

Abstract
The approval of venetoclax, a “BH3-mimetic” antagonist of the BCL-2 anti-apoptotic protein, for chronic lymphocytic leukemia represents a major milestone in translational apoptosis research. Venetoclax has already received “breakthrough” designation for acute myeloid leukemia, and is being studied in many other tumor types. However, resistance to BCL-2 inhibitor monotherapy may rapidly ensue. Several studies have shown that the other two major anti-apoptotic BCL-2 family proteins, BCL-XL and MCL-1, are the main determinants of resistance to venetoclax. This opens up possibilities for rationally combining venetoclax with other targeted agents to circumvent resistance. Here, we summarize the most promising combinations, and highlight those already in clinical trials. There is also increasing recognition that different tumors display different degrees of addiction to individual BCL-2 family proteins, and of the need to refine current “BH3 profiling” techniques. Finally, the successful clinical development of potent and selective antagonists of BCL-XL and MCL-1 is eagerly awaited.
Keywords: Venetoclax, resistance, BCL-2, MCL-1, BCL-XL, rational combinations
Introduction
The small-molecule “BH3 (BCL-2 homology domain 3)-mimetic” drug, venetoclax (Venclexta™), is currently approved by the Food and Drug Administration (FDA) for the treatment of patients with relapsed chronic lymphocytic leukemia (CLL) with a 17p deletion.[1] Venetoclax produces high overall response rates (ORRs) in heavily pre-treated, high risk subjects with CLL and, remarkably, leads to complete responses (CRs) in a substantial proportion (20%) of patients when administered as monotherapy, a feature that distinguishes it from small-molecule inhibitors of the B-cell receptor (BCR) pathway.[2] However, no patient in this phase I study had previously received BCR pathway inhibitors; additionally, in the pivotal trial in patients with relapsed or refractory CLL and deletion 17p which reported a much lower CR (or CR with incomplete count recovery, CRi) rate (8%), only five patients (5%) had previously been exposed to ibrutinib, idelalisib or other BCR pathway inhibitors.[1] An ongoing trial is investigating venetoclax monotherapy in patients with CLL who relapsed after or were refractory to ibrutinib and idelalisib.[3] Early results from this trial were recently presented: the ORR was 70% in the prior ibrutinib arm and 48% in the prior idelalisib arm; only 1 CRi was documented by independent review (in the prior ibrutinib arm).[3] Nevertheless, single-agent venetoclax can induce minimal residual disease (MRD)-negative CRs in some patients with relapsed/refractory CLL.[1, 2] MRD negativity has been shown to be a powerful predictor of long-term outcome in CLL, irrespective of line and type of therapy.[4]
Based on promising efficacy in patients with relapsed/refractory (R/R) acute myeloid leukemia (AML) as a single agent,[5] as well as highly encouraging results in the frontline setting in combination with hypomethylating agents (HMAs),[6] venetoclax, in combination with HMAs, has also received “breakthrough therapy designation” from the FDA for previously untreated AML in patients who cannot receive intensive chemotherapy.
Venetoclax is a bona fide “BH3-mimetic” (a small molecule capable of binding to and antagonizing BCL-2 family anti-apoptotic proteins by mimicking the BH3 domain of pro-apoptotic proteins). As discussed further below, many other compounds were initially considered to act as BH3-mimetics, but were found later to induce tumor cell death through other mechanisms.[7, 8] Efforts over many years to develop small-molecule inhibitors of protein-protein interactions culminated in the discovery of ABT-737, the first genuine BH3-mimetic and a specific antagonist of the anti-apoptotic proteins BCL-2, BCL-XL, and BCL-w.[9] A clinically relevant analog, ABT-263 (navitoclax), was soon developed,[10] but marked on-target (through BCL-XL inhibition) thrombocytopenia observed in clinical trials of this agent limited its clinical utility.[11–14] As BCL-XL is a critical pro-survival factor for platelets,[15] venetoclax (formerly ABT-199, GDC-0199) was rationally designed by reverse engineering using the chemical scaffold of navitoclax to generate a molecule which specifically targeted BCL-2 while sparing BCL-XL and, therefore, platelets.[16] Like ABT-737 and navitoclax, venetoclax also inhibits BCL-w. Figure 1 schematically depicts the mechanism of action of venetoclax. Venetoclax is well-absorbed orally (at least 65%), and primarily cleared by hepatic metabolism (approximately two-thirds of the administered dose), with the remainder (likely unabsorbed parent drug) excreted as the parent drug and its nitro reduction metabolite in feces.[17]
Figure 1.
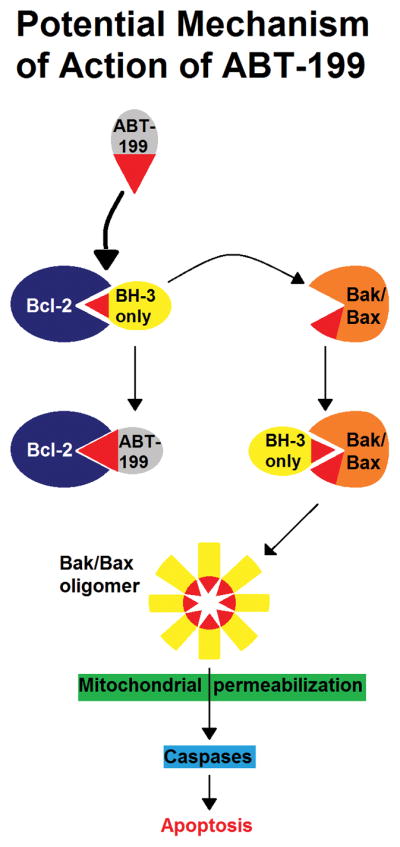
Potential mechanism of action of ABT-199 (venetoclax). Venetoclax binds to the BH3-binding groove of BCL-2 and displaces Bim and other BH3-only proteins that are normally sequestered by this pro-survival (anti-apoptotic) protein. These BH3-only proteins are thus freed to activate the apoptosis effectors, Bax and Bak. Bax/Bak activation then leads to their oligomerization and mitochondrial outer membrane permeabilization. This commits the cell to apoptosis through the mitochondrial pathway, which is triggered by the activation of caspases. Modified, with permission, from ref. [51].
Numerous preclinical studies have investigated mechanisms of resistance to ABT-737 and explored rational combinations to circumvent resistance to this agent. Although the lack of inhibition of BCL-XL by venetoclax is an important distinguishing feature between the two agents, many of these principles apply to venetoclax as well. Other studies have specifically evaluated resistance to venetoclax and combinations with other targeted agents. In this article, we review the major known mechanisms of resistance to venetoclax, and discuss synergistic strategies to overcome the same, some of which are already being explored in clinical trials.
The BCL-2 family, mechanism of action of BH3-mimetics and BH3 profiling
Overview of the BCL-2 family
The intrinsic (mitochondrial) pathway of apoptosis is controlled by the BCL-2 family of pro- and anti-apoptotic proteins. The apoptosis “effectors” within this family are represented by BAX and BAK, and their activation triggers mitochondrial outer membrane permeabilization (MOMP), committing the cell to apoptosis through the mitochondrial pathway.[18, 19] The anti-apoptotic proteins of the BCL-2 family are BCL-2, BCL-XL, MCL-1, BCL-w and BFL-1/A1. The anti-apoptotic (pro-survival) proteins bind to and sequester the apoptosis effectors, as well as the apoptosis “sensitizers”, BIM, BID, PUMA, NOXA, BAD, BMF, HRK and BIK.[20, 21] The five anti-apoptotic proteins and BAX/BAK all share four homologous domains referred to as BCL-2 homology 1, 2, 3 and 4 (BH1, BH2, BH3, BH4). In contrast, the eight apoptosis “sensitizers” contain only the BH3 domain and are, therefore, called “BH3-only” proteins.[18, 19] Although the major pro-apoptotic function of the BH3-only proteins is binding to the anti-apoptotic proteins, thus releasing BAX/BAK,[22] some of them (e.g., BIM, PUMA, BID) may also be able to directly activate BAX/BAK.[23–26]
Some BH3-only proteins bind to multiple anti-apoptotic proteins, e.g., BIM, PUMA, while others are more selective, e.g., BAD, NOXA.[27] Similarly, the sequestering of the apoptosis effectors by the major anti-apoptotic proteins is also somewhat selective, e.g., BAK is bound by MCL-1 and BCL-XL, but not by BCL-2.[28] BAX, on the other hand, is bound by BCL-2.[29]
Importantly, post-translational modifications, e.g., phosphorylation, significantly impact the function of different BCL-2 family proteins; thus, BAD, which antagonizes BCL-2, BCL-XL and BCL-w, must be dephosphorylated at two different serine residues by combined inhibition of mitogen activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K) signaling in order to perform its pro-apoptotic functions.[30] Furthermore, cellular survival signaling pathways can regulate BCL-2 family proteins, e.g., extracellular signal-regulated kinase (ERK) signaling promotes proteasomal degradation of BIM.[31]
BH3-mimetics
True BH3-mimetic compounds bind to the anti-apoptotic proteins by virtue of their structural similarity to the BH3 domains of the BH3-only proteins, displacing the latter, and inducing apoptosis through BAX/BAK activation.[32] A number of drugs (e.g., obatoclax, gossypol, AT101) that were initially considered to act as BH3-mimetics may not, in fact, do so in cells. For example, obatoclax induces autophagic cell death independent of BAX/BAK,[33] and a number of putative BH3-mimetics induce endoplasmic reticulum (ER) stress and thus up-regulate NOXA, which antagonizes MCL-1.[8] While this may certainly be a therapeutically useful attribute of these agents, it is not a “BH3-mimetic” effect.[34] ABT-737/navitoclax and venetoclax, on the other hand, represent genuine BH3-mimetics. Because of the exquisite dependence of CLL cells on BCL-2 for survival,[35] and of platelets on BCL-XL,[15] it has been proposed that true BH3-mimetic drugs must, at a minimum, rapidly induce apoptosis in CLL cells ex vivo if BCL-2-targeted (e.g., venetoclax[36]) and in platelets if BCL-XL-targeted.[37] Since NOXA binding leads to ubiquitination and degradation of MCL-1,[38–40] a BH3-mimetic targeting MCL-1 will, in fact, lead to rapid accumulation of MCL-1 by blocking this interaction, unless the agent also happens to inhibit the USP9X deubiquitinase.[37]
BH3 profiling
BH3 profiling is a technique that exposes permeabilized tumor cells to BH3-domain-containing peptides to measure MOMP.[41] The BH3 domains in these peptides are derived from the BH3-only proteins with known specificity for anti-apoptotic BCL-2 family proteins. The assay can help separate malignant cells into three major classes: those that have functional BAX/BAK but are not significantly sequestering pro-apoptotic proteins so that they respond to activator peptides but don’t respond or respond only weakly to sensitizer peptides (competent but unprimed), those that lack functional BAX/BAK (apoptotically incompetent, no response even to very high doses of activator peptides) and those that respond to both activator and sensitizer peptides (primed for apoptosis).[41] Apoptosis priming as measured by BH3 profiling correlates with chemotherapeutic success in AML,[42] and decreased apoptotic priming may underlie microenvironment-mediated resistance to chemotherapy in CLL.[43] Furthermore, BH3 profiling can help identify which anti-apoptotic BCL-2 family protein primed cells are most reliant on for survival, e.g., a response to the Bad peptide indicates dependence on BCL-2, and one to HRK peptide dependence on BCL-XL, while a response to NOXA peptide indicates dependence on MCL-1. The use of selective small-molecule antagonists of BCL-2 (e.g., venetoclax) or BCL-XL (discussed below) can further improve the precision of the assay in identifying the major pro-survival protein at play.
