-3′; lumican (LUM), 5′-GATGGCAATCGCATCTCAGA-3′,5′-TCGTTAGCAACACGTAGACATTCA-3′; and F-box-only 6(FBXO6), 5′-TCCTACG-GAGCCTGCATAGG-3′,5′-GCCATGCAAACATATCCTCTTCA-3′. Real-time PCR was carried out in a Mx3000P thermocycler (Stratagene) by using SYBR Green dye (ABgene, Epsom, U.K.). All samples were tested in a minimum of two independent experiments.
To confirm microarray data in experimental glioma, relative changes of expression were expressed for each target gene after normalization by the tubulin signal, according to the formula: 2ΔΔCt with ΔCt = |Cttubulin – Cttarget|, and ΔΔCt = (ΔCtD4 – ΔCtD2). Expression of selected genes in patients with GBM was compared with a pool of ASII by using ΔΔCt = (ΔCtGBM – ΔCtASII), with ΔCtASII calculated from the average of four to five ASIIs. The presence of a single PCR product was verified by melting point analysis. After the exclusion of the outliers, means of three qPCR experiments were used for plot generation (IL-6, n = 5 patients and FBXO6, n = 9 patients). For LUM (n = 12 patients) and CYR61 (n = 12 patients), values from one representative qPCR experiment were plotted, after statistical confirmation of the reproducibility between experiment replicates (R2 > 0.99).
Results
Experimental Glioma Recapitulate Hallmarks of Human GBM. U87 glioma cells deposited on the CAM form a solid, avascular tumor within the first day. Vascularization of the tumor steadily progresses as evidenced by biomicroscopy in vivo (Fig. 1a). At high magnification, tortuous and dilated vessels at the tumor surface were clearly visible (Fig. 1b), and blood flow could be observed (data not shown), providing direct evidence for functional tumor capillaries. Histological analysis confirms that nucleated chick erythrocytes are present in tumor capillaries (Fig. 1d).
Fig. 1.

Growth characteristics of experimental glioma on the CAM. (a) During the first 2 days after implantation, a well defined, solid tumor forms, which progressively becomes vascularized the following days. (b) Tortuous angiogenic tumor capillaries are visible at the surface of a D7 tumor. (c) Cross section of paraformaldehyde-fixed glioma in situ with adjacent CAM at D7. Areas of the tumor periphery and the center (rectangles) at higher magnification show angiogenesis (Ang), necrosis (Nec), and hemorrhage (Hem). (d) Histological analysis of D7 glioma reveals the presence of nucleated chick erythrocytes in tumor capillaries. (e) Glioma growth on the CAM is paralleled by the development of a large peritumoral edema (arrows, asterisk denotes tumor). CAMs close to the tumor are six times more swollen than controls. [Magnifications: a, ×10 (bar, 1 mm); b and c, ×63; d, ×100; and e, ×2.]
Coexistence of angiogenesis, necrosis, and hemorrhage is pathognomonic for human GBM. Transverse sections of 7-day-old tumors showed a highly heterogeneous composition: areas of dense vascularization were interspersed with white necrotic foci and hemorrhagic spots (Fig. 1c).
Glioma growth on the CAM also was accompanied by a large peritumoral edema. As early as D2, the CAM adjacent to the tumor implantation site was visibly swollen, about six times more than controls (6.5 ± 0.8 mm2 vs. 1.1 ± 0.2 mm2; n = 6 CAMs per group; P < 0.001; t = 6.813; df = 10) (Fig. 1e).
We next investigated whether blood vessels inside the tumor were formed through angiogenesis, a requirement for human tumor progression (13). Expression of VEGFR-2 is up-regulated in immature, new blood vessels during glioma development (14). From D2 on, capillaries invading the growing glioma were positive for transcripts of VEGFR-2, as revealed by in situ hybridization (n = 4/4 tumors) (Fig. 2 a–c). At higher magnification, VEGFR-2 mRNA could specifically be detected in capillary endothelial cells (Fig. 2 d–f). Vessels in the surrounding CAM were negative for VEGFR-2. By D4, the whole tumor was invaded by angiogenic, VEGFR-2-positive capillaries (n = 2/2 tumors) (Fig. 2b). At D7, VEGFR-2 signal was still present in capillaries throughout the tumor (n = 4/4 tumors).
Fig. 2.

