

Abstract:
Port wine stain (PWS) is a congenital, progressive vascular malformation. Many patients with PWS develop hypertrophy and discrete nodularity during their adult life, but the mechanism(s) remain incompletely understood. In this study, we attempted to investigate activation status of PKCα, PI3K, PDPK1 and PLC-γ and protein levels of PP2A and DAG to explore their potential roles in the formation of hypertrophic and nodular PWS lesions. We found phosphorylated levels of PKCα, PI3K, PDPK1, and PLC-γ and protein levels of PP2A and DAG showed moderate increases in the endothelial cells of hypertrophic PWS as compared to the adjacent normal skin. These increases extended throughout the entire stroma of blood vessels in PWS nodules. Many proliferating cells, such as fibroblasts, also showed strong activation of PKCα, PI3K, PDPK1 and PLC-γ and upregulations of PP2A and DAG in nodular PWS lesions. Our data showed that there is aberrant activation of PKCα, PI3K, PDPK1 and PLC-γ and upregulation of PP2A and DAG mainly in endothelial cells in hypertrophic PWS areas, but presenting in the entire vasculatures and surrounding fibroblasts in PWS nodules. Our data suggest that both PKCα and PI3K signaling pathways contribute to the development of hypertrophy and nodularity in adult PWS.
Key Words: port wine stain, PI3K, PKCα, PLC-γ, PDPK1, vascular malformation
INTRODUCTION
Port wine stain (PWS) is a congenital, progressive vascular malformation of human skin involving the superficial vascular plexus that occurs in estimated 3–5 children per 1,000 live births.1–3 Because most malformations occur on the face, PWS is a clinically significant problem in the majority of patients. Personality development is adversely influenced in virtually all patients by the negative reaction of others to a “marked” person. Detailed studies have documented lower self-esteem in such patients and problems with interpersonal relationships.4–6
In childhood, PWS are flat red macules, but lesions tend to darken progressively to purple and, by middle age, often become raised as a result of the development of vascular nodules.7,8 The late-stage cobblestoning appearance of PWS subjects is comprised by not only pronounced vascular ectasia with proliferation of thin and/or thick-walled vessels and their stroma, but also numerous epithelial, neural and mesenchymal hamartomatous abnormalities.9 Despite these histologic observations, the specific mechanisms involved in PWS nodular formation remains unclear. Our previous data have shown a sequential activation profile of various kinases during different stages of PWS.10 In one nodular PWS subject, we found that phosphatidylinositol 3-kinases (PI3K)/protein kinase B (AKT) and phosphoinositide phospholipase C γ subunit (PLC-γ) were activated in both hypertrophic areas and nodules within the lesion.10 These observations led us to hypothesize that the PI3K pathway may play an important role in nodular formation. In this study, we attempt to further investigate the phosphorylation levels of various kinases involved in the PI3K pathway, including 3-phosphoinositide-dependent protein kinase-1 (PDPK1), protein kinase C alpha (PKCα), protein phosphatase 2α (PP2A), PI3K, 1,2-Diacylglycerol (DAG) and PLC-γ, in hypertrophic and nodular PWS lesions.