Indeed, BH3 profiling has been used as a tool to identify the cause of acquired resistance to venetoclax in multiple myeloma (MM),[44] and predict sensitivity of MM cell lines and primary plasma cells from MM patients to both venetoclax and navitoclax,[45] and is being used to select patients for a clinical trial in AML investigating flavopiridol (alvocidib), a cyclin-dependent kinase 9 (CDK9) inhibitor that down-regulates MCL-1 (NCT02520011). In T-cell acute lymphoblastic leukemia (T-ALL), a disease with a poor prognosis and no currently approved targeted therapies, BH3 profiling reveals that while most cell lines and primary patient samples display dependence on BCL-XL, the early T-cell precursor (ETP) subtype depends on BCL-2,[46] providing rationale for targeted therapeutic intervention with venetoclax in this high-risk group of patients.[47] Indeed, the early immature T-ALL cell line, LOUCY, is exquisitely sensitive to venetoclax while more differentiated T-ALL cell lines are not, and strong synergism with cytarabine is seen.[48]
“Mitochondrial profiling” improves upon BH3 profiling by also measuring basal levels of pro-survival BCL-2 family proteins.[49] Using this technique, it has been demonstrated that the apoptogenic efficacy of venetoclax in AML cells is highly dependent on baseline levels of BCL-XL expression.[49] Indeed, while BH3 profiling appears to accurately predict BCL-2 dependence, it may not fully identify dependence on BCL-XL and MCL-1, since the latter appear to correlate better with protein expression of BCL-XL and MCL-1.[49]
Major determinants of venetoclax resistance: MCL-1 and BCL-XL
Resistance to ABT-737/navitoclax
As ABT-737 inhibits both BCL-2 and BCL-XL, it is not surprising that the main determinant of resistance to ABT-737 is MCL-1, the other major anti-apoptotic protein of the BCL-2 family.[50] Additionally, in AML cells, phosphorylated BCL-2 conferred resistance to ABT-737, and both inhibition of BCL-2 phosphorylation and reduction of MCL-1 expression restored sensitivity to ABT-737.[51] MCL-1 phosphorylation, resulting in enhanced stability of the protein and greater sequestration of BIM, has also been shown to contribute to ABT-737 resistance in human B-cell acute lymphoblastic leukemia (B-ALL) cell lines.[52] These observations resulted in a number of rational combinations being tested in preclinical systems, adding a variety of agents that down-regulate MCL-1 to ABT-737. MCL-1 is particularly critical to the development and maintenance of AML,[53] and being a short-lived protein, is sensitive to inhibition of transcription and translation.[54] Thus, CDK9 inhibitors,[55] sorafenib,[56] MEK inhibitors,[57] dual inhibitors of PI3K and mammalian target of rapamycin (mTOR)[58] and the combination of MEK and mTOR inhibitors[59] have all been shown to synergize with ABT-737 in induction of apoptosis in AML cell lines, primary AML samples and for some combinations, murine xenograft models of AML. In lymphoma cell lines that have developed resistance to ABT-737 through long-term exposure, increased levels of BFL-1/A1 and/or MCL-1 have been demonstrated.[60] Although pro-apoptotic BIM is still displaced by ABT-737 in these resistant cells, it is sequestered by the excess BFL-1/A1 and/or MCL-1 (partner swapping).[60] The TORC1/2 inhibitor, AZD8055, reduced MCL-1 protein levels and sensitized small-cell lung cancer (SCLC) cell lines as well as xenograft models of SCLC to navitoclax.[61]
Venetoclax resistance in CLL and rational combination strategies
Mature B-cells, both normal and leukemic (CLL), are highly sensitive to BCL-2 inhibition by venetoclax, which induces BAX/BAK-mediated apoptosis triggered mainly by BIM.[62] Although circulating CLL cells are extremely sensitive to ABT-737, those cultured in artificial systems that mimic the lymph node microenvironment quickly develop a high level of resistance to ABT-737 owing to de novo synthesis of BCL-XL and BCL-1/A1.[63] While BCL-XL-mediated resistance can be counteracted by higher concentrations of ABT-737,[63] this represents a problem with venetoclax, which lacks BCL-XL-inhibitory activity and indeed, is clinically less efficient at clearing nodal disease than blood and bone marrow.[2] The BCR signaling inhibitor, ibrutinib, on the other hand, is particularly effective at mobilizing CLL cells from lymph nodes[64, 65] and, in both ex vivo and in vitro studies in CLL, appears to synergize with venetoclax in inducing cytotoxicity, accompanied by decreases in levels of MCL-1 and BCL-XL.[66] Both BCL-XL and BFL-1/A1 are up-regulated in B-lymphocytes by the transcription factor, nuclear factor kappa B (NF-κB),[67] an important downstream target of ibrutinib.[68] The combination of venetoclax and ibrutinib is currently being studied in a number of ongoing clinical trials in CLL (NCT02758665, NCT02910583, NCT02756897).
Other groups have found that inhibitors of spleen tyrosine kinase (SYK), e.g., entospletinib, down-regulate MCL-1 more completely than other BCR pathway inhibitors such as ibrutinib and idelalisib, arguing for combining these agents with venetoclax in CLL.[69] However, the clinical efficacy of SYK inhibitors as single agents in CLL has been somewhat disappointing thus far.[70] In a high-content screening system that utilized an in vitro microenvironment model, the multi-kinase inhibitor sunitinib was identified as the clinically available kinase inhibitor most effective at overcoming venetoclax resistance (more than ibrutinib or idelalisib).[71] The mechanism was attributed to more efficient abrogation of microenvironment-mediated up-regulation of BCL-XL, MCL-1 and BFL-1/A1 by sunitinib.[71] Interleukin-4 (IL-4) signaling, which proceeds through the Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway, has been identified as a resistance mechanism to BCR pathway inhibition in CLL,[72] and the dual SYK/JAK inhibitor, cerdulatinib inhibits BCR- and IL-4-induced downstream signaling in CLL cells and induces apoptosis, particularly in IGHV-unmutated samples (characterized by greater BCR-signaling capacity and response to IL-4), and also prevents anti-IgM- and nurse-like cell (NLC)-mediated production of the chemokines CCL3/CCL4.[73] Cerdulatinib was able to overcome the protection afforded to CLL cells by anti-IgM, IL-4 or NLCs by preventing up-regulation of MCL-1- and BCL-XL, but did not affect BCL-2 levels.[73] In samples treated with IL-4, cerdulatinib synergistically induced apoptosis with venetoclax in vitro.[73]
Silencing of microRNA (miR) 377 may be an important mechanism underlying the BCL-XL overexpression that drives acquired therapeutic resistance to venetoclax in CLL and B-cell lymphomas.[74] Significantly higher BCL-XL and lower miR377 expression has also been observed in CLL cells from patients with prior exposure to chemotherapy.[74] Interestingly, it was recently demonstrated that in contrast to some other tumor types, hypoxia activates the stress kinase p38 MAPK in CLL cells, which in turn causes MCL-1 down-regulation, a high BIM:MCL-1 ratio and enhanced sensitivity to both ABT-737 and venetoclax.[75] Although none are currently in the clinic, a number of BCL-XL and MCL-1 inhibitors are being developed (discussed below).
