

Abstract
Ion mobility-mass spectrometry (IM-MS) has recently seen increased use in the analysis of small molecules, especially in the field of metabolomics, for increased breadth of information and improved separation of isomers. In this study, steroid epimers androsterone and trans-androsterone were analyzed with IM-MS to investigate differences in their relative mobilities. Although sodiated monomers exhibited very similar collision cross sections (CCS), baseline separation was observed for the sodiated dimer species (RS = 1.81), with measured CCS of 242.6 and 256.3 Å2, respectively. Theoretical modeling was performed to determine the most energetically stable structures of solution- and gas-phase monomer and dimer structures. It was revealed that these epimers differ in their preferred dimer binding mode in solution-phase: androsterone adopts a R=O - - Na+ - - OH—R′ configuration, whereas trans-androsterone adopts a R=O - - Na+ - - O=R′ configuration. This difference contributes to a significant structural variation, and subsequent CCS calculations based on these structures relaxed in the gas-phase were in agreement with experimentally measured values (ΔCCS ~ 5%). Additionally, these calculations accurately predicted the relative difference in mobility between the epimers. This study illustrates the power of combining experimental and theoretical results to better elucidate gas-phase structures.
Keywords: Ion Mobility – Mass Spectrometry (IM-MS), Isomers, Steroids, Theoretical Modeling, Collision Cross Sections (CCS)
Graphical Abstract
Introduction
Ion mobility spectrometry (IMS) is a technique used to measure the velocity of gas-phase ions subjected to an electric field [1–6]. Classical drift tube ion mobility involves introduction of a temporally confined ion packet into a uniform field drift tube, where these ions travel at a constant drift velocity (vd) proportional to their mobility constant (K) and the electric field strength (E), as vd = KE. As the ions move through the drift tube, they experience interactions with buffer gas molecules that ultimately dictate the ion’s drift time, with smaller ions undergoing fewer collisions and thus traveling more quickly. Separation of ions can be achieved based on differences in mass, shape, and charge, collectively referred to as the rotationally averaged collision cross section (CCS, Ω), a property which describes an ion’s size under given experimental conditions (e.g., drift gas composition, temperature, and pressure) [1, 4, 5]. Experimentally measured drift times can be converted to cross sections using the Mason-Shamp equation [1, 4, 5], below:
| (1) |
where ze is the charge, kB is the Boltzman constant, mI is the analyte ion mass, mB is the buffer gas molecule mass, td is the corrected drift time in milliseconds, E is the electric field strength in V·cm−2, L is the drift tube length in cm, P is the drift tube pressure in Torr, T is the drift tube temperature in Kelvin, and N is the drift tube number density in cm−3.
Hyphenation of IMS with other analytical techniques, especially mass spectrometry (IM-MS) [3], allows improved specificity of analysis. Although historically IM-MS has been used for the study of large biomolecules such as proteins [7–10], recently this technique has been used increasingly for small molecule analysis, primarily due to its rapid millisecond separation capabilities that allow coupling with liquid chromatography [11, 12] and improved analysis of complex samples with mass spectrometry [13, 14]. Additionally, IM-MS offers great potential in rapid separation of isomers that cannot be resolved with mass spectrometry alone. First demonstrated for structural isomers of polycyclic aromatic hydrocarbons [15], this potential has been shown across numerous classes of biological molecules including carbohydrates [16–24], peptides [25, 26], oligonucleotides [19], lipids [19, 27–29], amino acids [29–31], fatty acids [32], glycans and glycopeptides [33], and other small molecules [34]. However, diastereomers and especially epimers, which differ in stereochemistry at a single chiral center, have very subtle structural differences that may contribute to only minor variation in CCS. To improve separation, several novel IMS strategies have been employed, including pre-analysis derivatization [35–37], use of alternative drift gases and chiral modifiers [38, 39], alkali and transition metal complexation [16–18, 29, 40–42], and multimer formation [30, 43].
In combination with IM-MS, theoretical modeling can aid in elucidating gas-phase three-dimensional structure and improving experimental design in separation of isomers. Generated theoretical structures can be used to calculate theoretical CCS values based on one of several methods, including the projection approximation (PA) [44], exact hard-sphere scattering (EHSS) [45], and the trajectory method (TM) [46]. In the literature it has been shown that theoretical results obtained using TM most closely agree with experimentally obtained values [45, 46]. All these approaches have been gathered in a software program MOBCAL, used to predict CCS based on three-dimensional structure [44–46]. Because nitrogen gas has also become increasingly popular with ion mobility analyses due to lower cost and greater availability, recently the MOBCAL software was modified [47, 48] to allow for trajectory method calculations in N2 buffer gas, as well as to account for several other atoms found in biological molecules, such as fluorine.
Molecular modeling has been applied in several studies in the literature to compare theoretically- and experimentally-derived CCS values for the identification of gas-phase structures [48–51]. IM-MS experimental results provide information on the charge, atomic composition, and CCS of the molecules, but only a comparison of experimental and theoretical CCS can provide additional molecular structural information, which could not be achieved with experimental results alone. The CCS comparative approach has been used previously to determine structural information for small organic molecules such as crown ethers [52] and carbon clusters [53]; macromolecules such as polyethylene glycol polymers [7]; peptides including bradykinin [54–58]; proteins [7] and cytochrome C [8]; and small biologically-relevant molecules including vitamin D metabolites [51], amino acids [31, 59, 60], and carbohydrates [42]. Comparison of IMS and theoretical results can help identify isomer conformers that can lead to understanding of differences that contribute to separation, improving the potential for these applications. As examples, studies have compared theoretical and experimental IM-MS results for isomeric monosaccharide-transition metal complexes [42], amino acid-alkali metal complexes [31], and small molecule multimer complexes [18], each of which allowed resolution of isomers.
