

Abstract
Epigenetic deregulation is of importance in tumorigenesis. In particular CpG islands (CGI), are frequently hypermethylated. Here, genome-wide DNA-methylation profiles of 480,000 CpGs in lung cancer cells were generated. It was observed that intra- and intergenic CGI exhibited higher methylation compared to normal cells. The functional annotation of hypermethylated CGI revealed that the hypermethylation was associated with homeobox domain genes and targets marked by repressive histone modifications. The strongest methylation variation was observed in transitional areas of CGI, termed shores. 5′-shores of promoter-associated CGI in lung cancer cell lines were higher methylated than 3′-shores. Within two tandem-oriented genes, a significant hypermethylation of the downstream-located CGI promoters was revealed. Hypermethylation correlates with the length of the intergenic region between such tandem genes. As the RASSF1A tumor suppressor gene represents such a downstream tandem gene, its silencing was analyzed using an inducible system. It was determined that the induction of an upstream gene led to a repression of RASSF1A through a process involving histone deacetylases and CPSF1. A tumor-specific increase in expression of histone deacetylases and CPSF1 was detected in lung cancer. Our results suggest that the downstream gene could be susceptible to epigenetic silencing when organized in a tandem orientation.
Introduction
Inactivation of tumor suppressor genes (TSG) is frequently observed in lung cancer and is accomplished through genetic and epigenetic mechanisms including a repressive chromatin state of the TSG promoter (e.g. aberrant DNA methylation)1. Hypermethylation of promoter-associated CpG islands (CGI) in particular represents a fundamental event in the epigenetic silencing of TSG during lung carcinogenesis2. CGI are sequences greater than 500 bp comprised of GC-rich and CpG dense elements in the genome notably, approximately 70% of known genes harbor CGI within their transcription start site3. The most frequently epigenetically inactivated TSG in lung cancer are the Ras association domain family 1A (RASSF1A) and the cyclin-dependent kinase inhibitor 2A (CDKN2A/p16) genes4–7.
Mechanisms, to maintain the inactive epigenetic state of CGI promoters are well studied. DNA methylation patterns are maintained by DNA methyltransferase 1 (DNMT1) and the 5-methyl-cytosines in turn serve as binding sites for the methyl-CpG binding domain (MBD) proteins8. These MBD proteins are directly involved in transcriptional repression by creating a compacted chromatin environment together with histone deacetylases (HDACs), histone methyltransferases (e.g. SUV39H1), and/or ATP-dependent chromatin remodeling machines8, 9. For example, inactive TSG promoter regions exhibit a repressed chromatin structure that lacks H3/H4 acetylation1. These repressive chromatin- and DNA-modifications maintain CGI-associated TSG promoters in an inactive heterochromatic state that is mitotically heritable and basically irreversible.
The initial trigger for the repressive modification of a TSG promoter and the exact dynamics of its epigenetic silencing are not yet understood in detail10. One hypothesis is that certain DNA sequences have a higher susceptibility to de novo methylation by DNMT3A and 3B11. Another hypothesis states that a bivalent chromatin pattern may predispose TSG to epigenetic silencing12. Furthermore, depending on the location, shielding against positive or negative regulatory effects from neighboring chromatin or transcription machinery may be required and hence insulator and boundary models have also been proposed13–15.
In order to investigate novel mechanisms that are involved in the epigenetic inactivation of TSG, we performed a global DNA methylation analysis in lung cancer cells. Here we report that for genes organized in a tandem orientation the downstream gene may be susceptible to epigenetic inactivation. This effect was correlated with the distance between the transcription end site of the upstream gene and the transcription start site of the downstream gene.
Results
Increased CpG methylation in lung cancer cells
Epigenetic deregulation of tumor-associated genes is a frequent event in the pathology of lung cancer. However, distinct mechanisms that lead to aberrant DNA methylation are still under investigation. To dissect this further, we analyzed the DNA methylation patterns of over 480,000 CpG sites by bisulfite-based Illumina 450 K BeadChip arrays in normal human bronchial epithelial cells (NHBEC) and three non small cell lung cancer cell lines (NSCLC: A427, A549 and H322). CpGs on the array were annotated in CGI regions (CGIR), which consisted of a central CGI and 2 kb flanking shores and shelf regions (Fig. 1A). In NHBEC most CGIR-associated CpGs were rather weakly methylated (<20% methylation level) and only a few CGIR were strongly methylated (median methylation level 18%) (Fig. 1B). However, in the NSCLC a significant shift toward higher methylation levels of CGIR was observed (median methylation: 46%). For CpGs that were located outside of CGIR (no CGIR), this strong shift was not observed (Fig. 1B; median NHBEC: 82% and NSCLC: 83%). Genes associated with CGI that exhibited increased methylation in the lung cancer cell lines were subjected to a gene ontology (GO)-term analysis using different ontology data sets (Table S1). The GO-term analysis of hypermethylated genes revealed a significant enrichment of genes associated with homeobox domains and the function of sequence-specific DNA binding. Furthermore, the hypermethylated gene promoters were significantly enriched for genes that in ES cells harbor the repressive chromatin mark histone H3K27 trimethylation in their promoter sequences as well as for genes targeted by PRC2 and polycomb protein EED (Table S1).