Expression of VEGFR-2 during experimental glioma development. (a) During the second day after implantation, the tumor becomes vascularized from the base by CAM vessels positive for the transcript of VEGFR-2. (b) ByD4, the tumor is almost completely invaded by VEGFR-2-positive capillaries. (c) At D7, overall VEGFR-2 reactivity decreases. (d–f). Higher magnification shows endothelial cells positive for VEGFR-2 (arrows). (Scale bars: a–c, 0.5 mm; and d–f, 0.1 mm.)
Experimental glioma at D7 were further analyzed by immunohistochemistry and confocal microscopy. SNA-1 staining confirmed the presence of dilated tumor capillaries at the periphery of the tumor (Fig. 3a). Most angiogenic tumor capillaries were covered by pericytes. Coverage was heterogeneous and some desmin-positive “hot spots” were observed, which correspond to unregulated capillary growth areas resembling glomeruloid microvascular proliferations, which are typical of GBM (Fig. 3b). Two other mechanisms contributing to tumor vascularization were further evidenced in the course of experimental glioma progression: intussusceptive microvascular growth (15) (Fig. 3c) and vessel cooption (16). Invading glioma cells grow around normal CAM vessels, which subsequently become part of the tumor vasculature (Fig. 3 d and e).
Fig. 3.

Immunohistological and confocal microscopy analysis of experimental glioma. (a) Frozen sections of D7 tumors were stained with antivimentin antibody (tumor cells, T), antidesmin antibody (pericytes, P), and fluoresceincoupled SNA-1 lectin (endothelial cells, E). Note the high tumor cell density, the irregular and dilated vessels (arrows), and pericyte coating of most capillaries. (b) Tumor capillaries form glomeruloid-like microvascular proliferations, covered by pericytes. (c) The presence of intercapillary pillars (arrows) in tumor capillaries shows intussusceptive angiogenesis. (d) Cooption of larger, dilated CAM blood vessels at the base of the tumor. (e) Flexion and orientation of a coopted vessel toward the tumor. (f) The presence of TN-C in the extracellular matrix of glioma identified by species-specific mAbs. (g) Lowpower magnification illustrates the invasive behavior of the tumor (arrows mark the borders of the CAM). (h) Individual glioma cells invade the CAM adjacent to the implantation site and migrate along blood vessels. (i) Experimental glioma do not exhibit apoptosis as revealed by physiological distribution of cytochrome c around cell nuclei (arrows). (Scale bars: a, d, and e, 100 μm; b and f, 50 μm; c, 20 μm; and i, 10 μm.)
The extracellular matrix in human glioma is rich in the promigratory glycoprotein TN-C (17), and TN-C expression has been associated with a poor clinical outcome (18). We detected abundant immunoreactivity for TN-C of human and chick origin in the extracellular matrix of the tumor by using species-specific mAbs (Fig. 3f).
Glioma cell invasion into the CAM was clearly visible on immuno-stained sections. Blood vessels served as “guiding” structures for migration: numerous, elongated tumor cells were tightly wrapped around these vessels (Fig. 3 g and h).
Escape from apoptosis is typical of malignant tumors (13). We examined whether this important characteristic was preserved during experimental glioma growth. Permeabilization of mitochondrial membranes during apoptosis leads to the release of cytochrome c from the intermembrane space (19). Cytochrome c release together with nuclear fragmentation are reliable reporters of cells undergoing apoptosis. Almost all glioma cells inside tumors throughout development had intact nuclear morphology as evidenced by DAPI staining (data not shown) and showed a punctuate perinuclear distribution of cytochrome c (Fig. 3i), consistent with the absence of apoptosis.
Treatment of Experimental Glioma with Small-Molecule Receptor Tyrosine Kinase Inhibitors. We next tested whether selective blocking of growth factor receptors important for glioma progression, like VEGFR-2 (20) and platelet-derived growth factor receptors (21), could result in inhibition of experimental glioma.
Local treatment of size-matched glioma from D2 to D6 with single daily doses (50 μg per egg) of Gleevec, an antagonist of platelet-derived growth factor receptor function (22) or PTK787/ZK 222845 (23) markedly inhibited tumor growth as early as 2 days after the first application. Neither of the two molecules had visible effects on normal CAMs compared with solvent-treated controls, which displayed a very light fibrosis, but normal vascularization.