Here we compare theoretical and experimental IM-MS results for a model steroid epimer pair, androsterone and trans-androsterone. These endogenous androgens differ only in their chirality at the C3 position (Scheme 1), with androsterone containing an α-hydroxyl group and trans-androsterone a β-hydroxyl. Specifically, we address structural similarities and differences in the formation of the sodiated multimers of these isomers.
Scheme 1.

Androsterone (left) and trans-androsterone (right)
Experimental Methods
Standard Preparation
Androsterone and trans-androsterone were purchased from Sigma-Aldrich (St. Louis, MO). Solids were prepared as 10 μg/m solutions in Fisher Optima C-MS grade water (with 0.1% formic acid), purchased from Fisher Scientific (Pittsburgh, PA).
IM-MS Analysis
Standards were analyzed with an Agilent 6560 IM-QTOF ion mobility-mass spectrometer instrument (Santa Clara, CA). Standards were directly infused by syringe pump at a flow rate of 10 μ/min. An Agilent Jet Stream (AJS) source was used to perform electrospray ionization (ESI). The ESI source conditions were as follows: capillary voltage: +4000 V; nozzle voltage: +1000 V; drying gas: 325 °C at 5 L/min; sheath gas: 275 °C at 8 L/min.
Ion mobility analysis was performed with a 78 cm uniform field drift tube maintained at approximately 4 torr nitrogen drift gas and 32 °C. The IM trap fill time was 5000 μs and the trap release time was 150 μs. Drift time spectra were acquired over a 60 ms window. To calculate the corrected drift time (td), the field was varied over eight field strength steps from 9.6–18.6 V/cm (drift tube voltage 750–1450 V). Total drift time, tD, was recorded and plotted versus the inverse of the drift tube voltage. This plot provided two points of information: (a) the linearity of the plot indicated that all experiments were performed under low-field conditions, and (b) the y-intercept provided a correction factor (t0) to the drift time corresponding to the time ions spent in regions of the instrument outside of the drift tube. The Mason-Schamp equation (Eq. 1) [1, 4, 5], shown in Equation (1), was used to compute the collision cross section (Ω) for ions of interest based on corrected drift time. All drift spectra shown were acquired at 18.6 V/cm, unless otherwise noted; optimal peak resolving power was achieved at this field strength. Time of flight mass spectra were acquired in full scan high resolution mode over a range from m/z 100–1700. All IM-MS data processing was performed using Agilent IM-MS Browser B.06.00.
Theoretical Methods
Geometry optimizations and frequency calculations were performed at the B3LYP-D3/6-31G(d) level1 using the Gaussian 09 [61] program, where D3 means that the Grimme’s empirical dispersion correction [62] was added to the B3LYP functional. In order to account for solvent effects, we used the SMD solvation model designed by Truhlar and coworkers [63]. For both androsterone and trans-androsterone the sodiated monomer optimizations were performed by initially placing the sodium ion (Na+) close to the oxygen of either the ketone or the hydroxyl group (see Scheme 1). This allowed comparison of the energetically favorable “binding mode”, which indicates the preference for the sodium ion to be associated with either the ketone (R=O - - Na+) or the hydroxyl group (R—HO - - Na+) of the steroid. The sodiated dimer optimizations were performed by taking the sodiated monomer and then adding a second monomer to it. The initial dimer structures were created by arranging three different binding modes of Na+, relative to the ketone and/or the hydroxyl groups, as follows: R=O - - Na+ - - O=R′, R=O - - Na+ - - OH—R′, and R—HO - - Na+ - - OH—R′. For each case, more than 18 initial configurations with different torsion angles between the monomers were submitted to gas-phase geometry optimization. The gas-phase optimized structures were then submitted to new geometry optimizations in solution-phase, followed by frequency calculations in order to obtain the Gibbs free energies. All the gas-phase optimized geometries were submitted to collision cross section (CCS) calculations using the trajectory method (TM) in the MOBCAL software package [49]. These calculations were performed using N2 as the drift gas (used experimentally). For each structure, the atomic point charges needed for CCS calculations with TM were obtained using CHelpG [64] charges fitted to the electrostatic potential calculated at the B3LYP-D3/6-31+G(d,p) level using the Gaussian 09 [61] program.