MATERIALS AND METHODS
Patients and Tissue Samples
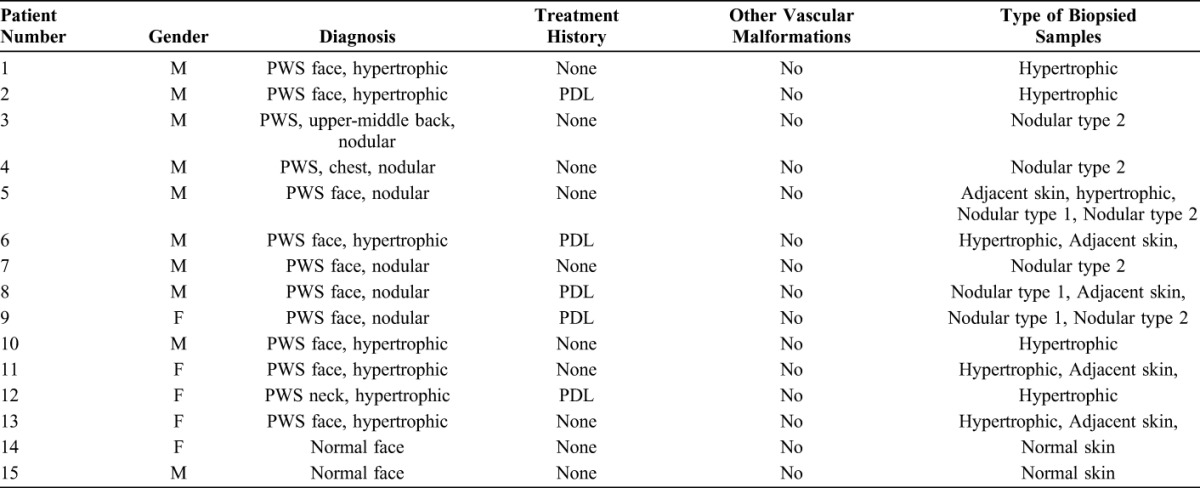
The study was approved by the Investigational Review Board at the Xijing Hospital, Xi'an, China. A total of 21 PWS biopsy samples, including 8 hypertrophic, 8 nodular PWS specimens, and 5 specimens from adjacent normal skin 0.5–1 cm away from PWS lesions, were obtained from 13 subjects and deidentified for this study. Two normal adult skin samples were retrieved from the skin biopsy tissue bank from our department to serve as normal controls. We categorize PWS nodules into 2 types based on their histopathological characterizations. Type 1 is mainly composed of large ectatic, thick or thin-walled blood vessels. Type 2 has vascular lobules as the vascular proliferative sites. It is composed of massive capillaries and venule-like vasculatures with both ectatic and/or nonectatic blood vessels. Nodular patients may present either one or both types of nodules within their lesions. The clinical history of PWS subjects and list of biopsy samples are showed in Table 1.
TABLE 1.
Clinical Description of Adult PWS Subjects and Types of Biopsied Samples
Histology
Skin specimens were fixed in Bouin's solution overnight. Then, they were dehydrated, cleaned, and embedded in paraffin and sectioned to 5 μm thick. The sections were stained with hematoxylin and eosin (H&E) and photographed under an optical microscope.
Immunohistochemistry
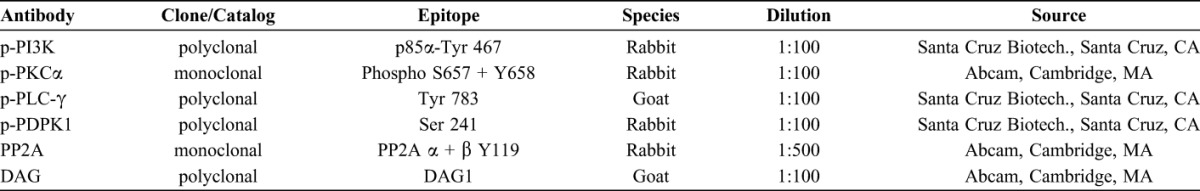
Routine immunohistochemistry procedures were used as follows. Briefly, the fresh biopsy tissue was fixed in 10% buffered formalin (Fisher Scientific, Pittsburgh, PA) and processed for permanent paraffin embedding on an ASP 300 tissue processor (Leica Microsystems, Bannockburn, IL). Approximately 6-μm-thick paraffin sections were cut and collected. Antigen retrieval was performed in 10 mM sodium citrate buffer (pH 6.0) at 97 °C for 4–5.5 hours. Sections were then incubated in a humidified chamber overnight at 4 °C with the following primary antibodies: anti-p-PI3K (Santa Cruz Biotech., Santa Cruz, CA), anti-p-PKCα (Abcam, Cambridge, MA), anti-p-PLC-γ (Santa Cruz Biotech), anti-p-PDPK1 (Santa Cruz Biotech), anti-PP2A (Abcam), and anti-DAG (Abcam). Biotinylated anti-mouse and anti-rabbit secondary antibodies were incubated with the sections for 2 hours at room temperature after the primary antibodies' reaction. An indirect biotin avidin diaminobenzidine (DAB) system (Dako, Glostrup, Denmark) was used for detection. The cellular immunoreactivity score was evaluated using a system previously reported by Populo et al.11 Briefly, four scales were used to evaluate the immunoreacive (IR) intensity scores: 0 (no staining), 1+ (weak), 2+ (intermediate), and 3+ (strong staining). Two scales were used to assess the percentage scores of IR cells: 1 (≤30%) and 2 (>30%). The final immunoreactivity scores of each antibody were estimated by multiplying the IR intensity and percentage scores, which were categorized as follows: 0 (negative), 1 (low), moderate (2, 3, and 4), and 6 (high).11 The summary of antibodies we used and their resources were listed in Table 2.