Regular vascularization, which consistently occurred in solvent-treated controls (n = 23), was never observed in Gleevec-treated tumors (n = 22), which appeared white because of a decrease in intratumoral blood flow and increased necrosis (Fig. 4a). Antivimentin reactivity was clearly decreased, confirming reduced tumor cell density (Fig. 4 b and c). Tumor vessel structure was markedly altered: dilated vessels, typical of control tumors, were not detected, and capillary lumina were irregular. Pericyte coating of angiogenic capillaries was reduced, as evidenced by a decrease in desmin immunoreactivity (Fig. 4c). Statistical analysis of Gleevec effects confirmed a significant decrease in relative tumor cell density (27.47 ± 5.28% vs. controls: 62.54 ± 6.64%; mean ± SEM), pericyte density (8.7 ± 1.87% vs. controls: 23.3 ± 4.84%), and significant reduction of angiogenic blood vessels (33.7 vessels ± 9.8 vs. controls: 88.3 vessels ± 3.6; P < 0.05 for all comparisons, Fig. 4e).
Fig. 4.

Topical treatment of experimental glioma with receptor tyrosine kinase inhibitors Gleevec and PTK787/ZK 222584. (a) Biomicroscopy follow-up of control and treated tumors. Control tumors show progressive growth and vascularization, whereas Gleevec- and PTK787/ZK 222584-treated tumors appear white and show signs of tissue damage. (b–d) Confocal microscopy analysis of triple-stained tumors at D7. Tumor cells (T) are blue, pericytes (P) are red, and the endothelium (E) is green. (b) Control tumors show densely packed tumor cells within a well established pericyte-covered vascular network. (c) Gleevec-treated tumors show a strong decrease in tumor cell, pericyte, and vessel density. The increase in green fluorescence is caused by nonspecific lectin binding to necrotic cells. (d) In PTK787/ZK 222584-treated tumors, only a few pericyte-covered capillaries are found after treatment, and almost all tumor cells have disappeared. As in Gleevec-treated tumors, nonspecific green fluorescence increases. (e) Quantification of receptor tyrosine kinase inhibitor effects on glioma development. Bars indicate means of relative cell density and vessels counts. (Magnification: a, ×10; scale bar: b–d, 100 μm.)
Treatment of experimental glioma with PTK787/ZK 222584 also abrogated progressive vascularization, as evidenced by in vivo biomicroscopy (n = 8 tumors) (Fig. 4d). Confocal microscopy of triple-stained tumor sections revealed almost complete absence of vimentin-positive tumor cells and strong reduction of blood vessels. Only capillaries covered by pericytes in the tumor center were visible after PTK787/ZK 222584 treatment (Fig. 4a).
Statistical evaluation of PTK787/ZK 222584 treatment confirmed a significant decrease in relative tumor cell density (11.16 ± 9.44% vs. controls: 62.54 ± 6.64%; P < 0.05) and blood vessel number (10.2 vessels ± 5.34 vs. controls: 88.3 vessels ± 3.6; P < 0.05), but not pericyte density (13.86 ± 5.55% vs. controls: 23.3 ± 4.84%; not significant) (Fig. 4e).
Gene Expression Profiling During Experimental Glioma Development. Fifty-mer oligonucleotide microarrays encompassing one-fourth of the human genome were used to determine gene expression changes during the transition from early-stage tumors at D2 to fully vascularized tumors at D4. Twenty-one genes were differentially regulated (>1.5-fold) between D2 and D4. Noteworthy, 6 of 21 identified genes have established functions in human GBM progression (IL-6, IL-8, CYR61, MYC, MYBL2, NT5E, all up-regulated, Table 1 and Table 2, which is published as supporting information on the PNAS web site) or are produced by malignant glial cells (CEBPG). To verify whether the expression changes could specifically be assigned to the human tumor cells, oligonucleotide sequences corresponding to the 21 genes on the array were compared with the chick genome database by using blast. For 18 genes, no significant homology was found, so that hybridization of avian transcripts could be excluded. Only avian c-MYC, CEF-10 (homolog to CYR61), and very low density lipoprotein/vitellogenin receptor (homolog to very low density lipoprotein receptor) genes shared sufficient similarity (>75%) for hybridization (24), so that the contribution of chick mRNA to the microarray signal could not be excluded (Table 2). Four genes were studied in more detail. IL-6 and CYR61 have important functions during human GBM development (25–27). Two other genes, LUM and FBXO6, are promising, yet uncharacterized candidates that might have distinct roles in tumor progression (28, 29).