Figure 1.
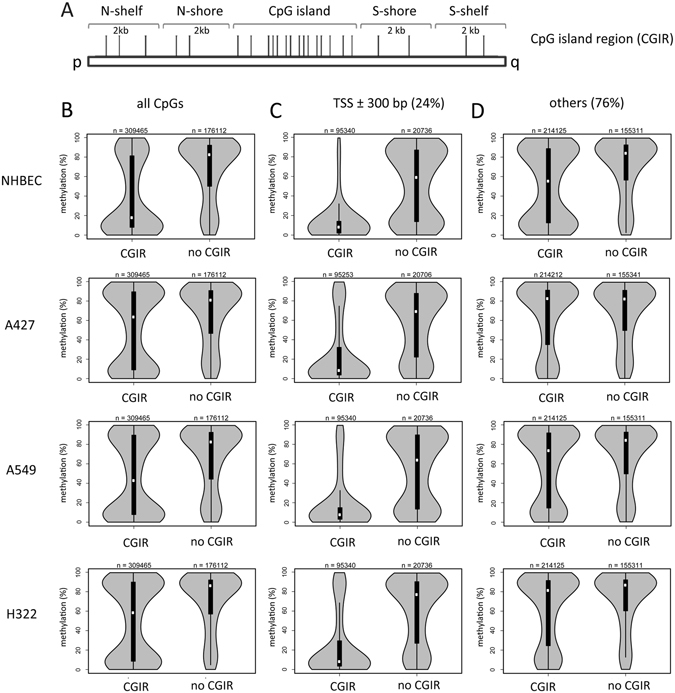
Increased methylation of CpG island-associated regions (CGIR) occurs in lung cancer cell lines. (A) GC and CpG rich genomic elements, termed CpG islands are flanked by 2 kb shore and 2 kb shelf regions. Depending on their orientation on the chromosome from the p- to q-arm these regions are denoted as N- or S-shores and shelves respectively. (B) Methylation levels of CpGs (n = 485,577) in CGIR (CpG island and flanking regions) and in no CGIR in normal human bronchial epithelial cells (NHBEC) and in three lung cancer cell lines (A427, A549 and H322) as quantified by 450 K bisulfite bead chip arrays and depicted in violin plots. (C) Methylation levels of transcription start site (TSS ± 300 bp flanking regions) associated CpGs (n = 116,076) were analyzed according to their location in CGI regions (CGIR) and no CGIR. (D) Methylation of CpGs outside of TSS (±300 bp) were analyzed according to the position in CGIR and no CGIR.
We further classified CpG sites as those associated with a transcription start site (TSS ± 300 bp) and others (no TSS-associated CpGs) (Fig. 1C and D). TSS-associated CGIR were generally unmethylated in NHBEC (median 8.1%) and NSCLC (median 8.2%). CpGs associated with TSS, but not located at CGI exhibited high methylation levels in NHBEC (median: 72%) and in NSCLC (median: 59%) compared to CpGs located at TSS-associated CGIR. Conversely CGIR, which are not located proximal to a TSS exhibited tumor-specific hypermethylation was found (Fig. 1D). An increase of 26% in the methylation of NSCLC (median: 82%) compared to NHBEC (median 56%) was detected (Fig. 1D).
Previously, hypermethylation of promoter-associated CGI (e.g. RASSF1A) has been reported in lung cancers2, 4. Therefore, we identified genes that exhibited the highest methylation levels in A427, A549 and H322 compared to NHBEC (Table 1). Hypermethylation has already been reported for PAX5, KCNAB3, MEIS2 and GFRA3 in different cancer entities16–20. However, we also identified other genes that may represent novel epigenetically inactivated lung cancer-related target genes (e.g. pyrimidinergic receptor P2RY6 and PLK5).
Table 1.