TABLE 2.
Summary of the Antibodies Used in This Study
RESULTS
Typical pathological features of vascular anomalies were observed in a series of biopsies from adjacent normal skin to hypertrophic areas to nodules within the lesions obtained from one facial nodular PWS subject (Fig. 1, Table 1, subject #5). Some capillaries and venule-like vasculatures in the reticular dermis in the normal adjacent skin showed mild enlargement (Fig. 1A), indicating the presence of some initial pathological changes. In the areas of PWS hypertrophy, there were numerous ectatic, thick or thin-walled vasculatures engorged with blood throughout the entire dermis (Fig. 1B). The nodules arising from PWS hypertrophied regions usually had one or both of the following types of structures: type 1 was mainly composed of large ectatic, thick or thin-walled blood vessels; and type 2 contained vascular lobules that appeared as proliferating vascular sites that were composed of massive capillaries and venule-like vasculatures, with both ectatic or nonectatic blood vessels. The proliferation of pericytes and fibroblasts was present in both cases, but more evident in type 2 (Fig. 1D) as compared to type 1 nodules (Fig. 1C).
FIGURE 1.
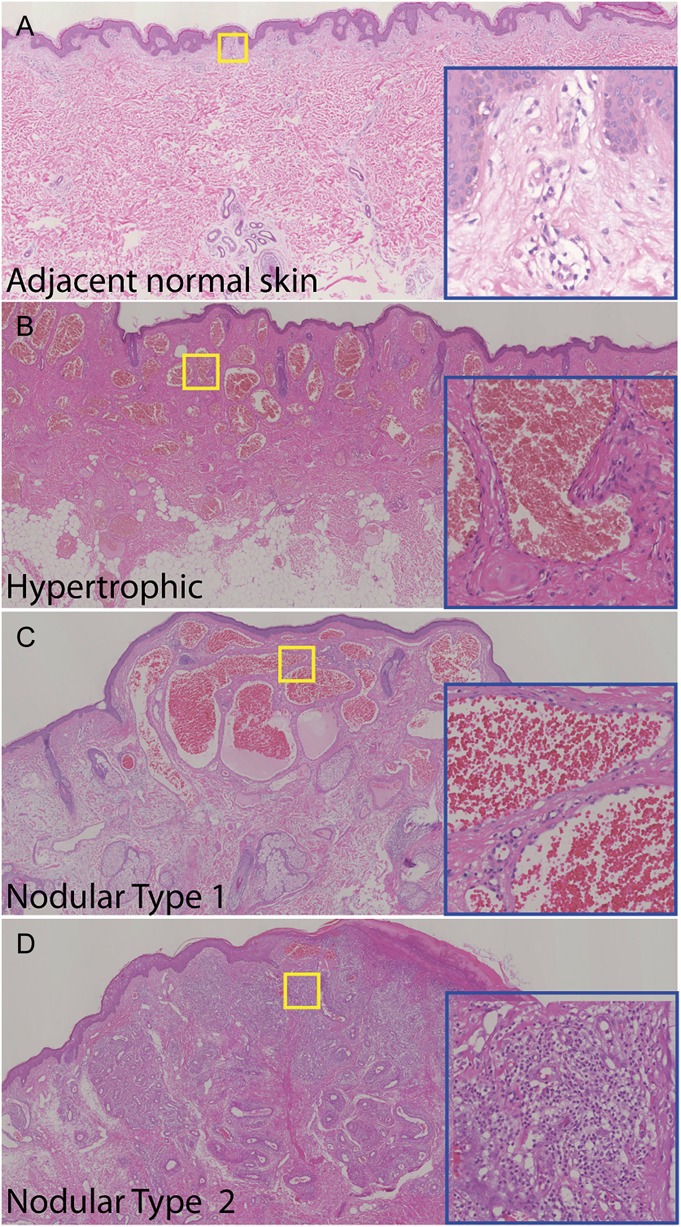
Histopathology of vascular anomalies in adjacent normal skin (A), hypertrophic area (B), and 2 types of nodules within the lesions from a PWS nodular subject (C, D) (#5). H&E staining. The blue insets are higher magnifications of the yellow boxed areas.