Table 1. Gene expression profiling of the angiogenic switch in experimental glioma by using MWG human 30K A oligonucleotide microarrays.
| GenBank accession no. | Gene | Expression levels, mean ± SEM |
|---|---|---|
| Angiogenesis | ||
| NM_000600 | IL-6 | 3.14 ± 0.37 |
| NM_000584 | IL-8 | 2.28 ± 0.25 |
| NM_001554 | CYR61 | 1.86 ± 0.15 |
| Cell growth control | ||
| NM_002467 | MYC/v-myc myelocytomatosis viral oncogene homolog (avian) | 2.37 ± 0.06 |
| NM_016447 | MPP6/maguk protein p55t; protein associated with lins 2 | 1.83 ± 0.18 |
| NM_002466 | MYBL2/v-myb myeloblastosis viral oncogene homolog (avian)-like 2 | 1.69 ± 0.04 |
| NM_002345 | LUM | 1.64 ± 0.06 |
| NM_002526 | NT5E/Ecto-5′ nucleotidase (CD73) | 1.59 ± 0.02 |
| NM_001806 | CEBPG/CCAAT enhancer binding protein gamma (C/EBP) | 1.55 ± 0.02 |
| NM_021145 | DMTF1/cyclin D binding myb-like transcription factor 1 | -2.23 ± 0.30 |
| NM_014553 | TFCP2L1/transcription factor CP2-like 1 (LBP-9; CRTR-1) | -1.95 ± 0.13 |
| NM_018438 | FBXO6 | -1.86 ± 0.20 |
| NM_002891 | RASGRF1/ras protein-specific guanine nucleotide-releasing factor 1 | -1.82 ± 0.16 |
| Neuronal differentiation | ||
| NM_003383 | VLDLR/very low density lipoprotein receptor | 1.67 ± 0.10 |
| Immune system | ||
| L17330 | 6H9A/pre-T/NK cell associated protein | -2.02 ± 0.18 |
| NM_001276 | CHI3L1/chitinase 3-like 1 (cartilage glycoprotein-39) | -1.92 ± 0.25 |
| NM_006187 | OAS3/2′-5′ oligoadenylate synthetase 3, 100 kDa | -1.76 ± 0.05 |
| Others | ||
| NM_020638 | FGF23/fibroblast growth factor 23 | -2.27 ± 0.22 |
| NM_000789 | ACE/angiotensin I converting enzyme | -1.84 ± 0.24 |
| NM_018671 | SMAP1/hypothetical protein; iro039700 | -1.75 ± 0.12 |
| NM_005588 | MEP1A/meprin a, alpha (PABA peptide hydrolase) | -1.61 ± 0.09 |
Twenty-one of 9,850 genes were found to be consistently regulated (>1.5-fold in three hybridization experiments) during the 48-h time lapse. Note that about one-third of regulated genes are known to be linked to glioma progression and angiogenesis, including IL-6, IL-8, and CYR61. Four selected genes that have been studied in more detail are marked in bold.
Differential expression levels of these four genes were determined by real-time qPCR in the same reverse-transcribed RNAs used for microarray hybridization, as well as in a second series of samples from an independent experimental glioma assay. Tumor cell-specific expression regulation between D2 and D4 was confirmed by using human-specific primers for all four genes in the two assays (LUM: 13.9/8-fold; CYR61: 4.5/2.3-fold; IL-6: 2.3/5.3-fold, and FBXO6: –1.5/–1.7-fold).
Gene Regulation in Experimental Glioma Predicts Expression Profiles in GBM Patients. World Health Organization grade II AS tumors have the potential to progress to GBM (secondary GBM). ASII tumors are poorly vascularized compared with highly angiogenic GBMs. Progression from ASII tumors to GBMs therefore might involve molecular mechanisms that are replicated in the course of experimental glioma. We compared expression levels of LUM, CYR61, IL-6, and FBXO6 between human GBM samples and ASII.
Expression levels of individual GBM samples were evaluated by qPCR for each of the four genes and normalized against the corresponding means calculated from the ASII pool (Fig. 5). All tumors tested for LUM showed up-regulation (12/12, median: 7.3), with a distribution ranging from 1.3- to 36.7-fold. Most GBMs showed CYR61 overexpression (8/12, median: 3.54), up to 42-fold. IL-6 was also up-regulated in all GBM samples (5/5, median: 11.55) and showed the highest relative transcription level, 143-fold. Down-regulation of FBXO6, as predicted by the model, was confirmed in seven of nine GBMs (median: 0.7), down to 0.18 compared with ASII tumors.