Top 20 lung cancer-specific hypermethylated CGI-associated genes.
| CpG-islands | associated gene | CpG-island promoter methylation [%] | ||||||
|---|---|---|---|---|---|---|---|---|
| A427 | A549 | H322 | Mean | NHBEC | methylation differences | |||
| 1 | chr11: 72975469–72975797 | P2RY6 | 99 | 97 | 99 | 98 | 2 | 96 |
| 2 | chr9: 37002489–37002957 | PAX5 | 99 | 96 | 99 | 98 | 5 | 93 |
| 3 | chr17: 7832532–7833164 | KCNAB3 | 97 | 97 | 96 | 97 | 4 | 93 |
| 4 | chr15: 37390175–37390380 | MEIS2 | 96 | 97 | 94 | 96 | 4 | 92 |
| 5 | chr5: 137610105–137610311 | GFRA3 | 96 | 94 | 96 | 95 | 4 | 91 |
| 6 | chr19: 1523705–1524565 | PLK5 | 94 | 94 | 89 | 92 | 2 | 90 |
| 7 | chr19: 18118741–18119553 | ARRDC2 | 95 | 93 | 94 | 94 | 4 | 90 |
| 8 | chr2: 228736230–228736544 | DAW1 | 94 | 94 | 94 | 94 | 4 | 90 |
| 10 | chr4: 682724–683079 | MFSD7 | 93 | 94 | 94 | 94 | 4 | 90 |
| 9 | chr4: 57371582–57372022 | ARL9 | 95 | 94 | 95 | 95 | 5 | 89 |
| 11 | chr14: 24837872–24838324 | NFATC4 | 96 | 94 | 95 | 95 | 6 | 89 |
| 12 | chr2: 27665251–27665670 | KRTCAP3 | 96 | 96 | 97 | 96 | 7 | 89 |
| 13 | chr17: 41177336–41177593 | RND2 | 93 | 87 | 97 | 92 | 3 | 89 |
| 14 | chr10: 102495116–102495613 | PAX2 | 95 | 95 | 92 | 94 | 5 | 89 |
| 15 | chr2: 54086776–54087266 | ASB3, GPR75 | 92 | 93 | 93 | 93 | 4 | 89 |
| 16 | chr12: 49487963–49488202 | DHH | 97 | 80 | 98 | 92 | 3 | 89 |
| 17 | chr20: 44540445–44540957 | PLTP | 94 | 93 | 80 | 89 | 1 | 88 |
| 19 | chrX: 100546063–100546550 | TAF7L | 96 | 96 | 96 | 96 | 8 | 86 |
| 18 | chr15: 72489478–72490119 | GRAMD2 | 93 | 93 | 94 | 93 | 6 | 87 |
| 20 | chr12: 124246524–124247254 | DNAH10 | 95 | 84 | 95 | 92 | 5 | 87 |
| 126 | chr3: 50377803–50378540 | RASSF1A | 91 | 93 | 91 | 92 | 13 | 79 |
To examine the aberrant methylation of CGIR in further detail, we analyzed the methylation level of the CGI and flanking shore and shelf regions in 69 NSCLC, 6 normal lung cell lines (NLC) and 5 normal lung tissues (NLT) (Fig. S1). The methylation profiles of these samples have been published previously21, 22. The methylation level of NSCLC was significantly higher in the shores compared to CGI. Furthermore, in NSCLC a significantly increased methylation of shores (median: 55%) compared to normal cells (median 35%; p = 2.2 × 10−16) was revealed (Fig. S1). This 20% tumor specific increase in methylation was not observed for CGI (NSCLC: median 15% and NLC: 11%) or and shelf regions (NSCLC: 77% and NLC: 73%).
Asymmetry in N- vs. S-shore methylation
To dissect the mechanism of aberrant promoter methylation, we analyzed the methylation level of CGI and flanking shore regions in further detail (Figs 2 and 3). In the utilized array, shores were annotated according to their chromosome orientation from the p- to q-arms as in N- and S-shores, respectively (Fig. 1A). Additionally, 92% of CpG sites (n = 447,186) were annotated depending on the gene specific orientation in TSS1500 (−1500 to −200), TSS200 (−200 to TSS), 5′-UTR, 1st exon, gene body and 3′-UTR (Fig. 2A). Initially, we analyzed whether the methylation levels in NHBEC and H322 were dependent on this gene specific annotation (Fig. 2B). In general, the methylation levels of the CGI at TSS1500, TSS200, 5′-UTR and 1st exon were low (median <10%). In contrast, CGI located in gene bodies exhibited a higher methylation level in NHBEC (median 17%) and an additional tumor-specific increase in methylation was observed in H322 (median 72%). This increased methylation was also detected for the 3′-UTR in H322 (Fig. 2B). Furthermore for the N- and S-shore, we observed significantly increased methylation for all gene-specific regions in H322 compared to NHBEC.
Figure 2.