In the normal skin adjacent to PWS, endothelial cells (ECs) in those nondilated blood vessels showed very mild IR signals for the p-PI3K, p-PKCα, p-PLC-γ, and p-PDPK1. Some scattered pericytes and fibroblasts with moderate IR signal for these antibodies were also observed. The early dilated blood vessels showed an increase in activation of these kinases (Figs. 2, 3). In hypertrophic PWS, ECs and a small portion of pericytes showed moderate to strong activation of PI3K, PKCα, PLC-γ, and PDPK1. The scattered IR-positive fibroblasts could also be observed. Strong activation of PI3K, PKCα, PLC-γ, and PDPK1 was shown in all ECs and the majority of replicated pericytes and fibroblasts that surrounded PWS blood vessels in the nodules (Figs. 2, 3). There were scattered individual ECs, pericytes, and fibroblasts with very mild to moderate activation of PI3K, PKCα, PLC-γ, and PDPK1 in normal skin. The relative IR intensity scores of p-PI3K, p-PKCα, p-PLC-γ, and p-PDPK1 showed a progressive increase from normal skin to hypertrophic to nodular PWS, which were positively correlated with PWS pathological progression (Fig. 4).
FIGURE 2.
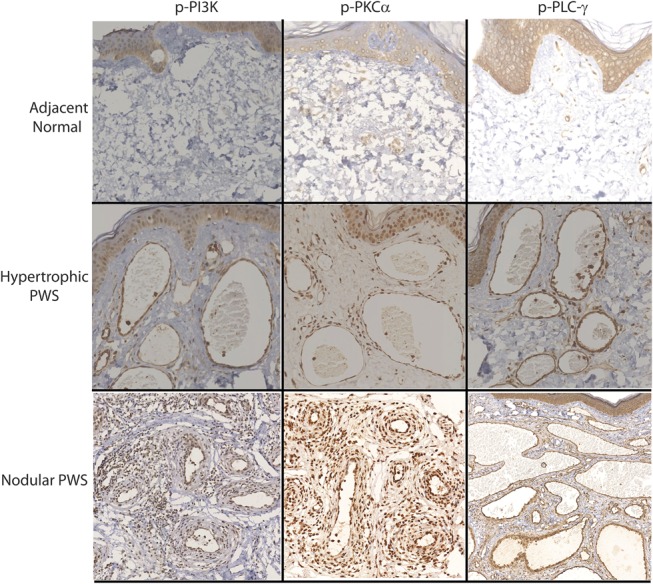
Activation of PI3K, PKCα, and PLC-γ in the blood vessels from different types of PWS lesions.
FIGURE 3.
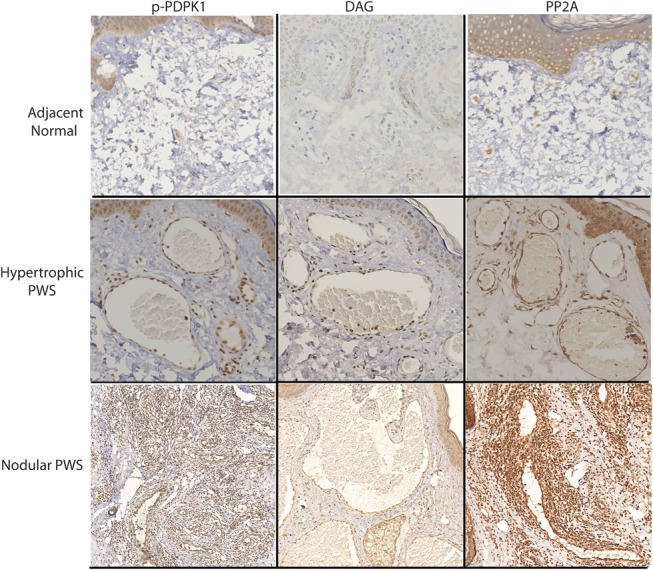
Activation of PDPK1 and expressions of DAG and PP2A in the blood vessels from different types of PWS lesions.
FIGURE 4.