Results and Discussions
Experimental Results
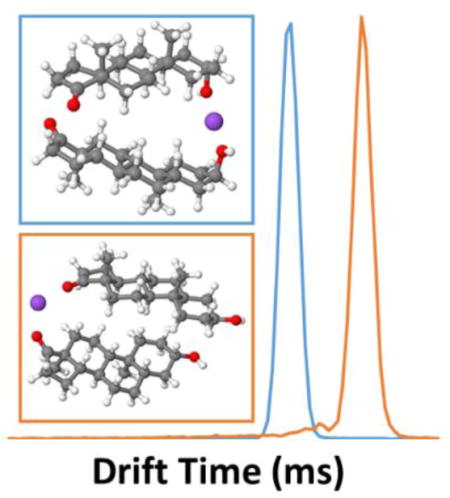
Ion mobility drift time spectra and mass spectra were acquired individually for androsterone and trans-androsterone. Because ionization efficiency for the [M+H]+ species of most steroids is typically low with ESI, the sodiated species were the focus of these experiments. Reconstruction of individual drift time spectra allowed comparison of these compounds at a given m/z value. An IM-MS spectrum of androsterone is shown in Figure 1a, indicating the primary sodiated species identified, with an extracted drift time spectrum overlay for the sodiated monomer of each species, [M+Na]+ at m/z 313.214 (Figure 1b), and the sodiated dimer of each species, [2M+Na]+ at m/z 603.439 (Figure 1c). Minimal separation (RS = 0.06) was observed between the primary drift peaks (tD ~ 24.50 ms) for the sodiated monomers (Figure 1b), indicating only very minor differences in their gas-phase structures. Collision cross sections (CCS) were measured as 197.1 ± 0.2 and 196.8 ± 0.2 Å2, respectively. All experimentally obtained CCS values are presented in Table 1. Although separation of the monomer drift peaks was minimal, differences in their overall drift spectra were observed. Notably, additional drift peaks were observed for each of the epimers at m/z 313.214. The presence of these drift peaks corresponds to unique drift tube species which are subsequently detected at that m/z; examples can include charge location isomers (more common with protonated species) which may differ in their CCS and drift time, or species that fragment to that given m/z after exiting the drift tube. The primary difference between the spectra of the epimers was the appearance of resolved drift peaks in the 30.0–33.0 ms range. Figure 2a shows an IM-MS spectrum with corresponding extracted mass spectrum for this drift time range (30.0–33.0 ms) indicating that these drift peaks correspond to the sodiated dimer [2M+Na]+ species at m/z 603.439 (Figure 2b). This instrumentation, which performs ion mobility analysis at intermediate pressure (4 torr) and roughly ambient temperature (~30 °C), represents a gentler environment than with those instruments that operate at atmospheric pressure and elevated temperature. Because of this, drift time detection of more labile species is possible, including dimers and trimers. However, these fragile species may fragment upon entering the high vacuum environment of the QTOF-MS, due to the pressure being much lower in this region (10−5 – 10−7 torr). This contributes to the presence of additional peaks in the drift spectra for lower m/z species.2 An overlay of the drift time spectra for the sodiated dimers (Figure 1c) shows that the epimers exhibit baseline separation (RS = 1.81). Differences in gas-phase structure for the sodiated dimers that account for this resolution are discussed in greater detail with the theoretical results in the next subsection.
Fig. 1.

(a) IM-MS spectrum of androsterone, indicating sodiated species at m/z 313.214 for [M+Na]+, 603.439 for [2M+Na]+, and 893.664 for [3M+Na]+. Extracted drift spectra overlays for androsterone and trans-androsterone are shown for the (b) sodiated monomer range (m/z 313–316) and (c) sodiated dimer range (m/z 603–606)
Table 1.
Measured collision cross sections (CCS) for all detected sodiated species of androsterone and trans-androsterone.
| Drift Tube Ion Species | Androsterone CCS (Å2) | trans-Androsterone CCS (Å2) |
|---|---|---|
| [M+Na]+ -- m/z 313.214 | 197.1 ± 0.2 | 196.8 ± 0.2 |
| [2M+Na]+ -- m/z 603.439 | 242.6 ± 0.3 | 256.3 ± 0.3 |
| [3M+Na]+ -- m/z 893.664 | 284.0 ± 0.3 | ---- |
| [3M+2Na]+2 -- m/z 458.327 | 315.0 ± 0.7 | ---- |
Fig. 2.

(a) IM-MS spectrum of androsterone with extracted mass spectra for the (b) 30.0–33.0 drift time range, corresponding to the sodiated dimer, and the (c) 35.5–35.7 ms drift time range, corresponding to the sodiated trimer
The drift spectrum for the androsterone sodiated dimer also exhibits unique drift peaks at 36.16 ms and 20.46 ms (Figure 1c); the latter is uniquely observed in the drift spectrum for the androsterone monomer as well (Figure 1b). Investigation of the mass spectrum for the 35.5–35.7 ms drift time range indicates that this peak corresponds to the singly sodiated trimer [3M+Na]+ species (Figure 2c) at m/z 893.664; the mass spectrum for the 21.0–21.1 ms drift time range indicates that this peak corresponds to the doubly sodiated trimer [3M+2Na]+2 species at m/z 458.327 (mass spectrum not shown). Neither species was detected for trans-androsterone. In addition, the [2M+2Na]+2 species was not observed for either compound. A summary of the identified drift tube ions for androsterone based on mass spectrometry, as well as the post-drift tube fragmentation patterns, is shown in Scheme 2. However, the unique presence of the sodiated trimer species for androsterone only will not be discussed further in this work. The relative stability of the sodiated dimer for both androsterone and trans-androsterone was investigated, based on several different experimental parameters, and these results are provided in the Supplementary Material. Overall, it was observed that the androsterone dimer was considerably more stable than that of trans-androsterone; this could contribute to the observation of larger complexes (i.e., singly and doubly sodiated trimers) for androsterone, but not for trans-androsterone.
Scheme 2.

Post-drift tube fragmentation of sodiated multimer species for androsterone
Theoretical versus Experimental Results: Monomers
To better understand the structural differences responsible for the mobility of the epimers, theoretical modeling was performed to determine the most energetically favorable ionic structures. Because of the connection between solution-phase ions and electrospray-formed gas-phase ions, optimized structures were determined for both solution- and gas-phase. The gas-phase energies and solution-phase Gibbs free energies for the optimized sodiated monomer structures of androsterone and trans-androsterone are reported in Table 2. The results indicate that the R=O - - Na+ binding mode is the most stable monomer configuration by ~0.3–0.5 kcal/mol for both epimers in solution. This is also the most stable binding mode in the gas-phase, with the energy difference between the two binding modes being ~4 kcal/mol for both epimers. Therefore, if we were to consider the sodiated monomers being formed in the gas-phase, only the binding mode in which Na+ binds to the ketone group would exist (approximately 99% of the sodiated monomer species would be of R=O - - Na+ type, as at room temperature kbT = 0.592 kcal/mol 3); however, in solution the difference in Gibbs free energies is lower than kbT; therefore, according to our theoretical results, both binding modes can be formed in solution.4 In this work we hypothesize that, once the molecules reach the drift tube in the gas-phase, they have enough time to relax their structures but they don’t change their solution binding mode. Changing the sodiated monomer binding mode in the gas-phase, for example, would involve a disassociation of the sodium ion followed by a reassociation on the other end of the molecule, which is unlikely in the gas-phase. Due to the fact that the gas-phase molecules are much more disperse than in solution, one can rationalize that the multimer formation is more likely in solution. Hence we hypothesize that all the multimers are being formed in solution.