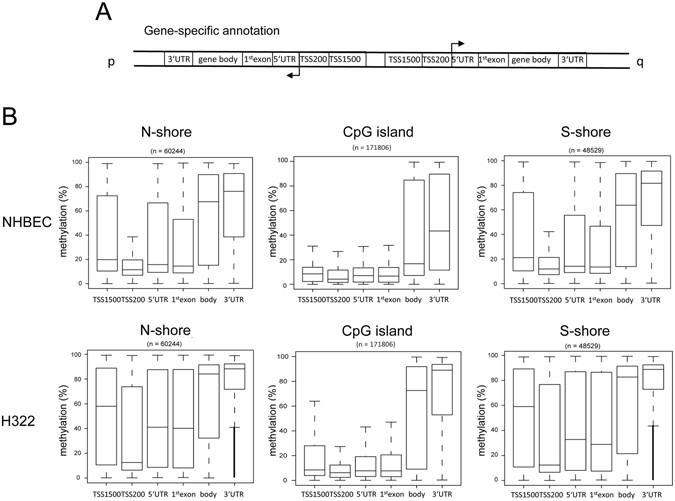
Methylation of gene specific CpGs in lung cancer cell lines. (A) CpG sites are annotated depending on the gene specific orientation in TSS1500 (−1500 to −200 bp), TSS200 (−200 bp to TSS), 5′-UTR, 1st exon, gene body and 3′-UTR. (B) Methylation levels of CpG islands and flanking N-and S-shores were analyzed in NHBEC and in the lung cancer cell line H322 by 450 K bisulfite bead chip arrays according to the gene specific annotation.
Figure 3.
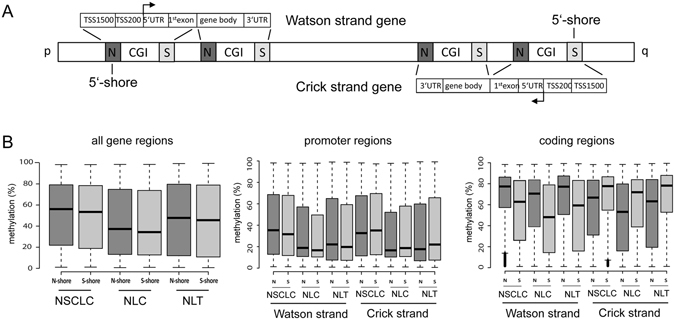
Increased 5′-shore methylation in promoter and coding regions. (A) Depending on the chromosome orientation genes are transcribed from the p- to the q arm (Watson strand genes) or in the opposite direction (Crick strand genes). For Watson strand genes the CGI flanking N-shores are the 5′-shores and for Crick strand genes S-shores are the 5′-shores. For further details are provided in Fig. 2. (B) Methylation levels of N- and S-shore gene specific regions in 69 NSCLC cell lines (ABC-1, SW1573, HOP18, HCC1171, LXFL529, H1703, H441, EBC-1, H322, A427, H1568, CHA-GO-K-1, H2087, H1975, H226, H2444, HCC515, H1299, H2030, H2405, HCC2935, H1373, H1666, RERF-LC-Ad1, Calu-6, H1155, H1651, H2347, H358, H838, H2228, HCC827, H2073, H1650, H23, H1993, Calu-3, H650, H460, H727, A549, H292, HCC4006, H2170, H1838, H820, H1355, RERF-LC-MS, H2122, H1793, H661, HOP62, HCC15, EKVX, H1792, H2110.1, Calu-1, H2110, HCC4017, H2009, HOP92, SK-MES-1, RERF-LC-KJ, H1437, H647, H2126, H2172, H1435 and H1755), six normal lung cell lines (NLC): gBEC1, gBEC1_UI, gSAC1, gSAC1_UI, gBEC and gSAC) and five normal lung tissues (NLT): GSM1264690, GSM1264711, GSM1264727, GSM1264764, GSM1264854). Methylation data were obtained from NLCBI-GEO-Accession: GSE36216 and GSE5240121, 22. Promoter regions are defined as the TSS1500 (−1500 to −200), TSS200 (−200 to TSS), 5′-UTR, 1st exon and coding regions consisted of the gene body and 3′-UTR.