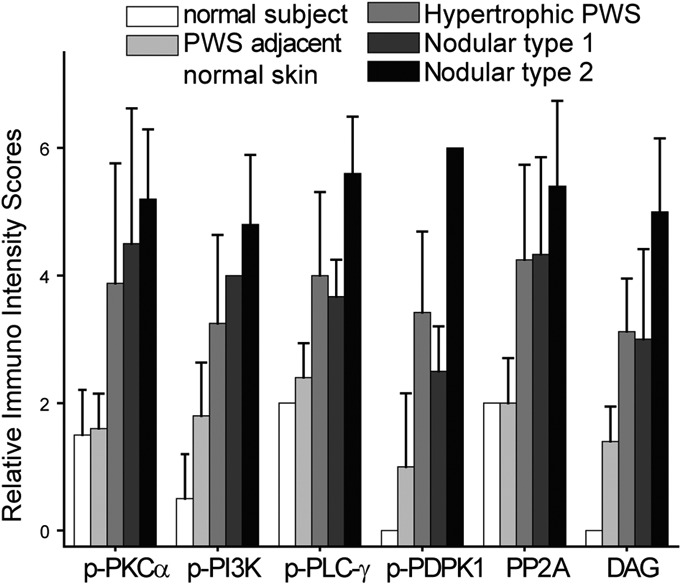
Relative IR intensity scores of p-PI3K, p-PKCα, p-PLC-γ, p-PDPK1, DAG, and PP2A in the blood vessels from different types of PWS lesions.
In normal skin adjacent to PWS lesions, there was no obvious IR signal observed. A mild IR signal for PP2A could be mainly observed in some scattered pericytes and fibroblasts (Fig. 3). A moderate expression of DAG was observed in approximately two-thirds of ECs and a few scattered pericytes in hypertrophic lesional areas. In nodules, all ECs and a few scattered pericytes and fibroblasts had a moderate-to-strong IR signal for DAG. PP2A showed a moderate expression level in all ECs and a few scattered pericytes in hypertrophic lesional areas, but it had a very strong IR signal throughout the entire thickened blood vessels, including ECs, replicated pericytes and fibroblasts, in nodules (Fig. 3). The increase in relative IR intensity scores of DAG and PP2A correlated with the pathological progression of PWS lesions (Fig. 4).
DISCUSSION
There have been 2 long-standing hypotheses regarding the pathogenesis of PWS birthmarks. One is a lack of nerve innervation to blood vessels contributing to development of PWS; the other is genetic mutations causing PWS, which is now favored by many researchers. Recent studies have suggested that sporadic somatic mutations of guanine nucleotide-binding protein, G alpha subunit q (Gnaq) (R183Q) and phosphatidylinositol 3-kinase catalytic, alpha polypeptide (Pi3kca) are linked to the vascular phenotypes of PWS.12–14 Shirley et al13 first discovered a low-frequency somatic mutation in the Gnaq gene (c.548 G→A, p.R183Q) present in PWS lesions. We and Cuoto et al further identified that Gnaq (R183Q) is primarily present in blood vessels (60%)15,16 and/or in connective tissue (30%)16 and hair follicle/glands (20%)16 in PWS lesions. These data suggest that pluripotent cells with Gnaq (R183Q) may give rise to multilineages in PWS.16 Another important candidate gene is Pi3kca where a mutation (p.G1049N) has been identified in PWS nodular lesions.12 Other somatic mutations, such as p.C420R, p.E542K, p.E545K, p.H1047R and p.H1047L, have been found in lymphatic and vascular malformation disorders.14 These findings suggest that Pi3kca alone or together with other genetic alterations, such as Gnaq mutations or environmental factors, contribute to the development of PWS. The mutation status of the Pi3kca and Gnaq genes in these PWS subjects studied here is unknown, which will be a subject of future study.
Finley proposed in 1984 that the cobblestoning appearance of the skin surface, thickening, and discrete nodular growths that develop during the adult life of PWS subjects was the result of pronounced vascular ectasia and proliferations of thin and/or thick-walled vessels and their stroma.17 However, the detailed molecular mechanism(s) underlying hypertrophy and the formation of nodules within PWS lesions remain obscure. Our previous data from limited nodular biopsies have indicated that PI3K and PLC-γ are activated in PWS nodular lesions. Furthermore, the Pi3kca mutation (p.G1049N) has been identified in nodular lesions from one PWS subject,12 which may contribute to the activation of PI3K. Moreover, Pi3kca mutations have been identified in many other vascular malformations, such as venous and lymphatic malformations.18,19 This evidence leads us to hypothesize that the PI3K and PLC-γ pathways play important roles in PWS nodular development. In this study, we performed a full investigation of the activation profiles of PI3K, PKCα, PLC-γ, and PDPK1 in PWS nodules. We found that all of them have strong activation in PWS ECs in ectatic thin or thick-walled blood vessels and in proliferated pericytes and fibroblasts, which directly supports our hypothesis. This study, together with our previous reports,10,20 is the first time that the molecular pathological signaling pathways underlying PWS hypertrophy and nodularity have been revealed.