As noted above, inhibitors of CDK9 down-regulate MCL-1 through transcriptional repression.[76] The pan-CDK inhibitor, dinaciclib, which exhibits significant single-agent activity in relapsed/refractory CLL,[77] also lowers MCL-1 stability via inhibition of cyclin E/CDK2, promoting its ubiquitination and degradation.[78] This releases BIM from MCL-1, resulting in striking synergism against CLL cell lines and primary patient samples in combination with either ABT-737 or venetoclax.[78]
Finally, CC-115, a novel inhibitor of mTOR kinase and DNA protein kinase, induced apoptosis in CLL cell lines and patient-derived CLL cells, irrespective of p53, ATM, NOTCH1 or SF3B1 mutational status and inhibited BCR signaling, including in idelalisib-resistant samples.[79] CC-115, which displayed evidence of clinical activity in 8 patients with relapsed/refractory ATM-deleted/mutated CLL/small lymphocytic lymphoma (SLL), was able to reverse CD40-mediated resistance to both fludarabine and venetoclax.[79]
Venetoclax resistance in other lymphoid neoplasms
Follicular lymphoma (FL)
The anti-apoptotic function of BCL-2 was discovered in the context of its overexpression in FL, which is characterized by a translocation between chromosomes 14 and 18 that juxtaposes the BCL-2 gene to the immunoglobulin heavy chain gene.[80, 81] The sensitivity of FL cells to venetoclax has been shown to correlate with their BCL-2/BIM ratio, with cells expressing higher levels of BIM being more susceptible.[82] Venetoclax-resistant cells had increased phospho-ERK, phospho-BIM and initially, increased phospho-AKT and markers of autophagy.[82] The acquisition of these resistance phenotypes could be prevented via selective ERK/AKT inhibition or anti-CD20 antibody treatment.[82] In FL cell lines, dexamethasone and BCR signaling inhibitors synergistically induce apoptosis, accompanied by down-regulation of BCL-XL, inhibition of ERK1/2 phosphorylation and accumulation of non-phosphorylated BIM.[83] High-BCL-2-expessing FL cell lines are less sensitive to these agents, a phenomenon that can be reversed by venetoclax.[83] However, BCL-XL overexpression can prevent the apoptotic response to dexamethasone, BCR pathway inhibitors and to venetoclax.[83]
Diffuse large B-cell lymphoma (DLBCL)
In venetoclax-resistant DLBCL cell lines generated via chronic exposure to venetoclax, substantial AKT activation and up-regulation of MCL-1 and BCL-XL (causing sequestration of BIM) were observed.[84] The dual PI3K/mTOR inhibitor, NVP-BEZ235, as well as the PI3Kδ inhibitor, idelalisib, down-regulated MCL-1 and re-sensitized these resistant cells to venetoclax.[84] A small interfering (si) RNA approach down-regulating AKT, MCL-1 and BCL-XL vindicated the role of each of these cell survival factors in mediating resistance to venetoclax.[84]
Acute lymphoblastic leukemia (ALL)
In xenograft studies of childhood ALL, concurrent inhibition of both BCL-2 and BCL-XL appears necessary for anti-leukemic efficacy and BCL-XL expression is a key predictor of poor response to venetoclax.[85] However, mixed lineage leukemia (MLL)-rearranged ALL has been shown to be a notable exception, retaining substantial susceptibility to BCL-2 inhibition alone.[85] Specifically, the MLL-AF4 fusion protein product of t(4;11) leads to high levels of BCL-2 expression and sensitivity to venetoclax, as well as synergism with standard induction-type chemotherapy in vivo.[86] The combination of BCR-ABL tyrosine kinase inhibitors (TKIs) and venetoclax is highly synergistic in vitro against Philadelphia chromosome positive (Ph+) ALL.[87] Furthermore, dasatinib and ponatinib, through their effects on the Lck/Yes novel (LYN) tyrosine kinase, may induce BIM and inhibit up-regulation of MCL-1, thus potentially circumventing the development of venetoclax resistance.[87] The dasatinib/venetoclax combination was synergistic against patient-derived Ph+ ALL samples and in xenograft models in mice, arguing for a clinical trial of this regimen in Ph+ ALL.[87]
Mantle cell lymphoma (MCL)
Primary MCL cells are highly sensitive to venetoclax and the BCL-2/(BCL-XL + MCL-1) messenger RNA (mRNA) ratio is highly predictive of venetoclax sensitivity.[88] Like in CLL, nodal microenvironment-mediated NF-κB activation and consequent BCL-XL up-regulation confer resistance to venetoclax.[88] Interestingly, the consequent loss of apoptotic priming can be overcome by the type II, glycoengineered anti-CD20 monoclonal antibody, obinutuzumab, which blocks BCL-XL induction through NF-κB inhibition.[89] As noted above, ibrutinib can mobilize malignant lymphocytes in CLL from their nodal microenvironmental niches, as well as block their subsequent tissue homing.[64] Ibrutinib is highly active in and FDA-approved for relapsed MCL,[90] and ibrutinib-mobilized MCL cells appear to be highly sensitive to venetoclax.[88] Accordingly, this combination is being explored in clinical trials (NCT02419560). A unique mechanism of venetoclax resistance that has been described in human MCL-like murine lymphoma cells involves the acquisition of mutations (F101C, F101L) within the BCL-2 BH3 domain that impair venetoclax binding.[91] Additionally, a mutation in BAX (G179E) that interferes with its anchoring to the mitochondrial membrane has been documented in a venetoclax-resistant human MCL cell line.[91]
Multiple myeloma (MM)
In MM, venetoclax lethality may be restricted to the subset harboring the same t(11;14) that is the hallmark of MCL and results in cyclin D1 overexpression.[92] MM is highly dependent on MCL-1 (although a subset is addicted to BCL-XL for survival),[93] and CDK9 inhibitors exert potent cytotoxicity against MM cells through MCL-1 down-regulation.[94] Although high levels of BCL-2 expression are common in MM, in vitro and xenograft studies using venetoclax, A-1331852 and A-1155463, selective inhibitors of BCL-XL, and the proteasome inhibitor, bortezomib (which leads to c-MYC-dependent induction of NOXA in malignant cells and consequent neutralization of MCL-1[95]) show that BCL-XL and MCL-1 may be more critical for survival of MM cells than BCL-2.[96] In these studies, sensitivity to venetoclax correlated with high BCL-2 to (BCL-XL or MCL-1) expression ratios.[92, 96] Indeed, MM cells co-expressing BCL-2 and BCL-XL were susceptible to A-1155463 but not to venetoclax, and venetoclax resistance in MM xenograft models co-expressing BCL-2 and MCL-1 could be mitigated by bortezomib.[96] MM xenograft models expressing all three major anti-apoptotic proteins were more sensitive to the combination of bortezomib and A-1331852 than to the combination of bortezomib and venetoclax.[96] In the setting of glutamine deprivation, MM cells induce BIM but its binding to BCL-2 is enhanced, enabling continued cell survival, but also priming them for apoptosis induction by venetoclax.[97] Thus, targeting glutamine metabolism represents an attractive synthetic lethal strategy to sensitize MM cells to venetoclax.[97]
Unlike navitoclax and venetoclax, which bind to the BH3 domain of BCL-2, BCL-2 IP3 Receptor Disruptor-2 (BIRD-2) is a decoy peptide that binds to the BH4 domain of BCL-2 and disrupts its interaction with inositol-1, 4, 5-triphosphate (IP3) receptors.[98] The BCL-2/IP3 interaction normally inhibits pro-apoptotic Ca++ signals, and BIRD-2 has been demonstrated to induce Ca++-mediated apoptosis in CLL, MM and FL cells, including those resistant to navitoclax, venetoclax or ibrutinib.[98] Furthermore, combining BIRD-2 with either navitoclax or venetoclax potentiates apoptosis induction by the BH3-mimetics alone.[98]
Venetoclax resistance in AML
Contrary to initial expectations, AML cell lines, primary patient samples and murine primary xenografts were found to be very sensitive to venetoclax.[99] In primary patient-derived cells, the median IC50 was approximately 10 nmol/L, and mitochondrial apoptosis occurred within 2 hours.[99] The ex vivo sensitivity of AML compared favorably with those observed in CLL models.[99] In a phase II study of single-agent venetoclax (800 mg daily) in patients with relapsed/refractory AML or previously untreated patients deemed unfit for intensive chemotherapy, the overall response rate (ORR) was 19%, and anti-leukemic activity not meeting International Working Group (IWG) criteria for response[100] was observed in another 19%.[5] Consistent with preclinical findings demonstrating particular susceptibility of isocitrate dehydrogenase 1/2 (IDH1/2)-mutated AML to BCL-2 inhibition,[101] 4 of 12 (33%) patients harboring IDH1/2 mutations achieved complete response (CR) or complete response with incomplete blood count recovery (CRi).[5] BH3 profiling demonstrated an on-target, mitochondrial mechanism of action of venetoclax and expectedly, identified BCL-XL or MCL-1 dependence as potential resistance mechanisms.[5] In a recent report, investigators characterized a panel of venetoclax-resistant myeloid leukemia cell lines derived through chronic exposure to venetoclax and found that acquired drug resistance was indeed driven by the up-regulation of MCL-1 and BCL-XL.[102] Interestingly, not only could the resistant AML cell lines be re-sensitized to venetoclax by targeting MCL-1 and BCL-XL, pre-emptively targeting one or both of the latter could actually delay or forestall the acquisition of venetoclax resistance.[102] These findings have obvious implications for selection of initial therapy for AML.