Notably, the methylation levels of flanking shores different among all analyzed lung cells (Figs 2 and 3). In lung cancer cell lines but also in normal lung, the methylation levels were significantly higher in N-shores than in S-shores, (Fig. 3B). To further elucidate the differential methylation of the N- and S-shores, we analyzed the methylation of the shores according to their gene context. For this purpose, we grouped genes transcribed from p- to q-arm direction as Watson strand genes and those genes transcribed from the opposite strand as Crick strand genes (Fig. 3A). The N- and S-shores represent the 5′-shore and for the Watson and Crick strand genes, respectively. In NSCLC we observed that promoter-associated 5′-shores (TSS1500, TSS200, 5′-UTR and 1st exon) exhibited a significantly higher methylation compared to 3′-shores (Fig. 3B). In normal lung the degree of promoter shore methylation was lower; however a similar tendency was revealed (Fig. 3B). We also analyzed the methylation of coding region-associated shores (gene body and 3′-UTR). As displayed in Fig. 2, we observed a high methylation of these regions in NSCLC but also normal lung samples. Additionally, we also found that the methylation level of 5′-shores is higher for the coding regions compared to the 3′-shores (Fig. 3B). To verify this finding for a specific gene, we analyzed the methylation of the RASSF1A CGI and flanking regions by bisulfite pyrosequencing (Fig. S2). Within the epigenetically inactivated NSCLC A427, A549 and H322, the RASSF1A TSS and its flanking regions were highly methylated. Notably, for the cell lines that harbored unmethylated TSS sites and expressed RASSF1A, the 5′-shore regions of the RASSF1A promoter exhibited higher methylation compared to the analyzed 3′-flanking region (Fig. S2).
Epigenetic silencing of downstream genes located in tandem orientation
Next, we were interested in analyzing the methylation level of CGI promoters that are located proximal to the termination end site (TES) of an upstream gene (Fig. 4). Thus we analyzed the methylation level of CGI-associated with the transcriptional start site (TSS) at 1, 2, 3, 4, 5 and 6 kb from the TES and TSS (Fig. 4A–C). Initially, CGI methylation was compared to all TSS-associated CGIs in A549 and H322 cells (Fig. 4B and C) which demonstrated that in general, TSS-associated CGIs exhibited low levels of methylation.
Figure 4.
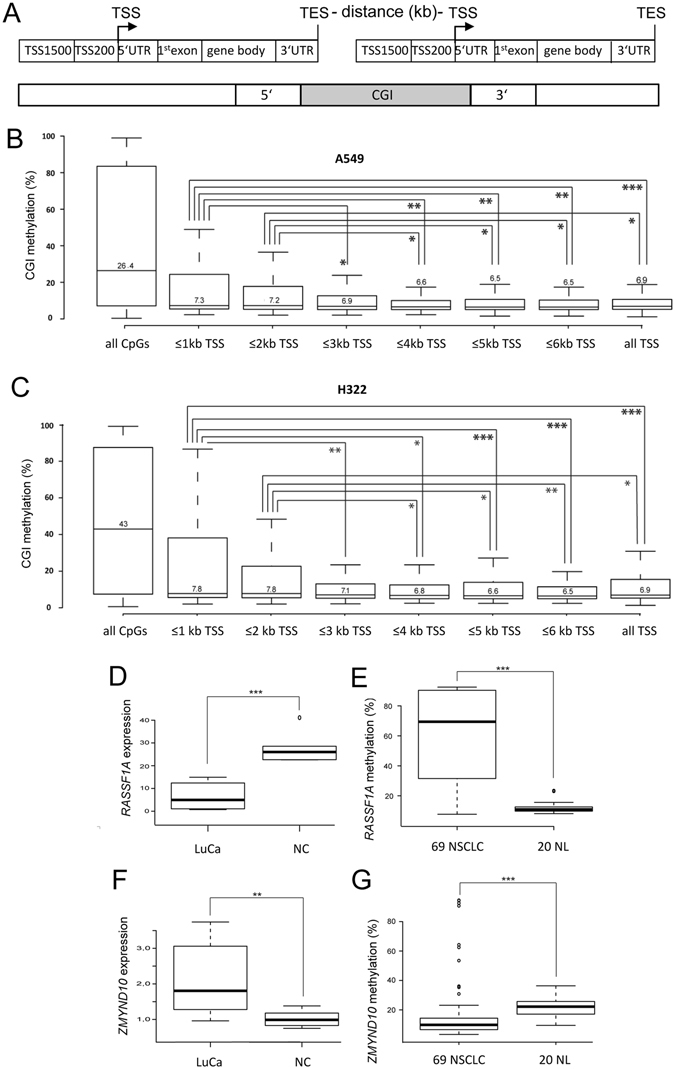
Epigenetic silencing of downstream genes located in tandem orientation. (A) Outline of the organization of tandem oriented genes with a CpG island at the transcriptional end site (TES) of the upstream gene and the transcriptional start site (TSS) of the downstream gene. (B) Methylation level of TSS-associated CGI in A549 cells was plotted depending on the distance between TES and TSS in kb (n = 258 to 297 CGI). Methylation as detected using 450 K bisulfite bead chip arrays. “All CpGs” and “all TSS” indicate the methylation level of CpGs located at CGI and TSS-associated CGI, respectively. (C) Methylation levels of tandem oriented downstream CGI in H322 cells. (D) RASSF1A expression levels were analyzed in lung cancer cell lines (LuCa: A427, A459, H322 and H358) and normal lung cells (NC: PAF and PASMC) by qRT-PCR. RASSF1A expression was normalized to the GAPDH level. (E) Methylation of RASSF1A was analyzed in 69 non small cell lung cancer (NSCLC) cell lines and 20 normal lung tissues (NL). For details see also Fig. 3. (F) ZMYND10 expression levels were analyzed in LuCa and NC as measured by qRT-PCR and normalized to GAPDH levels. (G) Methylation of ZMYND10 was analyzed in 69 NSCLC cell lines and 20 NL. *p < 0.05, **p < 0.01 and ***p < 0.001.