Fig. 5.

Expression levels of four selected genes identified in the microarray screen were tested in patient GBM samples. Log-transformed expression levels (x-fold over ASII pool) are shown for each patient GBM. LUM, IL-6, CYR61, and FBXO6 follow the same expression tendency in GBM patients compared with AS experimental glioma on D4 compared with D2. Bar lines indicate median.
Discussion
The CAM itself has been the subject of numerous studies addressing activity of proangiogenic or antiangiogenic factors, but even though the first tumor transplantation studies were performed ≈100 years ago (30) they are generally rare, which is surprising given the ease of access and the natural immunodeficient environment of the developing bird (for review see ref. 31).
In this article, we provide evidence that defined tumor growth with key features of human glioblastoma at cellular and molecular levels occurs in a highly reproducible manner after human GBM cell grafting on the CAM (experimental glioma).
Preclinical in vivo testing of anticancer drugs is performed mainly in adult rodents, which raises major ethical concerns. Data provided here strongly suggest that experimental glioma grown on the CAM exhibit sufficient similarity with fundamental aspects of the human disease. Topical treatment of experimental glioma with PTK787/ZK 222584 for 5 days potently inhibited progressive vascularization, associated with a strong decrease in glioma cell density. Most likely, these effects can be attributed to functional blocking of the avian VEGFR-2, which is strongly up-regulated in the course of experimental glioma progression.
Gleevec-treated tumors also exhibited consistent destruction of all cellular components of the tumor. Proliferation of U87 glioma cells depends on constitutively active platelet-derived growth factor receptors (PDGFR) signaling, which is inhibited by Gleevec (22), and PDGFR-mediated recruitment of pericytes to angiogenic capillaries is crucial for vessel stability (32).
These results demonstrate that the model allows successful testing of new anticancer drugs, designed to interfere with distinct molecular pathways important for tumor progression.
Experimental glioma display changes in gene expression similar to human GBM progression. IL-6 is produced by GBM cells in vitro and in vivo (33), and elevated IL-6 expression levels distinguish GBMs from low-grade AS (34). Recently, the prerequisite role of IL-6 for gliomagenesis has been demonstrated by crossbreeding GFAP-src mice, which develop GBM-like lesions, with IL-6-deficient mice, resulting in mice without GBMs (35). The function of IL-6 in tumor progression can be assigned to its proangiogenic properties (36). Up-regulation of IL-6 during the angiogenic switch in experimental glioma therefore fits well with current concepts on the role of IL-6 in gliomagenesis.
CYR61, which has a well established role in breast cancer progression, invasion, and angiogenesis (37), was up-regulated during experimental glioma development. CYR61 is overexpressed in 66% of patient GBMs compared with normal brain, and transfection of CYR61 into glioma cells resulted in faster tumor growth, associated with increased angiogenesis (27). The fact that upregulation of a second, emerging GBM-progression factor has been detected in our short-term model indicates that gene regulation of processes typical for GBM progression, such as angiogenesis, are mimicked.
Interestingly, we confirmed up-regulation of this gene in a variable manner in one-third of patient GBMs (8/12), a result similar to that of Xie et al. (27). The lack of CYR61 up-regulation or even its down-regulation in some GBM patients may identify a subset of GBM with better prognosis (26).
LUM and FBXO6, differentially regulated between D2 and D4, have yet unknown biological roles in glioma progression. LUM has been associated with tumor progression and invasion (28, 38), but recent insights also suggest a role as a negative growth regulator for melanoma cells (39). Invasion of glioma cells into the CAM is very active during the first days after implantation; LUM thus may participate in the regulation of the invasive process. Interestingly, all patient GBMs also showed overexpression of LUM when compared with ASII, suggesting a possible role of LUM in human GBM pathogenesis. This finding further provides evidence that the time frame chosen for microarray analysis of experimental glioma mimics a significant step in human GBM progression and validates the model as a rapid screening tool for identification of genes associated with human GBM growth.