Based on prior work showing that MEK inhibitors efficiently down-regulate MCL-1 in AML cell lines and primary AML cells (including CD34+38-123+ leukemia-initiating cells) and synergize in vivo with ABT-737 (which induces MCL-1 through ERK activation),[57] a phase I/II combination clinical trial of the MEK inhibitor, cobimetinib (Cotellic®), plus venetoclax has been initiated in patients with relapsed or refractory AML (NCT02670044).
In another arm of this trial, venetoclax is combined with idasanutlin, an inhibitor of murine double minute homolog 2 (MDM2), an important negative regulator of p53. Because of the mechanism of action of MDM2 inhibitors, i.e., activation of p53-dependent apoptosis,[103] patients must have functional, i.e., wild type TP53. Preclinical studies with Nutlin-3a, a first-generation MDM2 inhibitor, and ABT-737 in AML published a decade ago showed strikingly synergistic induction of mitochondrial apoptosis by the combination.[104] A key concept underlying these phenomena was p53-mediated apoptosis induction by Nutlin-3a predominantly in S-phase and G2/M cells, while ABT-737 induced apoptosis predominantly in G1, the cell cycle phase characterized by the lowest BCL-2 protein levels and BCL-2/BAX ratios, and absent BCL-2 phosphorylation.[104] Nutlin-3a also potentiates ABT-737-induced apoptosis in both proliferating and quiescent CD34+ chronic myeloid leukemia (CML) progenitor cells from patients in blast crisis (BC).[105] Most recently, the combination of idasanutlin and venetoclax has been studied in TP53-wild type AML cell lines (including in OCI-AML3 cells, which are relatively resistant to both venetoclax and idasanutlin, and are characterized by high levels of basal MCL-1 expression) and xenograft (subcutaneous and orthotopic) mouse models.[106, 107] The combination was synergistic both in vitro and in vivo, and inhibition of MCL-1 by the combination treatment was confirmed to be contributing to the superior activity of the regimen.[106, 107] Mechanistically, venetoclax was found to lead to MCL-1 stabilization and up-regulation through increased phosphorylation of ERK2, phenomena that were quickly reversed by p53 activation by idasanutlin.[107]
In combination with BCR-ABL TKIs, which partially down-regulate MCL-1 and BCL-XL, venetoclax synergistically targets quiescent stem/progenitor cells in CML-BC patient samples and BCR-ABL transgenic mice.[108]
Since fms-like tyrosine kinase 3 (FLT3) signals downstream to regulate multiple members of the BCL-2 family, e.g., via the PI3K/AKT/mTOR and MEK/ERK pathways, and sorafenib, besides inhibiting FLT3, also down-regulates MCL-1 through inhibition of translation[109] and induction of endoplasmic reticulum (ER) stress,[110] there is sound preclinical rationale to combine sorafenib with venetoclax in FLT3-mutated AML, and a clinical trial is planned. Synergism between sorafenib and ABT-737 in AML cell lines and primary AML samples, accompanied by up-regulation of BIM and down-regulation of MCL-1, X-linked inhibitor of apoptosis (XIAP) and survivin has already been documented.[56] Recently reported benefits of adding sorafenib to intensive chemotherapy in all patients with AML, regardless of FLT3 mutational status,[111] may extend the applicability of this approach beyond FLT3-mutated AML.
Preclinical studies using siRNA against the major anti-apoptotic proteins showed that in AML cell lines, silencing of BCL-XL and MCL-1, but not BCL-2, resulted in varying degrees of synergism with azacitidine.[112] These findings were recapitulated by studies using ABT-737 and venetoclax, where the former sensitized most myeloid cell lines and primary patient samples to azacitidine more potently compared with venetoclax, which synergized with azacitidine mostly at higher doses.[112] Silencing of BCL-XL and MCL-1 increased the activity of venetoclax.[112] In other studies, the combination of azacitidine and ABT-737 has been shown to synergistically induce apoptosis in primary AML cells in a p53-independent fashion, accompanied by azacitidine-induced down-regulation of MCL-1.[113] Highly promising results have been reported in an ongoing phase IB clinical trial (NCT02203773) of the combination of venetoclax with azacitidine or decitabine in patients 65 years of age or older with newly diagnosed AML unfit for intensive chemotherapy.[6] An ORR of 76% was reported for the first 39 patients; this was 88% and 82%, respectively, among patients with poor-risk cytogenetics and IDH1/2 mutations.[6] Median time to CR/CRi was 29.5 days.[6]
Glutamine levels control mitochondrial oxidative phosphorylation in AML cells, and pharmacologic inhibition (e.g., by CB-839) of glutaminase, the enzyme that catalyzes the rate-limiting conversion of glutamine to glutamate, induces arrest of proliferation and activation of mitochondrial apoptosis, which is synergistically enhanced by venetoclax.[114] AML with mutant IDH enzymes, which produce the oncogenic metabolite, 2-hydroxyglutarate (2-HG) instead of the normal alpha-ketoglutarate (α-KG), creating glutamine addiction, may be particularly susceptible to glutaminase inhibition,[115] as it is to BCL-2 inhibition.[101]
Finally, given the critical importance of MCL-1 in AML,[53] pharmacologic strategies to activate/up-regulate NOXA, such as induction of ER stress, may be therapeutically useful.[116] Pevonedistat, a first-in-class small-molecule inhibitor of protein neddylation with single-agent activity in AML,[117, 118] up-regulates NOXA by causing c-Myc accumulation and synergizes with both venetoclax and navitoclax in AML.[119] ONC201, a first-in-class imipridone that induces p53-independent apoptosis in primary MCL and AML samples and targets AML stem and progenitor cells, has been shown to increase translation of the transcription factor, ATF4, by triggering an atypical integrated stress response (ISR) not dependent on increased phosphorylation of the translation initiation factor, eIF2α.[120] ONC201 prominently down-regulates MCL-1, and BCL-2 overexpression was found to protect against ONC201-induced apoptosis.[120] Accordingly, ONC201 and venetoclax synergistically triggered apoptosis in AML and MCL cell lines.[120]
Venetoclax resistance in solid tumors
Venetoclax displaces BIM from BCL-2 and induces apoptosis through the mitochondrial pathway in neuroblastoma cells, but up-regulation of MCL-1 and sequestration of BIM by MCL-1 represent a major mechanism of resistance.[121] In colorectal carcinoma, targeting MCL-1 overcomes resistance to both ABT-737 and venetoclax, and gene therapy approaches have been studied in vitro and in vivo.[122] In both docetaxel-sensitive and -resistant prostate cancer, BCL-XL seems to be a key determinant of cell survival, as navitoclax, but not venetoclax, significantly augments the anti-tumor efficacy of docetaxel in human prostate cancer cells lines and xenograft mouse models.[123] Conversely, in estrogen receptor-expressing breast cancer, BCL-2 appears to be the crucial target as similar efficacy is observed with venetoclax or ABT-737 in xenografts, despite abundant BCL-XL expression.[124] Furthermore, BH3-mimetics have been shown to counteract tamoxifen-induced endometrial hyperplasia, as well as enhance its anti-tumor efficacy, and to synergize with dual PI3K/mTOR inhibitors in apoptosis induction in this setting.[124]
Novel compounds that inhibit BCL-XL or MCL-1