Nodules are estimated to occur in 44% of untreated patients with an average age onset of 22 years21; other reports show that the nodular percentage in mixed subjects with treated or untreated PWS is approximately 10% with a mean onset in the 20–39-year age group.22,23 These variations are probably due to early laser intervention, which can significantly delay the onset of PWS nodularity. Hypertrophy and nodularity respond poorly to most vascular lasers.24–28 Thus, new treatments are required for managing this challenging clinical problem. In our previous investigations, we have proposed that a better PWS therapeutic outcome might be achieved with PDL combined with the administration of antiangiogenesis agents.29–32 In this study, we have confirmed that PKCα, PI3K, PLC-γ, and PDPK1 are strongly activated in PWS nodular lesions, suggesting their pivotal roles in nodular formation. Therefore, laser combined with administration of inhibitors of these kinases, and their associated pathways, may be a potentially novel therapeutic strategy for nodular PWS subjects.
In summary, we have shown a progressive activation of PKCα, PDPK1, and PLC-γ and increases in the expression of PP2A and DAG in PWS hypertrophic and nodular lesions, which are positively correlated with the pathological progression of this disease. Our data have confirmed that both PKCα and PI3K signaling pathways play very critical roles in the development of PWS hypertrophy and nodularity, suggesting potential applications of antagonists to these kinases as a possible treatment strategy for hypertrophic and nodular PWS.
Footnotes
Supported by National Natural Scientific Foundation of China (81301355 to LG, 81430073 and 81220108016 to GW), and NIH AR063766 to WT.
R. Yin and L. Gao contributed equally to this work.
The authors have no conflict of interest to declare.
REFERENCES
- 1.Mulliken JB, Young AR. Vascular Birthmarks–Hemangiomas and Malformations. Philadelphia, PA: W.B. Saunders Co.; 1988. [Google Scholar]
- 2.Jacobs AH, Walton RG. The incidence of birthmarks in the neonate. Pediatrics. 1976;58:218–222. [PubMed] [Google Scholar]
- 3.Pratt AG. Birthmarks in infants. Arch Dermatol Syphilol. 1953;67:302–305. [DOI] [PubMed] [Google Scholar]
- 4.Heller A, Rafman S, Zvagulis I, et al. Birth-defects and psychosocial adjustment. Am J Dis Child. 1985;139:257–263. [DOI] [PubMed] [Google Scholar]
- 5.Malm M, Carlberg M. Port-wine stain—a surgical and psychological problem. Ann Plast Surg. 1988;20:512–516. [DOI] [PubMed] [Google Scholar]
- 6.Kalick SM. Toward an interdisciplinary psychology of appearances. Psychiatry. 1978;41:243–253. [PubMed] [Google Scholar]
- 7.Lever WF, Schaumburg-Lever G. Histopathology of the skin. 7th ed Philadelphia, PA: J.B. Lippincott Co; 1990. [Google Scholar]
- 8.Geronemus RG, Ashinoff R. The medical necessity of evaluation and treatment of port-wine stains. J Dermatol Surg Oncol. 1991;17:76–79. [DOI] [PubMed] [Google Scholar]
- 9.Sanchez-Carpintero I, Mihm MC, Mizeracki A, et al. Epithelial and mesenchymal hamartomatous changes in a mature port-wine stain: morphologic evidence for a multiple germ layer field defect. J Am Acad Dermatol. 2004;50:608–612. [DOI] [PubMed] [Google Scholar]
- 10.Tan W, Chernova M, Gao L, et al. Sustained activation of c-Jun N-terminal and extracellular signal-regulated kinases in port-wine stain blood vessels. J Am Acad Dermatol. 2014;71:964–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Populo H, Vinagre J, Lopes JM, et al. Analysis of GNAQ mutations, proliferation and MAPK pathway activation in uveal melanomas. Br J Ophthalmol. 2011;95:715–719. [DOI] [PubMed] [Google Scholar]
- 12.Lian CG, Sholl LM, Zakka LR, et al. Novel genetic mutations in a sporadic port-wine stain. JAMA Dermatol. 2014;150:1336–1340. [DOI] [PubMed] [Google Scholar]