FBXO6 encodes a member of the F-box protein family. F-box proteins are variable components of the Skp1/Cullin/F-box-type E3 ubiquitin ligase complex that function as receptors for phosphorylated proteins (40). Binding of F-box proteins to their target triggers a cascade, leading to the controlled degradation of the target protein by the proteasome. Interference with this process results in increased levels of critical proteins like cyclin E, Notch, or c-Myc and suggests that F-box proteins can function as tumor suppressors (41–43). Decreased expression of a given F-box protein thus may favor tumor progression. Additionally, dosage of F-box proteins may be critical for blood vessel formation (42). We observed consistent decrease in FBXO6 mRNA levels during experimental glioma growth by microarray and qPCR. More importantly, 9/12 patients with GBM had decreased levels of FBXO6 protein as compared with ASII, leading to the hypothesis that yet-unidentified target protein(s) of FBXO6 accumulate in GBM and might contribute to tumor progression. The potential implication of FBXO6 in the pathogenesis of CNS tumors is underscored by the recent findings that neuroblastoma with loss of heterozygosity on chromosome 1p36 are associated with down-regulation of FBXO6 (29), to a similar level as D4 compared with D2 experimental glioma. It would be interesting to see whether recently established experimental neuroblastoma grown on the CAM reproduce this kind of gene regulation (44).
We have simulated expression of significant cancer-related genes in a defined 48-h time interval in vivo. The critical time frame chosen for gene expression studies most likely favored identification of glioma progression genes. The angiogenic switch, a hallmark of tumor progression, was clearly identified in experimental glioma. The fast and reproducible development of experimental glioma with key features of the human disease makes the model a powerful tool for preclinical in vivo studies, in an ethically favorable context.
Experimental glioma is a hybrid tissue, in which only tumor cells are of human origin. It therefore should allow selective studies of gene expression during tumor progression by the simultaneous use of species-specific probes, like human and chick microarrays and qPCR primers. This model opens up the possibility to quickly screen for gene regulation occurring in the course of new anticancer treatments (e.g., chemoresistance) in a highly manageable environment.
Supplementary Material
Acknowledgments
We thank Prof. Mathias Chiquet (University of Bern, Bern, Switzerland) for providing TN-C antibodies, Prof. Joerg Wilting (University of Goettingen, Goettingen, Germany) for helpful suggestions during this project, Dr. Francesca De Giorgi-Ichas (Institut Europeen de Chimie et Biologie, Pessac, France) for help with the apoptosis analysis, Dr. Françoise Dellu-Hagedorn (Centre National de la Recherche Scientifique, Bordeaux, France) for statistical analysis, Prof. Hugues Loiseau (University Bordeaux 2, Bordeaux, France) for providing patient samples, and Drs. Jeannette Wood and Elisabeth Buchdungen (Novartis) for kindly providing PTK787/ZK 222584 and Imatinib (Gleevec). This work was supported by grants from the European Union (FP6, LSHC-CT-2003-5032, STROMA Consortium) and the Ligue Nationale Contre le Cancer, Equipe Labellisée (to A.B.).
Abbreviations: GBM, glioblastoma multiforme; AS, astrocytoma; CAM, chorio-allantoic membrane; VEGFR-2, vascular endothelial growth factor receptor 2; LUM, lumican; CYR61, cysteine-rich angiogenic inducer 61; FBXO6, F-box-only 6; D(n), day (n); qPCR, quantitative PCR; TN-C, tenascin C.
References
- 1.Maher, E. A., Furnari, F. B., Bachoo, R. M., Rowitch, D. H., Louis, D. N., Cavenee, W. K. & DePinho, R. A. (2001) Genes Dev. 15, 1311–1333. [DOI] [PubMed] [Google Scholar]
- 2.Dai, C. & Holland, E. C. (2001) Biochim. Biophys. Acta 1551, M19–M27. [DOI] [PubMed] [Google Scholar]
- 3.Holland, E. C., Celestino, J., Dai, C., Schaefer, L., Sawaya, R. E. & Fuller, G. N. (2000) Nat. Genet. 25, 55–57. [DOI] [PubMed] [Google Scholar]
- 4.Reilly, K. M., Loisel, D. A., Bronson, R. T., McLaughlin, M. E. & Jacks, T. (2000) Nat. Genet. 26, 109–113. [DOI] [PubMed] [Google Scholar]
- 5.Aldhous, P., Coghlan, A. & Copley, J. (1999) New Sci. 162, 26–31. [PubMed] [Google Scholar]
- 6.Russell, W. M. S. & Burch, R. L. (1992) The Principles of Humane Experimental Technique (Universities Federation for Animal Welfare, Potters Bar, U.K.).