The search for selective inhibitors of the anti-apoptotic functions of BCL-XL and MCL-1 has been ongoing for many years, although none of these compounds is in the clinic yet. Early efforts to identify selective antagonists of BCL-XL led to the description of the small-molecule, A-385358[125] and 072RB, a peptide derived from the BH3 domain of BIM.[126] Maritoclax was reported to be a selective MCL-1 antagonist able to disrupt the BIM-MCL-1 interaction and promote proteasomal degradation of MCL-1 that markedly enhanced the efficacy of ABT-737 against multiple hematologic malignancies,[127] but subsequent studies have shown that the compound likely also acts through mechanisms independent of MCL-1.[128] Of the early putative MCL-1 inhibitors, TW-37 is one that induces apoptosis in a BAK-dependent manner in cells that require MCL-1 for survival, but apoptosis induced by TW-37 is also, in part, dependent on NOXA.[129] (-)BI97D6 is a pan-BCL-2 family inhibitor that was shown to disrupt Mcl-1/Bim and Bcl-2/Bax interactions and induce BAX/BAK-dependent apoptosis in AML cells, overcoming MCL-1-mediated resistance as well as extrinsic, microenvironmental resistance.[130]
UMI-77 is a novel, selective MCL-1 inhibitor that blocks the heterodimerization of MCL-1 with BAX and BAK by binding to the BH3-binding groove of MCL-1, and induces BAX/BAK-dependent apoptosis in pancreatic cancer cells; it also effectively inhibited tumor growth in a xenograft model of pancreatic cancer.[131] These investigators also reported the discovery of a 3-Substituted-N-(4-hydroxynaphthalen-1-yl)arylsulfonamide compound that binds selectively to MCL-1, disrupts the interaction between endogenous MCL-1 and biotinylated NOXA-BH3 peptide and causes BAX/BAK-dependent cell death in human leukemia cell lines and Emu-myc lymphomas overexpressing MCL-1, but not in those overexpressing BCL-2.[132] The development and optimization of potent and selective BCL-XL inhibitors also continues to be pursued.[133, 134] In particular, BCL-XL may be a more relevant therapeutic target than BCL-2 in lung cancer, and the BCL-XL-selective inhibitors, BXI-61 and BXI-72, were found to exhibit greater potency against human lung cancer than ABT-737 with less thrombocytopenia in vivo.[135]
BCL-2 is important for neutrophil survival,[136] and neutropenia is therefore an important on-target adverse effect of venetoclax.[1, 2] Navitoclax, which inhibits both BCL-2 and BCL-XL, causes significant worsening of neutropenia when combined with docetaxel in patients, but this problem is avoided in preclinical studies using the BCL-XL-selective inhibitors, A-1155463 and A-1331852, in combination with docetaxel against a range of solid tumors, while maintaining high efficacy.[137]
A-1210477 is a potent and selective inhibitor of MCL-1 that binds to MCL-1 with sufficient affinity to disrupt BIM-MCL-1 complexes and induce apoptosis via the mitochondrial pathway in MM and non-small cell lung cancer (NSCLC) cell lines shown to be MCL-1-dependent by BH3 profiling or using siRNA.[138] A-1210477 synergizes with navitoclax to kill a variety of cancer cell lines.[138] A-1210477 synergizes with venetoclax in BCL-2-high non-Hodgkin’s lymphoma (NHL) cell lines, and with navitoclax in BCL-2-low NHL cell lines, and chemical segregation studies using venetoclax or A-1155463 show that BCL-XL inhibition is the principal driver of synergy in the latter.[139] S63845 is a more recently described MCL-1 inhibitor that potently induces BAX/BAK-dependent mitochondrial apoptosis in MM, leukemia and lymphoma cells, and exhibits potent in vivo anti-tumor activity against a number of different tumor types with good tolerability in mice.[140] Other compounds that inhibit MCL-1 at picomolar concentrations and cause MOMP and caspase activation in engineered cell lines, as well as inhibition of proliferation of primary MM and AML cells, have been discovered, laying the foundation for the development of clinical candidates.[141]
Thus, the (preclinical) availability of potent and selective small-molecule antagonists of all three major pro-survival BCL-2 family proteins now allows us to carefully dissect the anti-apoptotic protein dependencies of individual tumor types and select optimal drug combinations to test in the clinic. In the case of kinase inhibitors, the oncogenic kinase pathway dependence of leukemia cells can be identified in 70% of specimens in just three days using a rapid inhibitor screen.[142] Once other BH3-mimetics targeting BCL-XL and MCL-1 become available for clinical use, it is likely that BH3-profiling and similar assays will be widely used to predict tumor susceptibilities to the individual agents.
Conclusions
Venetoclax is the first BH3-mimetic to receive regulatory approval and its use is likely to expand to additional tumor types in the coming years. Predictably, high levels of BCL-XL and MCL-1, the two major anti-apoptotic proteins of the BCL-2 family not inhibited by venetoclax, are key determinants of both acquired and intrinsic resistance to venetoclax. This paves the way for a vast array of rational combinations as outlined in this article, both with emerging, selective antagonists of BCL-XL and/or MCL-1, and with other classes of targeted agents. Table 1 lists clinical trials combining venetoclax with both conventional and other novel agents in diverse hematologic tumor types. At present, there are no clinical trials evaluating this agent in solid tumors. Finally, improved techniques to assess apoptotic priming (e.g., mitochondrial profiling) and a more robust toolkit of potent and selective BH3-mimetics will help us better understand the pro-survival protein addiction patters of individual tumors and tailor therapy accordingly.
Table 1.
Ongoing and planned combination clinical trials involving venetoclax.
| Clinicaltrials.gov identifier | Disease state(s) | Partner drug(s) | Phase |
|---|---|---|---|
| NCT03000660 | R/R AL Amyloidosis | Dexamethasone | I |
| NCT02756897 | CLL; R/R or high risk, treatment-naive | Ibrutinib | II |
| NCT02951117 | R/R MM | ABBV-838 (ADC against CS-1); dex | IB |
| NCT02899052 | R/R MM | Carfilzomib; dex | II |
| NCT02966782 | MDS (HMA failures) | Azacitidine | IB |
| NCT02993523 | AML (elderly; treatment-naïve) | Azacitidine (vs. aza plus placebo) | III |
| NCT02942290 | MDS (previously untreated) | Azacitidine (vs. aza alone) | II |
| NCT02877550 | FL, treatment-naive | Obinutuzumab | I |
| NCT02758665 | Previously untreated CLL with TP53 deletion and/or mutation | Ibrutinib plus obinutuzumab | II |
| NCT02992522 | R/R B-NHL | Lenalidomide plus obinutuzumab | I |
| NCT02670044 | R/R AML | Cobimetinib (MEK inhibitor) or idasanutlin (HDM2 inhibitor) | IB/II |
| NCT02611323 | R/R FL or DLBCL | Obinutuzumab plus polatuzumab vedotin (ADC against CD79b) | Ib/II |
| NCT02987400 | R/R DLBCL | Obinutuzumab | II |
| NCT02910583 | Treatment-naïve CLL/SLL | Ibrutinib | II |
| NCT02755597 | MM (proteasome inhibitor candidates) | Bortezomib plus dex (vs. bortezomib plus dex plus placebo) | III |
| NCT02956382 | R/R FL | Ibrutinib | I/II |
| NCT01671904 | CLL; R/R or previously untreated | BR or bendamustine plus obinutuzumab | IB |
| NCT02950051 | Fit patients with previously untreated CLL without del17p or TP53 mutation | Rituximab or obinutuzumab or obinutuzumab plus ibrutinib (vs. FCR/BR without venetoclax) | III |
| NCT02427451 | CLL; R/R or previously untreated | Ibrutinib plus obinutuzumab | I/II |
| NCT02287233 | AML (elderly; treatment-naïve) | LDAC | I/II |
| NCT02419560 | R/R MCL | Ibrutinib | I/IB |
Abbreviations: R/R, relapsed/refractory; CLL, chronic lymphocytic leukemia; MM, multiple myeloma; ADC, antibody-drug conjugate; MDS, myelodysplastic syndrome; HMA, hypomethylating agent; AML, acute myeloid leukemia; FL, follicular lymphoma; B-NHL, B-cell non Hodgkin’s lymphoma; DLBCL, diffuse large B-cell lymphoma; MEK, mitogen activated protein kinase kinase; HDM2, human double minute 2; SLL, small lymphocytic lymphoma; LDAC, low dose cytarabine; MCL, mantle cell lymphoma; BR, bendamustine plus rituximab; FCR, fludarabine, cyclophosphamide and rituximab; dex, dexamethasone; aza, azacitidine.
Supplementary Material
Acknowledgments
This work was supported in part by the MD Anderson Cancer Center Support Grant P30 CA016672 from the National Institutes of Health. The authors thank Pankit Vachhani, M.D., Roswell Park Cancer Institute, Buffalo, NY, for his assistance with preparing the figure.