Table 2.
Gibbs free energies (solution-phase) and energy differences (gas-phase) of the androsterone and trans-androsterone sodiated monomers, obtained at B3LYP-D3/6-31G(d) level. The differences are reported in kcal/mol and are with respect to the most stable sodiated monomer.
| Binding mode | Gibbs Free Energy difference (Solution-Phase) | Energy difference (Gas-phase) |
|---|---|---|
| Androsterone | ||
| Na+ binding at ketone | 0.000 | 0.000 |
| Na+ binding at hydroxyl | 0.276 | 4.016 |
| trans-androsterone | ||
| Na+ binding at ketone | 0.000 | 0.000 |
| Na+ binding at hydroxyl | 0.522 | 4.275 |
Table 3 presents collision cross sections (CCS) calculated with MOBCAL using the optimized geometries for the gas-phase sodiated monomers. These results indicate that the predicted CCS for the most stable binding modes show good agreement with the experimentally measured results. We see the theoretical CCS values underestimate the experimental ones by around 5% for both epimers. Despite the minor absolute difference between experimental and theoretical CCS, the CCS ratio of androsterone to trans-androsterone is very similar (within 0.2%) for experimental (1.0015) and theoretical (1.0038) results, demonstrating the ability for this approach to accurately predict relative differences in CCS by looking at the most stable binding modes.
Table 3.
Comparison of experimentally-measured and trajectory method-calculated gas-phase CCS. As several structures were optimized for the dimers with different initial torsional angles, the dimer results are averages weighted by the Boltzmann factors for T = 298 K with the respective energy of each optimized structure. Experimental errors and theoretical standard deviations are also shown.
| Binding mode | Androsterone | Trans-Androsterone | ||
|---|---|---|---|---|
|
| ||||
| Theoretical CCS (Å2) | Experimental CCS (Å2) | Theoretical CCS (Å2) | Experimental CCS (Å2) | |
| Results for Sodiated Monomers: | ||||
| Na+ binding at ketone | 186.9 ± 2.1 | 197.1 ± 0.2 | 186.2 ± 1.2 | 196.8 ± 0.2 |
| Na+ binding at hydroxyl | 180.8 ± 0.9 | 190.2 ± 1.4 | ||
| Results for Sodiated Dimers: | ||||
| R=O - - Na+ - - O=R′ | 250.4 ± 1.7 | 242.6 ± 0.3 | 245.7 ± 2.1 | 256.3 ± 0.3 |
| R=O - - Na+ - - OH—R′ | 234.7 ± 2.3 | 246.0 ± 2.2 | ||
| R—HO - - Na+ - - OH—R′ | 237.4 ± 3.2 | 257.4 ± 2.2 | ||
Another interesting aspect to notice is that, although the CCS values agree for the R=O - - Na+ binding mode, there is an evident difference between the CCS values for the R=OH - - Na+ binding mode. Androsterone and trans-androsterone differ from each other on the positioning of the hydroxyl group. Although this structural difference does not affect the R=O - - Na+ binding mode CCS values, when sodium binds at the hydroxyl group this difference leads the sodium to be placed in a significantly different position around the molecule (for androsterone sodium stays perpendicular to the molecular plane, and for trans-androsterone it stays inside the molecular plane), leading thus to different R=OH - - Na+ binding mode CCS values. Structures for the R=OH - - Na+ binding mode are shown in Figure S3.
Theoretical versus Experimental Results: Dimers
Consideration of sodium bound dimers first involved investigation of the solution-phase binding mode: Na+ binding at both ketone groups (R=O - - Na+ - - O=R′), Na+ binding at a ketone group and at a hydroxyl group (R=O - - Na+ - - OH—R′), or Na+ binding at both hydroxyl groups (R—HO - - Na+ - - OH—R′). The gas-phase optimized geometries for the sodiated dimers are shown in Figure 3. As shown in Table 5, the most stable solution-phase androsterone sodiated dimer is R—HO - - Na+ - - OH—R′, with the R=O - - Na+ - - OH—R′ being close in free energy. The most stable trans-androsterone sodiated dimer is R=O - - Na+ - - O=R′, with the R=O - - Na+ - - OH—R′ being close in free energy. The other sodiated dimer binding modes (R=O - - Na+ - - O=R′ for androsterone, and androsterone, and R—HO - - Na+ - - OH—R′ for trans-androsterone) are unlikely to be formed due to the high free energy difference in comparison to the most stable binding modes. However, the gas-phase results presented in Table 4 show that only the R=O - - Na+ - - OH—R′ binding mode for androsterone and the R=O - - Na+ - - O=R′ binding mode for trans-androsterone are energetically favorable in the gas-phase, due to the high free energy for the structures of the other binding modes. This comparison displays the importance of performing theoretical modeling in both solution- and gas-phase, especially when done in combination with ESI experiments.
Fig. 3.

Optimized gas-phase geometries for androsterone and trans-androsterone sodiated dimers
Table 4.