- 7.Sherman, A., Dawson, A., Mather, C., Gilhooley, H., Li, Y., Mitchell, R., Finnegan, D. & Sang, H. (1998) Nat. Biotechnol. 16, 1050–1053. [DOI] [PubMed] [Google Scholar]
- 8.Pekarik, V., Bourikas, D., Miglino, N., Joset, P., Preiswerk, S. & Stoeckli, E. T. (2003) Nat. Biotechnol. 21, 93–96. [DOI] [PubMed] [Google Scholar]
- 9.Hagedorn, M., Balke, M., Schmidt, A., Bloch, W., Kurz, H., Javerzat, S., Rousseau, B., Wilting, J. & Bikfalvi, A. (2004) Dev. Dyn. 230, 23–33. [DOI] [PubMed] [Google Scholar]
- 10.Hagedorn, M., Zilberberg, L., Wilting, J., Canron, X., Carrabba, G., Giussani, C., Pluderi, M., Bello, L. & Bikfalvi, A. (2002) Cancer Res. 62, 6884–6890. [PubMed] [Google Scholar]
- 11.Eichmann, A., Marcelle, C., Breant, C. & Le Douarin, N. M. (1993) Mech. Dev. 42, 33–48. [DOI] [PubMed] [Google Scholar]
- 12.Schenk, S., Muser, J., Vollmer, G. & Chiquet-Ehrismann, R. (1995) Int. J. Cancer 61, 443–449. [DOI] [PubMed] [Google Scholar]
- 13.Hanahan, D. & Weinberg, R. A. (2000) Cell 100, 57–70. [DOI] [PubMed] [Google Scholar]
- 14.Plate, K. H., Breier, G., Millauer, B., Ullrich, A. & Risau, W. (1993) Cancer Res. 53, 5822–5827. [PubMed] [Google Scholar]
- 15.Patan, S., Munn, L. L. & Jain, R. K. (1996) Microvasc. Res. 51, 260–272. [DOI] [PubMed] [Google Scholar]
- 16.Holash, J., Maisonpierre, P. C., Compton, D., Boland, P., Alexander, C. R., Zagzag, D., Yancopoulos, G. D. & Wiegand, S. J. (1999) Science 284, 1994–1998. [DOI] [PubMed] [Google Scholar]
- 17.Chiquet-Ehrismann, R. (1993) Semin. Cancer Biol. 4, 301–310. [PubMed] [Google Scholar]
- 18.Herold-Mende, C., Mueller, M. M., Bonsanto, M. M., Schmitt, H. P., Kunze, S. & Steiner, H. H. (2002) Int. J. Cancer 98, 362–369. [DOI] [PubMed] [Google Scholar]
- 19.Kroemer, G. & Reed, J. C. (2000) Nat. Med. 6, 513–519. [DOI] [PubMed] [Google Scholar]
- 20.Millauer, B., Shawver, L. K., Plate, K. H., Risau, W. & Ullrich, A. (1994) Nature 367, 576–579. [DOI] [PubMed] [Google Scholar]
- 21.Strawn, L. M., Mann, E., Elliger, S. S., Chu, L. M., Germain, L. L., Niederfellner, G., Ullrich, A. & Shawver, L. K. (1994) J. Biol. Chem. 269, 21215–21222. [PubMed] [Google Scholar]
- 22.Kilic, T., Alberta, J. A., Zdunek, P. R., Acar, M., Iannarelli, P., O'Reilly, T., Buchdunger, E., Black, P. M. & Stiles, C. D. (2000) Cancer Res. 60, 5143–5150. [PubMed] [Google Scholar]
- 23.Wood, J. M., Bold, G., Buchdunger, E., Cozens, R., Ferrari, S., Frei, J., Hofmann, F., Mestan, J., Mett, H., O'Reilly, T., et al. (2000) Cancer Res. 60, 2178–2189. [PubMed] [Google Scholar]
- 24.Kane, M. D., Jatkoe, T. A., Stumpf, C. R., Lu, J., Thomas, J. D. & Madore, S. J. (2000) Nucleic Acids Res. 28, 4552–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weissenberger, J., Loeffler, S., Kappeler, A., Kopf, M., Lukes, A., Afanasieva, T. A., Aguzzi, A. & Weis, J. (2004) Oncogene 23, 3308–3316. [DOI] [PubMed] [Google Scholar]
- 26.Xie, D., Yin, D., Wang, H. J., Liu, G. T., Elashoff, R., Black, K. & Koeffler, H. P. (2004) Clin. Cancer Res. 10, 2072–2081. [DOI] [PubMed] [Google Scholar]