References
- 1.Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: A multicentre, open-label, phase 2 study. Lancet Oncol. 2016 doi: 10.1016/S1470-2045(16)30019-5. [DOI] [PubMed] [Google Scholar]
- 2.Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:311–22. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones J, Choi MY, Mato AR, et al. Venetoclax (VEN) monotherapy for patients with chronic lymphocytic leukemia (CLL) who relapsed after or were refractory to ibrutinib or idelalisib. Blood. 2016;128:637. [Google Scholar]
- 4.Kwok M, Rawstron AC, Varghese A, et al. Minimal residual disease is an independent predictor for 10-year survival in CLL. Blood. 2016;128:2770–3. doi: 10.1182/blood-2016-05-714162. [DOI] [PubMed] [Google Scholar]
- 5.Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016 doi: 10.1158/2159-8290.CD-16-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pollyea DA, Dinardo CD, Thirman MJ, et al. Results of a phase 1b study of venetoclax plus decitabine or azacitidine in untreated acute myeloid leukemia patients ≥ 65 years ineligible for standard induction therapy. J Clin Oncol. 2016;34:7009. [Google Scholar]
- 7.Vogler M, Weber K, Dinsdale D, et al. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ. 2009;16:1030–9. doi: 10.1038/cdd.2009.48. [DOI] [PubMed] [Google Scholar]
- 8.Albershardt TC, Salerni BL, Soderquist RS, et al. Multiple BH3 mimetics antagonize antiapoptotic MCL1 protein by inducing the endoplasmic reticulum stress response and up-regulating BH3-only protein NOXA. J Biol Chem. 2011;286:24882–95. doi: 10.1074/jbc.M111.255828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 10.Tse C, Shoemaker AR, Adickes J, et al. ABT-263: A potent and orally bioavailable bcl-2 family inhibitor. Cancer Res. 2008;68:3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 11.Gandhi L, Camidge DR, Ribeiro de Oliveira M, et al. Phase I study of navitoclax (ABT-263), a novel bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–16. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson WH, O’Connor OA, Czuczman MS, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11:1149–59. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rudin CM, Hann CL, Garon EB, et al. Phase 2 study of single agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012 doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts AW, Seymour JF, Brown JR, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30:488–96. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mason KD, Carpinelli MR, Fletcher JI, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–86. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 16.Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013 doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 17.Liu H, Michmerhuizen MJ, Lao Y, et al. Absorption, metabolism, and excretion of a novel bcl-2 inhibitor venetoclax in humans. Drug Metab Dispos. 2016 doi: 10.1124/dmd.116.071613. [DOI] [PubMed] [Google Scholar]
- 18.Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 19.Letai AG. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8:121–32. doi: 10.1038/nrc2297. [DOI] [PubMed] [Google Scholar]
- 20.Cheng EH, Levine B, Boise LH, Thompson CB, Hardwick JM. Bax-independent inhibition of apoptosis by bcl-XL. Nature. 1996;379:554–6. doi: 10.1038/379554a0. [DOI] [PubMed] [Google Scholar]
- 21.Cheng EH, Wei MC, Weiler S, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–11. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 22.Willis SN, Fletcher JI, Kaufmann T, et al. Apoptosis initiated when BH3 ligands engage multiple bcl-2 homologs, not bax or bak. Science. 2007;315:856–9. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 23.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–92. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 24.Wei MC, Lindsten T, Mootha VK, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–71. [PMC free article] [PubMed] [Google Scholar]
- 25.Kuwana T, Bouchier-Hayes L, Chipuk JE, et al. BH3 domains of BH3-only proteins differentially regulate bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–35. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Certo M, Del Gaizo Moore V, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 27.Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 28.Willis SN, Chen L, Dewson G, et al. Proapoptotic bak is sequestered by mcl-1 and bcl-xL, but not bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, bax, that accelerates programmed cell death. Cell. 1993;74:609–19. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 30.She QB, Solit DB, Ye Q, O’Reilly KE, Lobo J, Rosen N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell. 2005;8:287–97. doi: 10.1016/j.ccr.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, bim. J Biol Chem. 2003;278:18811–6. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- 32.Koss B, Ryan J, Budhraja A, et al. Defining specificity and on-target activity of BH3-mimetics using engineered B-ALL cell lines. Oncotarget. 2016;7:11500–11. doi: 10.18632/oncotarget.7204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCoy F, Hurwitz J, McTavish N, et al. Obatoclax induces Atg7-dependent autophagy independent of beclin-1 and BAX/BAK. Cell Death Dis. 2010;1:e108. doi: 10.1038/cddis.2010.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soderquist RS, Danilov AV, Eastman A. Gossypol increases expression of the pro-apoptotic BH3-only protein NOXA through a novel mechanism involving phospholipase A2, cytoplasmic calcium, and endoplasmic reticulum stress. J Biol Chem. 2014;289:16190–9. doi: 10.1074/jbc.M114.562900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–21. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson MA, Deng J, Seymour JF, et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood. 2016;127:3215–24. doi: 10.1182/blood-2016-01-688796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soderquist SR, Eastman A. BCL2 inhibitors as anticancer drugs: A plethora of misleading BH3 mimetics. Mol Cancer Ther. 2016;15:2011–7. doi: 10.1158/1535-7163.MCT-16-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Czabotar PE, Lee EF, van Delft MF, et al. Structural insights into the degradation of mcl-1 induced by BH3 domains. Proc Natl Acad Sci U S A. 2007;104:6217–22. doi: 10.1073/pnas.0701297104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomez-Bougie P, Menoret E, Juin P, Dousset C, Pellat-Deceunynck C, Amiot M. Noxa controls mule-dependent mcl-1 ubiquitination through the regulation of the mcl-1/USP9X interaction. Biochem Biophys Res Commun. 2011;413:460–4. doi: 10.1016/j.bbrc.2011.08.118. [DOI] [PubMed] [Google Scholar]
- 40.Yan J, Zhong N, Liu G, et al. Usp9x- and noxa-mediated mcl-1 downregulation contributes to pemetrexed-induced apoptosis in human non-small-cell lung cancer cells. Cell Death Dis. 2014;5:e1316. doi: 10.1038/cddis.2014.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–85. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 42.Vo TT, Ryan J, Carrasco R, et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. 2012;151:344–55. doi: 10.1016/j.cell.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davids MS, Deng J, Wiestner A, et al. Decreased mitochondrial apoptotic priming underlies stroma-mediated treatment resistance in chronic lymphocytic leukemia. Blood. 2012;120:3501–9. doi: 10.1182/blood-2012-02-414060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dousset C, Maiga S, Gomez-Bougie P, et al. BH3 profiling as a tool to identify acquired resistance to venetoclax in multiple myeloma. Br J Haematol. 2016 doi: 10.1111/bjh.14251. [DOI] [PubMed] [Google Scholar]
- 45.Touzeau C, Ryan J, Guerriero J, et al. BH3 profiling identifies heterogeneous dependency on bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia. 2016;30:761–4. doi: 10.1038/leu.2015.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chonghaile TN, Roderick JE, Glenfield C, et al. Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov. 2014;4:1074–87. doi: 10.1158/2159-8290.CD-14-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jain N, Lamb AV, O’Brien S, et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: A high-risk subtype. Blood. 2016;127:1863–9. doi: 10.1182/blood-2015-08-661702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anderson NM, Harrold I, Mansour MR, et al. BCL2-specific inhibitor ABT-199 synergizes strongly with cytarabine against the early immature LOUCY cell line but not more-differentiated T-ALL cell lines. Leukemia. 2014;28:1145–8. doi: 10.1038/leu.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishizawa J, Kojima K, McQueen T, et al. Mitochondrial profiling of acute myeloid leukemia in the assessment of response to apoptosis modulating drugs. PLoS One. 2015;10:e0138377. doi: 10.1371/journal.pone.0138377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Delft MF, Wei AH, Mason KD, et al. The BH3 mimetic ABT-737 targets selective bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if mcl-1 is neutralized. Cancer Cell. 2006;10:389–99. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Konopleva M, Contractor R, Tsao T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–88. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 52.Mazumder S, Choudhary GS, Al-Harbi S, Almasan A. Mcl-1 phosphorylation defines ABT-737 resistance that can be overcome by increased NOXA expression in leukemic B-cells. Cancer Res. 2012 doi: 10.1158/0008-5472.CAN-11-4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Glaser SP, Lee EF, Trounson E, et al. Anti-apoptotic mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012;26:120–5. doi: 10.1101/gad.182980.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gores GJ, Kaufmann SH. Selectively targeting mcl-1 for the treatment of acute myelogenous leukemia and solid tumors. Genes Dev. 2012;26:305–11. doi: 10.1101/gad.186189.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing bak activation and bax translocation. Cancer Res. 2007;67:782–91. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- 56.Zhang W, Konopleva M, Ruvolo VR, et al. Sorafenib induces apoptosis of AML cells via bim-mediated activation of the intrinsic apoptotic pathway. Leukemia. 2008;22:808–18. doi: 10.1038/sj.leu.2405098. [DOI] [PubMed] [Google Scholar]