Gibbs free energies (solution-phase) and energy differences (gas-phase) of the androsterone and trans-androsterone sodiated dimers, obtained at B3LYP-D3/6-31G(d) level. The differences are reported in kcal/mol and are with respect to the most stable sodiated dimer.
| Binding mode | Gibbs Free Energy difference (Solution-Phase) | Energy difference (Gas-phase) |
|---|---|---|
| Androsterone | ||
| R=O - - Na+ - - O=R′ | 5.885 | 2.609 |
| R=O - - Na+ - - OH—R′ | 0.560 | 0.000 |
| R—HO - - Na+ - - OH—R′ | 0.000 | 6.497 |
| trans-androsterone | ||
| R=O - - Na+ - - O=R′ | 0.000 | 0.000 |
| R=O - - Na+ - - OH—R′ | 0.539 | 3.516 |
| R—HO - - Na+ - - OH—R′ | 4.893 | 11.266 |
It is interesting to note that the solution- and gas-phase most stable binding modes for androsterone are not the same. The relative energies have components of internal energy, interactions with solvent, and entropic effects coming from internal vibrations. Given all these variables, the correlation between energy ordering in the solution and gas phases is non-trivial, and there is nothing that requires them to be similar. On the Gaussian program [61], the entropic contribution to the Gibbs Free Energy is computed using information from the frequency calculation. Therefore, even disregarding major structural changes, the entropic contribution may significantly alter the stability in solution in respect to the stability in the gas-phase. Our results show that the difference ΔG-ΔE between the R—HO - - Na+ - - OH—R′ and R=O - - Na+ - - OH—R′ androsterone binding modes in solution is mostly entropic (TΔS=−5.183 kcal/mol). This large entropic difference for androsterone (that leads to a stability reordering) is possibly because the two structures are packed differently.
Table 3 presents CCS for the gas-phase sodiated dimers. As with the monomer calculations, the results indicate that the predicted CCS for the most stable binding modes show good agreement with the experimentally measured results, with theoretical values underestimating the experimental ones by 5% for both epimers. Again, the ratio of androsterone to trans-androsterone CCS is very similar (within 0.2%) for experimental (0.9662) and theoretical (0.9465) results, based on the values corresponding to the most stable binding modes in solution. This shows that the theoretical modeling approach can both accurately predict the significant difference in dimer CCS that is observed experimentally, while also determining the most energetically favorable structures and binding modes that contribute to this difference. It is also interesting to note that the binding modes that are close in free energy also have similar CCS, while the binding mode that is higher in free energy has a larger predicted CCS; this indicates that the CCS results are in agreement with the stability obtained in solution, not in the gas-phase.
Conclusions
Experimentally, androsterone and trans-androsterone sodiated monomers exhibited very similar mobility drift time and CCS; in contrast, baseline separation was observed for the sodiated dimers. Theoretical calculations were performed to determine the most stable conformations in solution- and gas-phase for the sodiated monomer and dimer species, which revealed that these epimers have different energetically favorable sodiated dimer binding modes. Calculation of CCS based on these energetically favorable structures was in good agreement with experimentally obtained values (ΔCCS ~ 5%). Additionally, the modeling approach was able to predict even more accurately the relative differences in CCS between the epimers, revealing the structural differences and binding mode preferences that contributed to significant separation between dimers in the experimental results. This work is an example of how the combination of experimental results and theoretical calculations is able to provide a higher level of understanding of the molecules studied that neither experimentation or theoretical modeling alone are able to achieve.
Supplementary Material
Fig. S1 Instrumental schematic of the Agilent 6560 IM-QTOF, detailing instrumental parameters that were varied to change the energy applied for measure of dimer stability
Fig. S2 MS/MS spectra for androsterone and trans-androsterone, with the parent sodiated dimer at m/z 603 isolated. Fragmentation of the dimer can be seen (a,b) in the absence of collision energy, (c,d) at CE = 10 ev, and (e,f) at CE = 20 eV
Fig. S3 Androsterone and trans-androsterone structures for the R=OH - - Na+ binding mode
Table S1 Experimental parameters varied for measurement of dimer stability, including the standard experimental value and the range measured.
Acknowledgments
The authors gratefully acknowledge financial support from Agilent Technologies, the University of Florida Graduate Fellowship, and CAPES (Brazil).
Footnotes
Some of the calculations were also performed at the B3LYP-D3/6-31+G(d,p) level and yielded results with trends similar to those obtained at the B3LYP-D3/6-31G(d) level.
It is important to note that identification of unique drift tube species by mass spectrometry is more straightforward if some fraction of these species remains intact upon entering the high vacuum environment of the mass spectrometer, such that they can also be detected at their native m/z (as well as that of the fragment m/z) at equivalent drift time.
Considering the Boltzmann distribution, it can be shown that in solution and in the gas-phase, where PMNa,hyd and PMNa, ket are partial pressures. Thus for androsterone [MNa]hyd/[MNa]ket = 0.627 and PMNa,hyd/PMNa,ket =0.00113, and for trans-androsterone [MNa]hyd/[MNa]ket =0.414 and PMNa/,hyd/PMNa,ket =0.00073.
This statement also has an impact on the sodiated dimer formation if we assume that the sodiated dimers are formed from sodiated monomers. According to our results we would expect to see only the R=O - - Na+ - - O=R′ dimer binding mode if formed in the gas-phase because only R=O - - Na+ monomers would exist, however if the sodiated dimer is formed in solution then other binding modes can exist.