- 13.Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luks VL, Kamitaki N, Vivero MP, et al. Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J Pediatr. 2015;166:1048–1054. e1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Couto JA, Huang L, Vivero MP, et al. Endothelial cells from capillary malformations are enriched for somatic GNAQ mutations. Plast Reconstr Surg. 2016;137:77e–82e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan W, Nadora DM, Gao L, et al. The somatic GNAQ mutation (R183Q) is primarily located in port wine stain blood vessels. J Am Acad Dermatol. 2016;74:380–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finley JL, Noe JM, Arndt KA, et al. Port-wine stains. Morphologic variations and developmental lesions. Arch Dermatol. 1984;120:1453–1455. [DOI] [PubMed] [Google Scholar]
- 18.Boscolo E, Coma S, Luks VL, et al. AKT hyper-phosphorylation associated with PI3K mutations in lymphatic endothelial cells from a patient with lymphatic malformation. Angiogenesis. 2015;18:151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Limaye N, Kangas J, Mendola A, et al. Somatic activating PIK3CA mutations cause venous malformation. Am J Hum Genet. 2015;97:914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan W, Zakka LR, Gao L, et al. Pathological alterations involve the entire skin physiological Milieu in infantile and early childhood port wine stain. Br J Dermatol. 2016. DOI: 10.1111/bjd.15068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JW, Chung HY, Cerrati EW, et al. The natural history of soft tissue hypertrophy, bony hypertrophy, and nodule formation in patients with untreated head and neck capillary malformations. Dermatol Surg. 2015;41:1241–1245. [DOI] [PubMed] [Google Scholar]
- 22.Mills CM, Lanigan SW, Hughes J, et al. Demographic study of port wine stain patients attending a laser clinic: family history, prevalence of naevus anaemicus and results of prior treatment. Clin Exp Dermatol. 1997;22:166–168. [PubMed] [Google Scholar]
- 23.Klapman MH, Yao JF. Thickening and nodules in port-wine stains. J Am Acad Dermatol. 2001;44:300–302. [DOI] [PubMed] [Google Scholar]
- 24.Izikson L, Nelson JS, Anderson RR. Treatment of hypertrophic and resistant port wine stains with a 755 nm laser: a case series of 20 patients. Lasers Surg Med. 2009;41:427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kono T, Frederick Groff W, Chan HH, et al. Long-pulsed neodymium: yttrium-aluminum-garnet laser treatment for hypertrophic port-wine stains on the lips. J Cosmet Laser Ther. 2009;11:11–13. [DOI] [PubMed] [Google Scholar]
- 26.Chen D, Hu XJ, Lin XX, et al. Nodules arising within port-wine stains: a clinicopathologic study of 31 cases. Am J Dermatopathol. 2011;33:144–151. [DOI] [PubMed] [Google Scholar]
- 27.del Pozo J, Fonseca E. Port-wine stain nodules in the adult: report of 20 cases treated by CO2 laser vaporization. Dermatol Surg. 2001;27:699–702. [DOI] [PubMed] [Google Scholar]
- 28.Tierney EP, Hanke CW. Treatment of nodules associated with port wine stains with CO2 laser: case series and review of the literature. J Drugs Dermatol. 2009;8:157–161. [PubMed] [Google Scholar]
- 29.Gao L, Phan S, Nadora DM, et al. Topical rapamycin systematically suppresses the early stages of pulsed dye laser-induced angiogenesis pathways. Lasers Surg Med. 2014;46:679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson JS, Jia W, Phung TL, et al. Observations on enhanced port wine stain blanching induced by combined pulsed dye laser and rapamycin administration. Lasers Surg Med. 2011;43:939–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan W, Jia W, Sun V, et al. Topical rapamycin suppresses the angiogenesis pathways induced by pulsed dye laser: molecular mechanisms of inhibition of regeneration and revascularization of photocoagulated cutaneous blood vessels. Lasers Surg Med. 2012;44:796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao L, Nadora DM, Phan S, et al. Topical axitinib suppresses angiogenesis pathways induced by pulsed dye laser. Br J Dermatol. 2015;172:669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]