- 57.Konopleva M, Milella M, Ruvolo P, et al. MEK inhibition enhances ABT-737-induced leukemia cell apoptosis via prevention of ERK-activated MCL-1 induction and modulation of MCL-1/BIM complex. Leukemia. 2012;26:778–87. doi: 10.1038/leu.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 58.Rahmani M, Aust MM, Attkisson E, Williams DC, Jr, Ferreira-Gonzalez A, Grant S. Dual inhibition of bcl-2 and bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and bim-dependent mechanism. Cancer Res. 2012 doi: 10.1158/0008-5472.CAN-12-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang W, Ruvolo VR, Gao C, et al. Evaluation of apoptosis induction by concomitant inhibition of MEK, mTOR, and bcl-2 in human acute myelogenous leukemia cells. Mol Cancer Ther. 2014;13:1848–59. doi: 10.1158/1535-7163.MCT-13-0576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115:3304–13. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Faber AC, Farago AF, Costa C, et al. Assessment of ABT-263 activity across a cancer cell line collection leads to a potent combination therapy for small-cell lung cancer. Proc Natl Acad Sci U S A. 2015;112:E1288–96. doi: 10.1073/pnas.1411848112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khaw SL, Merino D, Anderson MA, et al. Both leukaemic and normal peripheral B lymphoid cells are highly sensitive to the selective pharmacological inhibition of prosurvival bcl-2 with ABT-199. Leukemia. 2014;28:1207–15. doi: 10.1038/leu.2014.1. [DOI] [PubMed] [Google Scholar]
- 63.Vogler M, Butterworth M, Majid A, et al. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood. 2009;113:4403–13. doi: 10.1182/blood-2008-08-173310. [DOI] [PubMed] [Google Scholar]
- 64.Ponader S, Chen SS, Buggy JJ, et al. The bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119:1182–9. doi: 10.1182/blood-2011-10-386417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cervantes-Gomez F, Lamothe B, Woyach JA, et al. Pharmacological and protein profiling suggests venetoclax (ABT-199) as optimal partner with ibrutinib in chronic lymphocytic leukemia. Clin Cancer Res. 2015;21:3705–15. doi: 10.1158/1078-0432.CCR-14-2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee HH, Dadgostar H, Cheng Q, Shu J, Cheng G. NF-kappaB-mediated up-regulation of bcl-x and bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc Natl Acad Sci U S A. 1999;96:9136–41. doi: 10.1073/pnas.96.16.9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Herman SE, Gordon AL, Hertlein E, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117:6287–96. doi: 10.1182/blood-2011-01-328484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bojarczuk K, Sasi BK, Gobessi S, et al. BCR signaling inhibitors differ in their ability to overcome mcl-1-mediated resistance of CLL B cells to ABT-199. Blood. 2016;127:3192–201. doi: 10.1182/blood-2015-10-675009. [DOI] [PubMed] [Google Scholar]
- 70.Sharman J, Hawkins M, Kolibaba K, et al. An open-label phase 2 trial of entospletinib (GS-9973), a selective spleen tyrosine kinase inhibitor, in chronic lymphocytic leukemia. Blood. 2015;125:2336–43. doi: 10.1182/blood-2014-08-595934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oppermann S, Ylanko J, Shi Y, et al. High-content screening identifies kinase inhibitors that overcome venetoclax resistance in activated CLL cells. Blood. 2016;128:934–47. doi: 10.1182/blood-2015-12-687814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aguilar-Hernandez MM, Blunt MD, Dobson R, et al. IL-4 enhances expression and function of surface IgM in CLL cells. Blood. 2016 doi: 10.1182/blood-2015-11-682906. [DOI] [PubMed] [Google Scholar]
- 73.Blunt MD, Koehrer S, Dobson R, et al. The dual Syk/JAK inhibitor cerdulatinib antagonises B-cell receptor and microenvironmental signaling in chronic lymphocytic leukemia. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Al-Harbi S, Choudhary GS, Ebron JS, et al. miR-377-dependent BCL-xL regulation drives chemotherapeutic resistance in B-cell lymphoid malignancies. Mol Cancer. 2015;14:185,015-0460-8. doi: 10.1186/s12943-015-0460-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huelsemann MF, Patz M, Beckmann L, et al. Hypoxia-induced p38 MAPK activation reduces mcl-1 expression and facilitates sensitivity towards BH3 mimetics in chronic lymphocytic leukemia. Leukemia. 2015;29:981–4. doi: 10.1038/leu.2014.320. [DOI] [PubMed] [Google Scholar]
- 76.Bose P, Simmons GL, Grant S. Cyclin-dependent kinase inhibitor therapy for hematologic malignancies. Expert Opin Investig Drugs. 2013;22:723–38. doi: 10.1517/13543784.2013.789859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Flynn J, Jones J, Johnson AJ, et al. Dinaciclib is a novel cyclin-dependent kinase inhibitor with significant clinical activity in relapsed and refractory chronic lymphocytic leukemia. Leukemia. 2015;29:1524–9. doi: 10.1038/leu.2015.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Choudhary GS, Tat TT, Misra S, et al. Cyclin E/Cdk2-dependent phosphorylation of mcl-1 determines its stability and cellular sensitivity to BH3 mimetics. Oncotarget. 2015;6:16912–25. doi: 10.18632/oncotarget.4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thijssen R, Ter Burg J, Garrick B, et al. Dual TORK/DNA-PK inhibition blocks critical signaling pathways in chronic lymphocytic leukemia. Blood. 2016;128:574–83. doi: 10.1182/blood-2016-02-700328. [DOI] [PubMed] [Google Scholar]
- 80.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–2. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 81.Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–6. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 82.Bodo J, Zhao X, Durkin L, et al. Acquired resistance to venetoclax (ABT-199) in t(14;18) positive lymphoma cells. Oncotarget. 2016 doi: 10.18632/oncotarget.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Adem J, Ropponen A, Eeva J, Eray M, Nuutinen U, Pelkonen J. Differential expression of bcl-2 family proteins determines the sensitivity of human follicular lymphoma cells to dexamethasone-mediated and anti-BCR-mediated apoptosis. J Immunother. 2016;39:8–14. doi: 10.1097/CJI.0000000000000102. [DOI] [PubMed] [Google Scholar]
- 84.Choudhary GS, Al-Harbi S, Mazumder S, et al. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015;6:e1593. doi: 10.1038/cddis.2014.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Khaw SL, Suryani S, Evans K, et al. Venetoclax responses of pediatric ALL xenografts reveal sensitivity of MLL-rearranged leukemia. Blood. 2016;128:1382–95. doi: 10.1182/blood-2016-03-707414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Benito JM, Godfrey L, Kojima K, et al. MLL-rearranged acute lymphoblastic leukemias activate BCL-2 through H3K79 methylation and are sensitive to the BCL-2-specific antagonist ABT-199. Cell Rep. 2015;13:2715–27. doi: 10.1016/j.celrep.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Leonard JT, Rowley JS, Eide CA, et al. Targeting BCL-2 and ABL/LYN in philadelphia chromosome-positive acute lymphoblastic leukemia. Sci Transl Med. 2016;8:354ra114. doi: 10.1126/scitranslmed.aaf5309. [DOI] [PubMed] [Google Scholar]
- 88.Chiron D, Dousset C, Brosseau C, et al. Biological rational for sequential targeting of bruton tyrosine kinase and bcl-2 to overcome CD40-induced ABT-199 resistance in mantle cell lymphoma. Oncotarget. 2015;6:8750–9. doi: 10.18632/oncotarget.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chiron D, Bellanger C, Papin A, et al. Microenvironment-dependent proliferation and mitochondrial priming loss in mantle cell lymphoma is overcome by anti-CD20. Blood. 2016 doi: 10.1182/blood-2016-06-720490. [DOI] [PubMed] [Google Scholar]
- 90.Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369:507–16. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fresquet V, Rieger M, Carolis C, Garcia-Barchino MJ, Martinez-Climent JA. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood. 2014;123:4111–9. doi: 10.1182/blood-2014-03-560284. [DOI] [PubMed] [Google Scholar]
- 92.Touzeau C, Dousset C, Le Gouill S, et al. The bcl-2 specific BH3 mimetic ABT-199: A promising targeted therapy for t(11;14) multiple myeloma. Leukemia. 2014;28:210–2. doi: 10.1038/leu.2013.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gong JN, Khong T, Segal D, et al. Hierarchy for targeting pro-survival BCL2 family proteins in multiple myeloma: Pivotal role of MCL1. Blood. 2016 doi: 10.1182/blood-2016-03-704908. [DOI] [PubMed] [Google Scholar]
- 94.Raje N, Kumar S, Hideshima T, et al. Seliciclib (CYC202 or R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates activity via down-regulation of mcl-1 in multiple myeloma. Blood. 2005;106:1042–7. doi: 10.1182/blood-2005-01-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nikiforov MA, Riblett M, Tang WH, et al. Tumor cell-selective regulation of NOXA by c-MYC in response to proteasome inhibition. Proc Natl Acad Sci U S A. 2007;104:19488–93. doi: 10.1073/pnas.0708380104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Punnoose EA, Leverson JD, Peale F, et al. Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist venetoclax in multiple myeloma models. Mol Cancer Ther. 2016;15:1132–44. doi: 10.1158/1535-7163.MCT-15-0730. [DOI] [PubMed] [Google Scholar]
- 97.Bajpai R, Matulis SM, Wei C, et al. Targeting glutamine metabolism in multiple myeloma enhances BIM binding to BCL-2 eliciting synthetic lethality to venetoclax. Oncogene. 2016;35:3955–64. doi: 10.1038/onc.2015.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lavik AR, Zhong F, Chang MJ, et al. A synthetic peptide targeting the BH4 domain of bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the BH3-binding pocket of bcl-2. Oncotarget. 2015;6:27388–402. doi: 10.18632/oncotarget.4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pan R, Hogdal LJ, Benito JM, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014 doi: 10.1158/2159-8290.CD-13-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the international working group for diagnosis, standardization of response criteria, treatment outcomes, and reporting standards for therapeutic trials in acute myeloid leukemia. J Clin Oncol. 2003;21:4642–9. doi: 10.1200/JCO.2003.04.036. [DOI] [PubMed] [Google Scholar]