References
- 1.Mason EA, Schamp HW. Mobility of gaseous ions in weak electric fields. Ann Phys. 1958;4:233–270. [Google Scholar]
- 2.McDaniel EW. Collisional phenomena in ionized gases. John Wiley & Sons; New York: 1964. [Google Scholar]
- 3.Cohen MJ, Karasek FW. Plasma Chromatography™ – A new dimension for gas chromatography and mass spectrometry. J Chrom Sci. 1970;8:330–337. [Google Scholar]
- 4.Revercomb HE, Mason EA. Theory of plasma chromatography/gaseous electrophoresis- A review. Anal Chem. 1975;47:970–983. [Google Scholar]
- 5.Mason EA, McDaniel EW. Transport properties of ions in gases. Wiley; New York: 1988. [Google Scholar]
- 6.Eiceman GA, Karpas Z. Ion Mobility Spectrometry. Taylor & Francis; New York: 2005. [Google Scholar]
- 7.Von Helden G, Wyttenbach T, Bowers MT. Conformation of macromolecules in the gas phase: Use of matrix-assisted laser desorption methods in ion chromatography. Science. 1995;267:1483–1485. doi: 10.1126/science.267.5203.1483. [DOI] [PubMed] [Google Scholar]
- 8.Clemmer DE, Hudgins RR, Jarrold MF. Naked protein conformations: cytochrome C in the gas phase. J Am Chem Soc. 1995;117:10141–10142. [Google Scholar]
- 9.Valentine SJ, Counterman AE, Clemmer DE. Conformer-dependent proton-transfer reactions of ubiquitin ions. J Am Soc Mass Spectrom. 1997;8:954–961. [Google Scholar]
- 10.Shelimov KB, Jarrold MF. Conformations, unfolding, and refolding of apomyoglobin in vacuum: an activation barrier for gas-phase protein folding. J Am Chem Soc. 1997;119:2987–2994. [Google Scholar]
- 11.McGinn DG, Kinzer JA, Shumate CB, Siems WF, Hill HH. Ion mobility detection following liquid chromatographic separation. J Microcol. 1990 Sep;2:188–192. [Google Scholar]
- 12.Hill HH, Siems WF, Eatherton RL, StLouis RH, Morrissey MA, Shumate CB, McGinn DG. Instrumentation for Trace Organic Monitoring. CRC Press; New York: 1991. Gas supercritical fluid, and liquid chromatographic detection of trace organics by ion mobility spectrometry; pp. 49–64. [Google Scholar]
- 13.May JC, Goodwin CR, Lareau NM, Leaptrot KL, Morris CB, Kurulugama RT, Mordehai A, Klein C, Barry W, Darland E, Overney G, Imatani K, Stafford GC, Fjeldsted JC, McLean JA. Conformational ordering of biomolecules in the gas phase: Nitrogen collision cross sections measured on a prototype high resolution drift tube ion mobility-mass spectrometer. Anal Chem. 2014;86:2107–2116. doi: 10.1021/ac4038448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valentine SJ, Kulchania M, Srebalus Barnes CA, Clemmer DE. Multidimensional separations of complex peptide mixtures: A combined high-performance liquid chromatography/ion mobility/time-of-flight mass spectrometry approach. Int J Mass Spectrom. 2001;212:97–109. [Google Scholar]
- 15.Hagen DF. Characterization of isomeric compounds by gas and plasma chromatography. Anal Chem. 1979;51:870–874. [Google Scholar]
- 16.Carlesso V, Afonso C, Fournier F, Tabet JC. Stereochemical effects from doubly-charged iron clusters for the structural elucidation of diastereomeric monosaccharides using ESI/IT-MS. Int J Mass Spectrom. 2002;219:559–575. [Google Scholar]
- 17.Clowers BH, Hill HH. Influence of cation adduction on the separation characteristics of flavonoid diglycoside isomers using dual gate-ion mobility-quadrupole ion trap mass spectrometry. J Mass Spectrom. 2006;41:339–351. doi: 10.1002/jms.994. [DOI] [PubMed] [Google Scholar]
- 18.Domalain V, Tognetti V, Hubert-Roux M, Lange CM, Joubert L, Baudoux J, Rouden J, Afonso C. Role of cationization and multimers formation for diastereomers differentiation by ion mobility-mass spectrometry. J Am Soc Mass Spectrom. 2013;24:1437–1445. doi: 10.1007/s13361-013-0690-1. [DOI] [PubMed] [Google Scholar]
- 19.Fenn LS, Kliman M, Mahsut A, Zhao SR, McLean JA. Characterizing ion mobility-mass spectrometry conformation space for the analysis of complex biological samples. Anal Bioanal Chem. 2009;394:235–244. doi: 10.1007/s00216-009-2666-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fenn LS, McLean JA. Structural resolution of carbohydrate positional and structural isomers based on gas-phase ion mobility-mass spectrometry. Phys Chem Chem Phys. 2011;13:2196–2205. doi: 10.1039/c0cp01414a. [DOI] [PubMed] [Google Scholar]
- 21.Clowers BH, Dwivedi P, Steiner WE, Hill HH, Bendiak B. Separation of sodiated isobaric disaccharides and trisaccharides using electrospray ionization-atmospheric pressure ion mobility-time of flight mass spectrometry. J Am Soc Mass Spectrom. 2005;16:660–669. doi: 10.1016/j.jasms.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 22.Williams JP, Grabenauer M, Holland RJ, Carpenter CJ, Wormald MR, Giles K, Harvey DJ, Bateman RH, Scrivens JH, Bowers MT. Characterization of simple isomeric oligosaccharides and the rapid separation of glycan mixtures by ion mobility mass spectrometry. Int J Mass Spectrom. 2010;298:119–127. [Google Scholar]