Notably, we observed that TSS located ≤2 kb downstream of TES displayed a significantly increased methylation compared to that at TSS located more than 4 kb downstream of TES (Fig. 4B and C). This data suggested that CGI promoters in close proximity to a TES exhibited increased methylation. Next, we annotated the 20 top downstream genes that showed a lung cancer specific hypermethylation in A549, H322 and A427 compared to normal lung tissues (Table 2). In particular, the tumor suppressor genes RASSF1A and CDKN1C, which have previously been described as epigenetically inactivated, were found among the identified genes located in this type of configuration2, 4, 23–25. The top annotated genes (Table 2) also include AOX1 and CPT1C, which have previously been found to be hypermethylated in cancer as well26, 27. To verify the epigenetic inactivation of RASSF1A in lung cancer, we analyzed its CGI methylation and expression (Fig. 4D and E). RASSF1A expression in lung cancer was significantly downregulated compared to that in normal cells (Fig. 4D). Consequently the promoter methylation of RASSF1A was significantly higher in the 69 analyzed NSCLC vs. the to 20 normal lung samples analyzed (Fig. 4E). To understand the mechanism underlying this aberrant methylation, we examined the epigenetic status of the upstream-located ZMYND10 gene (Fig. 4F and G), for which an inverse level of expression and methylation was observed in the analyzed samples. Specifically, in lung cancer the ZMYND10 expression was elevated (Fig. 4F) and its promoter exhibited reduced methylation compared to normal tissue samples (Fig. 4G). These results suggested that the epigenetic silencing of RASSF1A may be associated with an activation of the upstream tandem gene ZMYND10.
Table 2.
Top 20 tandem-oriented hypermethylated downstream genes.
| downstream CpG-island | downstream gene | Downstream CpG island methylation (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A549 | H322 | A427 | mean LuCa | NLu1 | NLu2 | mean normal | methylation differences | |||
| 1 | chr2:201450526–201451027 | AOX1 | 82 | 94 | 94 | 90 | 6 | 5 | 6 | 85 |
| 2 | chr3:50377803–50378540 | RASSF1A | 93 | 91 | 91 | 92 | 7 | 8 | 8 | 84 |
| 3 | chr14:24803678–24804353 | ADCY4 | 90 | 93 | 92 | 92 | 12 | 7 | 10 | 82 |
| 4 | chr17:40932329–40933299 | WNK4 | 92 | 94 | 93 | 93 | 15 | 15 | 15 | 78 |
| 5 | chr19:50193020–50194798 | CPT1C | 92 | 90 | 92 | 91 | 14 | 13 | 14 | 77 |
| 6 | chr3:49314437–49314815 | C3orf62 | 53 | 95 | 96 | 81 | 5 | 5 | 5 | 77 |
| 7 | chr11:64066757–64068741 | TEX40 | 81 | 79 | 85 | 82 | 12 | 11 | 12 | 70 |
| 8 | chr17:72931729–72932601 | OTOP30 | 78 | 90 | 88 | 86 | 19 | 16 | 17 | 68 |
| 9 | chr11:66045211–66045708 | CNIH2 | 28 | 97 | 86 | 70 | 3 | 4 | 3 | 67 |
| 10 | chr1:36771830–36773009 | SH3D21 | 37 | 93 | 92 | 74 | 8 | 8 | 8 | 66 |
| 11 | chr1:21043832–21044771 | KIF17 | 60 | 75 | 66 | 67 | 8 | 9 | 9 | 59 |
| 12 | chr17:72855621–72858012 | GRIN2C | 35 | 48 | 93 | 59 | 8 | 8 | 8 | 51 |
| 13 | chr17:6679205–6679710 | FBXO39 | 23 | 88 | 87 | 66 | 20 | 14 | 17 | 49 |
| 14 | chr12:7023261–7024089 | ENO2 | 38 | 54 | 92 | 61 | 13 | 13 | 13 | 48 |
| 15 | chr11:64509433–64513826 | RASGRP2 | 21 | 69 | 59 | 49 | 6 | 6 | 6 | 43 |
| 16 | chr17:46670522–46671458 | PHPT1 | 23 | 51 | 72 | 49 | 7 | 6 | 6 | 43 |