- 27.Xie, D., Yin, D., Tong, X., O'Kelly, J., Mori, A., Miller, C., Black, K., Gui, D., Said, J. W. & Koeffler, H. P. (2004) Cancer Res. 64, 1987–1996. [DOI] [PubMed] [Google Scholar]
- 28.Naito, Z., Ishiwata, T., Kurban, G., Teduka, K., Kawamoto, Y., Kawahara, K. & Sugisaki, Y. (2002) Int. J. Oncol. 20, 943–948. [PubMed] [Google Scholar]
- 29.Janoueix-Lerosey, I., Novikov, E., Monteiro, M., Gruel, N., Schleiermacher, G., Loriod, B., Nguyen, C. & Delattre, O. (2004) Oncogene 23, 5912–5922. [DOI] [PubMed] [Google Scholar]
- 30.Murphy, J. B. & Rous, P. (1912) J. Exp. Med. 15, 119–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hagedorn, M. & Wilting, J. (2004) in Methods in Endothelial Cell Biology, ed. Augustin, H. (Springer, Berlin), pp. 247–262.
- 32.Lindahl, P., Johansson, B. R., Leveen, P. & Betsholtz, C. (1997) Science 277, 242–245. [DOI] [PubMed] [Google Scholar]
- 33.Van Meir, E., Sawamura, Y., Diserens, A. C., Hamou, M. F. & de Tribolet, N. (1990) Cancer Res. 50, 6683–6688. [PubMed] [Google Scholar]
- 34.Rolhion, C., Penault-Llorca, F., Kemeny, J. L., Lemaire, J. J., Jullien, C., Labit-Bouvier, C., Finat-Duclos, F. & Verrelle, P. (2001) J. Neurosurg. 94, 97–101. [DOI] [PubMed] [Google Scholar]
- 35.Tchirkov, A., Rolhion, C., Bertrand, S., Dore, J. F., Dubost, J. J. & Verrelle, P. (2001) Br. J. Cancer 85, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Motro, B., Itin, A., Sachs, L. & Keshet, E. (1990) Proc. Natl. Acad. Sci. USA 87, 3092–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsai, M. S., Bogart, D. F., Castaneda, J. M., Li, P. & Lupu, R. (2002) Oncogene 21, 8178–8185. [DOI] [PubMed] [Google Scholar]
- 38.Leygue, E., Snell, L., Dotzlaw, H., Hole, K., Hiller-Hitchcock, T., Roughley, P. J., Watson, P. H. & Murphy, L. C. (1998) Cancer Res. 58, 1348–1352. [PubMed] [Google Scholar]
- 39.Vuillermoz, B., Khoruzhenko, A., D'Onofrio, M. F., Ramont, L., Venteo, L., Perreau, C., Antonicelli, F., Maquart, F. X. & Wegrowski, Y. (2004) Exp. Cell Res. 296, 294–306. [DOI] [PubMed] [Google Scholar]
- 40.Kipreos, E. T. & Pagano, M. (2000) Genome Biol. 1, 3002.1–3002.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moberg, K. H., Bell, D. W., Wahrer, D. C., Haber, D. A. & Hariharan, I. K. (2001) Nature 413, 311–316. [DOI] [PubMed] [Google Scholar]
- 42.Tetzlaff, M. T., Yu, W., Li, M., Zhang, P., Finegold, M., Mahon, K., Harper, J. W., Schwartz, R. J. & Elledge, S. J. (2004) Proc. Natl. Acad. Sci. USA 101, 3338–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yada, M., Hatakeyama, S., Kamura, T., Nishiyama, M., Tsunematsu, R., Imaki, H., Ishida, N., Okumura, F., Nakayama, K. & Nakayama, K. I. (2004) EMBO J. 23, 2116–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hecht, M., Papoutsi, M., Tran, H. D., Wilting, J. & Schweigerer, L. (2004) Cancer Res. 64, 6109–6118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.