- 23.Hill HH, Bendiak B, Clowers B, Zhu M. Ion mobility-mass spectrometry analysis of isomeric carbohydrate precursor ions. Anal Bioanal Chem. 2009;394:1853–1867. doi: 10.1007/s00216-009-2865-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pagel K, Harvey DJ. Ion mobility-mass spectrometry of complex carbohydrates: Collision cross sections of sodiated N-linked glycans. Anal Chem. 2013;85:5138–5145. doi: 10.1021/ac400403d. [DOI] [PubMed] [Google Scholar]
- 25.Srebalus Barnes CA, Hilderbrand AE, Valentine SJ, Clemmer DE. Resolving isomeric peptide mixtures: A combined HPLC/ion mobility-TOFMS analysis of a 4000-component combinatorial library. Anal Chem. 2002;74:26–36. doi: 10.1021/ac0108562. [DOI] [PubMed] [Google Scholar]
- 26.Wu C, Siems WF, Klasmeier J, Hill HH. Separation of isomeric peptides using electrospray ionization/high-resolution ion mobility spectrometry. Anal Chem. 2000;72:391–395. doi: 10.1021/ac990601c. [DOI] [PubMed] [Google Scholar]
- 27.Kliman M, May JC, McLean JA. Lipid analysis and lipidomics by structurally selective ion mobility-mass spectrometry. Biochim Biophys Acta - Mol Cell Biol Lipids. 2011;1811:935–945. doi: 10.1016/j.bbalip.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Damen CWN, Isaac G, Langridge J, Hankemeier T, Vreeken RJ. Enhanced lipid isomer separation in human plasma using reversed-phase UPLC with ion-mobility/high-resolution MS detection. J Lipid Res. 2014;55:1772–1783. doi: 10.1194/jlr.D047795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paglia G, Kliman M, Claude E, Geromanos S, Astarita G. Applications of ion-mobility mass spectrometry for lipid analysis. Anal Bioanal Chem. 2015;407:4995–5007. doi: 10.1007/s00216-015-8664-8. [DOI] [PubMed] [Google Scholar]
- 30.Domalain V, Hubert-Roux M, Tognetti V, Joubert L, Lange CM, Rouden J, Afonso C. Enantiomeric differentiation of aromatic amino acids using traveling wave ion mobility-mass spectrometry. Chem Sci. 2014;5:3234. [Google Scholar]
- 31.Flick TG, Campuzano IDG, Bartberger MD. Structural resolution of 4-substituted proline diastereomers withion mobility spectrometry via alkali metal ion cationization. Anal Chem. 2015;87:3300–3307. doi: 10.1021/ac5043285. [DOI] [PubMed] [Google Scholar]
- 32.Zhang F, Guo S, Zhang M, Zhang Z, Guo Y. Characterizing ion mobility and collision cross section of fatty acids using electrospray ion mobility mass spectrometry. J Mass Spectrom. 2015;50:906–913. doi: 10.1002/jms.3600. [DOI] [PubMed] [Google Scholar]
- 33.Both P, Green AP, Gray CJ, Sardzik R, Voglmeir J, Fontana C, Austeri M, Rejzek M, Richardson D, Field RA, Widmalm G, Flitsch SL, Eyers CE. Discrimination of epimeric glycans and glycopeptides using IM-MS and its potential for carbohydrate sequencing. Nat Chem. 2014;6:65–74. doi: 10.1038/nchem.1817. [DOI] [PubMed] [Google Scholar]
- 34.Baker ES, Hong JW, Gidden J, Bartholomew GP, Bazan GC, Bowers MT. Diastereomer assignment of an olefin-linked bi-paracyclophane by ion mobility mass spectrometry. J Am Chem Soc. 2004;126:6255–6257. doi: 10.1021/ja039486k. [DOI] [PubMed] [Google Scholar]
- 35.Ahonen L, Fasciotti M, Gennas GB, Kotiaho T, Daroda RJ, Eberlin M, Kostiainen R. Separation of steroid isomers by ion mobility mass spectrometry. J Chrom A. 2013;1310:133–137. doi: 10.1016/j.chroma.2013.08.056. [DOI] [PubMed] [Google Scholar]
- 36.Ochoa ML, Harrington PB. Detection of methamphetamine in the presence of nicotine using in situ chemical derivatization and ion mobility spectrometry. Anal Chem. 2004;76:985–991. doi: 10.1021/ac035123r. [DOI] [PubMed] [Google Scholar]
- 37.Fenn LS, McLean JA. Enhanced carbohydrate structural selectivity in ion mobility-mass spectrometry analyses by boronic acid derivatization. Chem Commun (Camb) 2008;43:5505–5507. doi: 10.1039/b810421b. [DOI] [PubMed] [Google Scholar]
- 38.Dwivedi P, Wu C, Matz LM, Clowers BH, Siems WF, Hill HH., Jr Gas-phase chiral separations by ion mobility spectrometry. Anal Chem. 2006;78:8200–8206. doi: 10.1021/ac0608772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asbury G, Hill H. Using different drift gases to change separation factors (alpha) in ion mobility spectrometry. Anal Chem. 2000;72:580–4. doi: 10.1021/ac9908952. [DOI] [PubMed] [Google Scholar]
- 40.Jackson P, Attalla MI. N-Nitrosopiperazines form at high pH in post-combustion capture solutions containing piperazine: a low-energy collisional behaviour study. Rapid Commun Mass Spectrom. 2010;24:3567–3577. doi: 10.1002/rcm.4815. [DOI] [PubMed] [Google Scholar]
- 41.Domalain V, Hubert-Roux M, Lange CM, Baudoux J, Rouden J, Afonso C. Use of transition metals to improve the diastereomers differentiation by ion mobility and mass spectrometry. J Mass Spectrom. 2014;49:423–427. doi: 10.1002/jms.3349. [DOI] [PubMed] [Google Scholar]
- 42.Leavell MD, Gaucher SP, Leary JA, Taraszka JA, Clemmer DE. Conformational studies of Zn-ligand-hexose diastereomers using ion mobility measurements and density functional theory calculations. J Am Soc Mass Spectrom. 2002;13:284–293. doi: 10.1016/S1044-0305(01)00362-2. [DOI] [PubMed] [Google Scholar]