| 17 | chr2:74729399–74731166 | LBX2 | 59 | 36 | 86 | 60 | 19 | 17 | 18 | 42 |
| 18 | chr6:30881533–30882296 | VARS2 | 23 | 58 | 67 | 49 | 10 | 9 | 10 | 40 |
| 19 | chr11:62368454–62370491 | MTA2 | 53 | 37 | 55 | 48 | 12 | 20 | 16 | 33 |
| 20 | chr11:2907308–2907675 | CDKN1C | 51 | 38 | 51 | 47 | 17 | 15 | 16 | 31 |
Activation of ZMYND10 results in inactivation of the RASSF1A promoter
To dissect the mechanism involved in the epigenetic silencing of a tandem oriented downstream promoter, we cloned a 2.3 kb fragment of the ZMYND10-RASSF1A locus that included the TES of ZMYND10 and the full length RASSF1A promoter in an inducible reporter system (Fig. 5). The TES of ZYMND10 and the TSS of RASSF1A are in close proximity (170 bp). The obtained construct (TO-EGFP-2,3-RLuc) allowed for the inducible expression of a GFP-ZMYND10 fusion protein under the control of a tetracycline/doxycycline (Dox) regulated Tet-On (TO) promoter (Fig. 5A and B). TO-EGFP-2,3-RLuc was stably transfected in TREx-293 cells, which continuously express the tetracycline repressor. In this system, the activity of the downstream RASSF1A promoter was analyzed by a luciferase (RLuc) assay (Fig. 5B and C). After 4 days of Dox treatment with different concentrations (4 to 16 mM) we observed the decrease in the RLuc activity under the control of the RASSF1A promoter (Fig. 5C). Additionally, we observed a decreased expression of RLuc in a time-dependent manner from 1 day (40% reduction) to 6 days (80% reduction) with 4 mM Dox treatment (Fig. 5D). Subsequently, we generated two TREx clones (B7 and A7) and analyzed the expression of these tandem genes by western blot and luciferase assay (Fig. 5E). Both clones expressed the EGFP-ZYMND10 fusion protein after Dox treatment (Fig. 5E). RLuc expression was reduced after induction of the upstream gene at the protein level along with a significant reduction of luciferase activity (Fig. 5E) inducting of the upstream located TO promoter induced occlusion of the downstream RASSF1A promoter.
Figure 5.
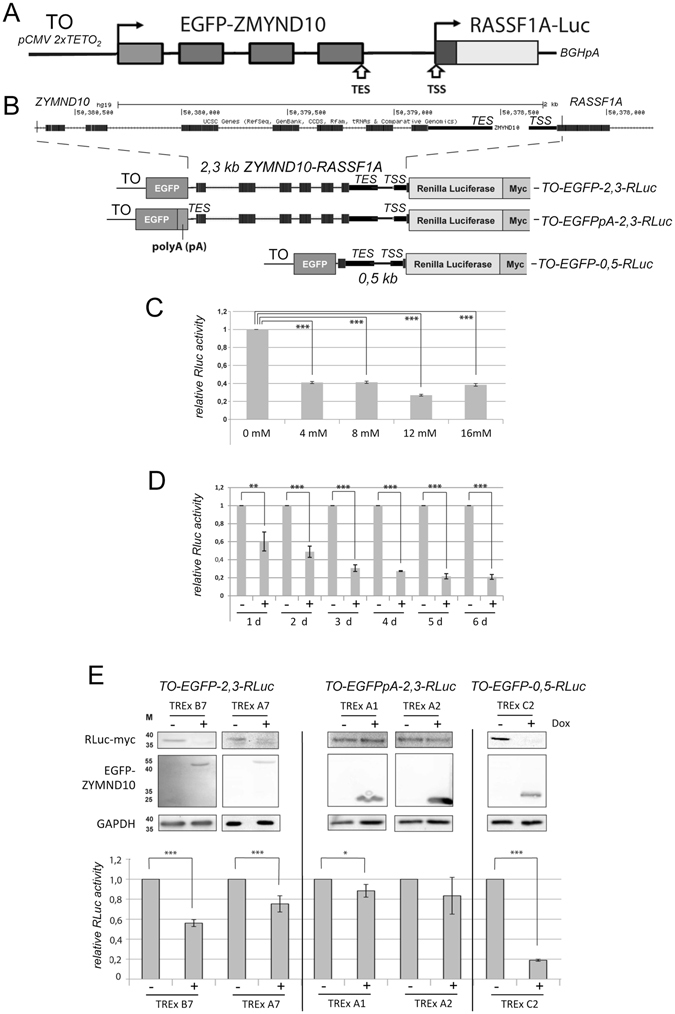
Induction of an upstream gene correlates with the downregulation of a tandem oriented downstream promoter. (A) Outline of the tandem oriented reporter gene construct. The ZMYND10 - RASSF1A promoter including the ZYMND10 transcriptional end site (TES) and the RASSF1A promoter and transcriptional start site were cloned in a tetracycline inducible vector system (Tet -On/TO). The EGFP-ZYMND fusion gene was under the control of the pCMV 2xTETO2 promoter and the 500 bp RASSF1A promoter including 5′-UTR and 17 bp of Exon1α was ligated in frame to Myc tagged Renilla luciferase (RLuc). (B) UCSC genome browser view of the 2.3. kb ZMYND10 - RASSF1A promoter locus. Black boxes represent exons. Outline of the genetic structure of the tandem reporter TO-EGFP-2,3-RLuc. In the construct TO-EGFPpA-2,3-RLuc a 300 bp SV40 poly A site (pA) was inserted at the 3′-end of EGFP. To generate the tandem reporter TO-EGFP-0,5-RLuc a 1.8 kb fragment of ZYMND10 was deleted. (C) Luciferase assay of the TO-EGFP-2,3-RLuc construct. The tandem reporter was stably transfected into TREx293 cells and induced for four days with the indicated concentration of doxycycline (Dox). RLuc activity was analyzed as described in the methods section. (D) Time dependent