- 101.Chan SM, Thomas D, Corces-Zimmerman MR, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015;21:178–84. doi: 10.1038/nm.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lin KH, Winter PS, Xie A, et al. Targeting MCL-1/BCL-XL forestalls the acquisition of resistance to ABT-199 in acute myeloid leukemia. Sci Rep. 2016;6:27696. doi: 10.1038/srep27696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kojima K, Konopleva M, Samudio IJ, et al. MDM2 antagonists induce p53-dependent apoptosis in AML: Implications for leukemia therapy. Blood. 2005;106:3150–9. doi: 10.1182/blood-2005-02-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kojima K, Konopleva M, Samudio IJ, Schober WD, Bornmann WG, Andreeff M. Concomitant inhibition of MDM2 and bcl-2 protein function synergistically induce mitochondrial apoptosis in AML. Cell Cycle. 2006;5:2778–86. doi: 10.4161/cc.5.23.3520. [DOI] [PubMed] [Google Scholar]
- 105.Carter BZ, Mak PY, Mak DH, et al. Synergistic effects of p53 activation via MDM2 inhibition in combination with inhibition of bcl-2 or bcr-abl in CD34+ proliferating and quiescent chronic myeloid leukemia blast crisis cells. Oncotarget. 2015;6:30487–99. doi: 10.18632/oncotarget.5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lehmann C, Friess T, Birzele F, Kiialainen A, Dangl M. Superior anti-tumor activity of the MDM2 antagonist idasanutlin and the bcl-2 inhibitor venetoclax in p53 wild-type acute myeloid leukemia models. J Hematol Oncol. 2016;9:50,016-0280-3. doi: 10.1186/s13045-016-0280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pan R, Ruvolo V, Mu H, et al. BCL-2 inhibition by ABT-199 (Venetoclax/GDC-0199) and p53 activation by RG7388 (idasanutlin) reciprocally overcome leukemia apoptosis resistance to either strategy alone: Efficacy and mechanisms. Blood. 2015;126:673. [Google Scholar]
- 108.Carter BZ, Mak PY, Mu H, et al. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci Transl Med. 2016;8:355ra117. doi: 10.1126/scitranslmed.aag1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rahmani M, Davis EM, Bauer C, Dent P, Grant S. Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of mcl-1 through inhibition of translation. J Biol Chem. 2005;280:35217–27. doi: 10.1074/jbc.M506551200. [DOI] [PubMed] [Google Scholar]
- 110.Rahmani M, Davis EM, Crabtree TR, et al. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol Cell Biol. 2007;27:5499–513. doi: 10.1128/MCB.01080-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rollig C, Serve H, Huttmann A, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16:1691–9. doi: 10.1016/S1470-2045(15)00362-9. [DOI] [PubMed] [Google Scholar]
- 112.Bogenberger JM, Kornblau SM, Pierceall WE, et al. BCL-2 family proteins as 5-azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia. 2014;28:1657–65. doi: 10.1038/leu.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tsao T, Shi Y, Kornblau S, et al. Concomitant inhibition of DNA methyltransferase and BCL-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells. Ann Hematol. 2012;91:1861–70. doi: 10.1007/s00277-012-1537-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jacque N, Ronchetti AM, Larrue C, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood. 2015;126:1346–56. doi: 10.1182/blood-2015-01-621870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Emadi A, Jun SA, Tsukamoto T, Fathi AT, Minden MD, Dang CV. Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp Hematol. 2014;42:247–51. doi: 10.1016/j.exphem.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 116.Wang Q, Mora-Jensen H, Weniger MA, et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc Natl Acad Sci U S A. 2009;106:2200–5. doi: 10.1073/pnas.0807611106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Swords RT, Kelly KR, Smith PG, et al. Inhibition of NEDD8-activating enzyme: A novel approach for the treatment of acute myeloid leukemia. Blood. 2010;115:3796–800. doi: 10.1182/blood-2009-11-254862. [DOI] [PubMed] [Google Scholar]
- 118.Swords RT, Erba HP, DeAngelo DJ, et al. Pevonedistat (MLN4924), a first-in-class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: A phase 1 study. Br J Haematol. 2015;169:534–43. doi: 10.1111/bjh.13323. [DOI] [PubMed] [Google Scholar]
- 119.Knorr KL, Schneider PA, Meng XW, et al. MLN4924 induces noxa upregulation in acute myelogenous leukemia and synergizes with bcl-2 inhibitors. Cell Death Differ. 2015;22:2133–42. doi: 10.1038/cdd.2015.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ishizawa J, Kojima K, Chachad D, et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci Signal. 2016;9:ra17. doi: 10.1126/scisignal.aac4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bate-Eya LT, den Hartog IJ, van der Ploeg I, et al. High efficacy of the BCL-2 inhibitor ABT199 (venetoclax) in BCL-2 high-expressing neuroblastoma cell lines and xenografts and rational for combination with MCL-1 inhibition. Oncotarget. 2016;7:27946–58. doi: 10.18632/oncotarget.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mattoo AR, Zhang J, Espinoza LA, Jessup JM. Inhibition of NANOG/NANOGP8 downregulates MCL-1 in colorectal cancer cells and enhances the therapeutic efficacy of BH3 mimetics. Clin Cancer Res. 2014;20:5446–55. doi: 10.1158/1078-0432.CCR-14-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tamaki H, Harashima N, Hiraki M, et al. Bcl-2 family inhibition sensitizes human prostate cancer cells to docetaxel and promotes unexpected apoptosis under caspase-9 inhibition. Oncotarget. 2014;5:11399–412. doi: 10.18632/oncotarget.2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vaillant F, Merino D, Lee L, et al. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer Cell. 2013;24:120–9. doi: 10.1016/j.ccr.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 125.Shoemaker AR, Oleksijew A, Bauch J, et al. A small-molecule inhibitor of bcl-XL potentiates the activity of cytotoxic drugs in vitro and in vivo. Cancer Res. 2006;66:8731–9. doi: 10.1158/0008-5472.CAN-06-0367. [DOI] [PubMed] [Google Scholar]
- 126.Ponassi R, Biasotti B, Tomati V, et al. A novel bim-BH3-derived bcl-XL inhibitor: Biochemical characterization, in vitro, in vivo and ex-vivo anti-leukemic activity. Cell Cycle. 2008;7:3211–24. doi: 10.4161/cc.7.20.6830. [DOI] [PubMed] [Google Scholar]
- 127.Doi K, Li R, Sung SS, et al. Discovery of marinopyrrole A (maritoclax) as a selective mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting mcl-1 for proteasomal degradation. J Biol Chem. 2012;287:10224–35. doi: 10.1074/jbc.M111.334532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Varadarajan S, Poornima P, Milani M, et al. Maritoclax and dinaciclib inhibit MCL-1 activity and induce apoptosis in both a MCL-1-dependent and -independent manner. Oncotarget. 2015;6:12668–81. doi: 10.18632/oncotarget.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Varadarajan S, Vogler M, Butterworth M, Dinsdale D, Walensky LD, Cohen GM. Evaluation and critical assessment of putative MCL-1 inhibitors. Cell Death Differ. 2013;20:1475–84. doi: 10.1038/cdd.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pan R, Ruvolo VR, Wei J, et al. Inhibition of mcl-1 with the pan-bcl-2 family inhibitor (-)BI97D6 overcomes ABT-737 resistance in acute myeloid leukemia. Blood. 2015;126:363–72. doi: 10.1182/blood-2014-10-604975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Abulwerdi F, Liao C, Liu M, et al. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther. 2014;13:565–75. doi: 10.1158/1535-7163.MCT-12-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Abulwerdi FA, Liao C, Mady AS, et al. 3-substituted-N-(4-hydroxynaphthalen-1-yl)arylsulfonamides as a novel class of selective mcl-1 inhibitors: Structure-based design, synthesis, SAR, and biological evaluation. J Med Chem. 2014;57:4111–33. doi: 10.1021/jm500010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lessene G, Czabotar PE, Sleebs BE, et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat Chem Biol. 2013;9:390–7. doi: 10.1038/nchembio.1246. [DOI] [PubMed] [Google Scholar]
- 134.Tao ZF, Hasvold L, Wang L, et al. Discovery of a potent and selective BCL-XL inhibitor with in vivo activity. ACS Med Chem Lett. 2014;5:1088–93. doi: 10.1021/ml5001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Park D, Magis AT, Li R, et al. Novel small-molecule inhibitors of bcl-XL to treat lung cancer. Cancer Res. 2013;73:5485–96. doi: 10.1158/0008-5472.CAN-12-2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Croker BA, O’Donnell JA, Nowell CJ, et al. Fas-mediated neutrophil apoptosis is accelerated by bid, bak, and bax and inhibited by bcl-2 and mcl-1. Proc Natl Acad Sci U S A. 2011;108:13135–40. doi: 10.1073/pnas.1110358108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Leverson JD, Phillips DC, Mitten MJ, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. 2015;7:279ra40. doi: 10.1126/scitranslmed.aaa4642. [DOI] [PubMed] [Google Scholar]
- 138.Leverson JD, Zhang H, Chen J, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax) Cell Death Dis. 2015;6:e1590. doi: 10.1038/cddis.2014.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Phillips DC, Xiao Y, Lam LT, et al. Loss in MCL-1 function sensitizes non-hodgkin’s lymphoma cell lines to the BCL-2-selective inhibitor venetoclax (ABT-199) Blood Cancer J. 2015;5:e368. doi: 10.1038/bcj.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kotschy A, Szlavik Z, Murray J, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477–82. doi: 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- 141.Lee T, Bian Z, Zhao B, et al. Discovery and biological characterization of potent myeloid cell leukemia-1 inhibitors. FEBS Lett. 2016 doi: 10.1002/1873-3468.12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tyner JW, Yang WF, Bankhead A, 3rd, et al. Kinase pathway dependence in primary human leukemias determined by rapid inhibitor screening. Cancer Res. 2013;73:285–96. doi: 10.1158/0008-5472.CAN-12-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.