- 43.Dwivedi P, Bendiak B, Clowers BH, Hill HH. Rapid resolution of carbohydrate isomers by electrospray ionization ambient pressure ion mobility spectrometry-time-of-flight mass spectrometry (ESI-APIMS-TOFMS) J Am Soc Mass Spectrom. 2007;18:1163–1175. doi: 10.1016/j.jasms.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 44.Shvartsburg AA, Jarrold MF. An exact hard-spheres scattering model for the mobilities of polyatomic ions. Chem Phys Lett. 1996;261:86–91. [Google Scholar]
- 45.Mesleh MF, Hunter JM, Shvartsburg AA, Schatz GC, Jarrold MF. Structural information from ion mobility measurements: effects of the long-range potential. J Phys Chem. 1996;100:16082–16086. [Google Scholar]
- 46.Shvartsburg AA, Schatz GC, Jarrold MF. Mobilities of carbon cluster ions: Critical importance of the molecular attractive potential. J Chem Phys. 1998;108:2416. [Google Scholar]
- 47.Campuzano I, Bush MF, Robinson CV, Beaumont C, Richardson K, Kim H, Kim HI. Structural characterization of drug-like compounds by ion mobility mass spectrometry: Comparison of theoretical and experimentally derived nitrogen collision cross sections. Anal Chem. 2012;84:1026–1033. doi: 10.1021/ac202625t. [DOI] [PubMed] [Google Scholar]
- 48.Kim H, Kim HI, Johnson PV, Beegle LW, Beauchamp JL, Goddard WA, Kanik I. Experimental and theoretical investigation into the correlation between mass and ion mobility for choline and other ammonium cations in N2. Anal Chem. 2008;80:1928–1936. doi: 10.1021/ac701888e. [DOI] [PubMed] [Google Scholar]
- 49.Mesleh MF, Hunter JM, Shvartsburg AA, Schatz GC, Jarrold MF. Structural information from ion mobility measurements: Effects of the long-range potential. J Phys Chem A. 1997;101:968–968. [Google Scholar]
- 50.Zakharova NL, Crawford CL, Hauck BC, Quinton JK, Seims WF, Hill HH, Clark AE. An assessment of computational methods for obtaining structural information of moderately flexible biomolecules from ion mobility spectrometry. J Am Soc Mass Spectrom. 2012;23:792–805. doi: 10.1007/s13361-012-0339-5. [DOI] [PubMed] [Google Scholar]
- 51.Chouinard CD, Cruzeiro VWD, Beekman CR, Roitberg A, Yost RA. Investigating Differences in Gas-Phase Conformations of 25-Hydroxyvitamin D3 Sodiated Epimers using Ion Mobility-Mass Spectrometry and Theoretical Modeling. Submitted to Anal Chem. 2016 doi: 10.1007/s13361-017-1673-4. [DOI] [PubMed] [Google Scholar]
- 52.Lee S, Wyttenbach T, Von Helden G, Bowers MT. Gas Phase Conformations of Li+, Na+, K+, and Cs+ Complexed with 18-Crown-6. J Am Chem Soc. 1995;117:10159–10160. [Google Scholar]
- 53.Von Helden G, Hsu MT, Gotts N, Bowers MT. Carbon cluster cations with up to 84 atoms: structures, formation mechanism, and reactivity. J Phys Chem. 1993;97:8182–8192. [Google Scholar]
- 54.Kinnear BS, Kaleta DT, Kohtani M, Hudgins RR, Jarrold MF. Conformations of unsolvated valine-based peptides. J Am Chem Soc. 2000;122:9243–9256. [Google Scholar]
- 55.Hudgins RR, Mao Y, Ratner MA, Jarrold MF. Conformations of GlynH+ and AlanH+ peptides in the gas phase. Biophysics (Oxf) 1999;76:1591–1597. doi: 10.1016/S0006-3495(99)77318-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taraszka JA, Li J, Clemmer DE. Metal-mediated peptide ion conformations in the gas phase. J Phys Chem B. 2000;104:4545–4551. [Google Scholar]
- 57.Henderson SC, Valentine SJ, Counterman AE, Clemmer DE. ESI/ion trap/ion mobility/time-of-flight mass spectrometry for rapid and sensitive analysis of biomolecular mixtures. Anal Chem. 1999;71:291–301. doi: 10.1021/ac9809175. [DOI] [PubMed] [Google Scholar]
- 58.Wyttenbach T, Von Helden G, Bowers MT. Gas-phase conformation of biological molecules: bradykinin. J Am Chem Soc. 1996;118:8355–8364. [Google Scholar]
- 59.Wyttenbach T, Witt M, Bowers MT. On the stability of amino acid zwitterions in the gas phase: the influence of derivatization, proton affinity, and alkali ion addition. J Am Chem Soc. 2000;122:3458–3464. [Google Scholar]
- 60.Wyttenbach T, Witt M, Bowers MT. On the question of salt bridges of cationized amino acids in the gas phase: glycine and arginine. Int J Mass Spectrom. 1999;182/183:243–252. [Google Scholar]
- 61.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09 Revision. 2009. p. A.02. [Google Scholar]
- 62.Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 63.Marenich AV, Cramer CJ, Truhlar DG. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 64.Breneman CM, Wiberg KB. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J Comput Chem. 1990;11:361–373. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Instrumental schematic of the Agilent 6560 IM-QTOF, detailing instrumental parameters that were varied to change the energy applied for measure of dimer stability
Fig. S2 MS/MS spectra for androsterone and trans-androsterone, with the parent sodiated dimer at m/z 603 isolated. Fragmentation of the dimer can be seen (a,b) in the absence of collision energy, (c,d) at CE = 10 ev, and (e,f) at CE = 20 eV
Fig. S3 Androsterone and trans-androsterone structures for the R=OH - - Na+ binding mode
Table S1 Experimental parameters varied for measurement of dimer stability, including the standard experimental value and the range measured.