downregulation of RLuc activity. TREx293 cells stably transfected with TO-EGFP-2,3-RLuc were induced with 4 mM Dox (+) or uninduced (−) for the indicated days (d) and RLuc activity was analyzed. (E) Analysis of different tandem constructs by western blot and luciferase assay. Two TREx clones (B7 and A7) of the stable transfected TO-EGFP-2,3-RLuc were induced with 4 mM Dox for four days and the protein levels were subsequently analyzed by western blot. Protein lysates were separated on SDS PAGE and blotted. Full-length blots are included in the supplementary information file. For detection of the indicated proteins primary antibody against myc, GFP and GAPDH were utilized. In parallel RLuc activity was analyzed with a luciferase assay. RLuc luciferase activity was normalized to transient transfected firefly luciferase. Additionally the expression of RLuc and EGFP were analyzed in two TO-EGFPpA-2,3-RLuc TREx clones (A1 and A2) and the C2 clone of TO-EGFP-0,5-RLuc by western blot and luciferase assay. *p < 0.05, **p < 0.01 and ***p < 0.001.
To analyze this mechanism in detail, we cloned the SV40 polyA (pA) site proximal to EGFP in order to generate a novel TES 2 kb upstream of the RASSF1A TSS (Fig. 5B). The expression of this construct termed TO-EGFPpA-2,3-RLuc was analyzed in two different TREx clones (A1 and A2) by western blot and luciferase assay (Fig. 5E). Under these conditions we observed Dox induced expression of EGFP, but only a weak reduction in RLuc activity and expression (Fig. 5E). Interestingly, this effect was significantly less pronounced compared to the original constructs that contained a distance of 170 bp between the TES and TSS. Thereafter, we deleted 1.8 kb of the coding region of ZYMND10, which resulted in a close proximity (0.5 kb) between the TO promoter and the RASSF1A promoters (Fig. 5B). This construct TO-EGFP-0,5-RLuc was analyzed without and with Dox induction of the upstream promoter (Fig. 5E). Here, we also found a drastic downregulation of RLuc expression on protein and activity levels. These results indicating that the occlusion of the RASSF1A promoter correlated with the distance to the upstream promoter and the TES.
Epigenetic deregulation of promoter sequences in lung cancer
We next analyzed the epigenetic inactivation of the RLuc promoter by ChIP (Fig. 6). As histone acetylation (ac) represents the key epigenetic hallmark promoter activity, we analyzed the histone H3K9ac and H4ac at the TO/EGFP and RASSF1A promoters with and without induction of the upstream promoter in the tandem reporter assay by ChIP (Fig. 6A and B). Following Dox induction of the TO promoter, significantly increased acetylation of histone H3 and H4 were detected within this region. In contrast, at the RASSF1A promoter a significant reduction in histone H3K9ac and H4ac was found (Fig. 6B). The deactetylation of these histones was inactive of the epigenetic inactivation of the downstream promoter. To identify factors involved in this effect, we overexpressed different histone deacetylases (HDAC1/2/3/4/5/6/8/10 or Sirt1) and analyzed the expression of the RLuc promoter using the luciferase assay (Fig. S3). We found that overexpression of all analyzed histone deacetylases significantly repressed the activity of the RASSF1A promoter (Fig. S3).
Figure 6.
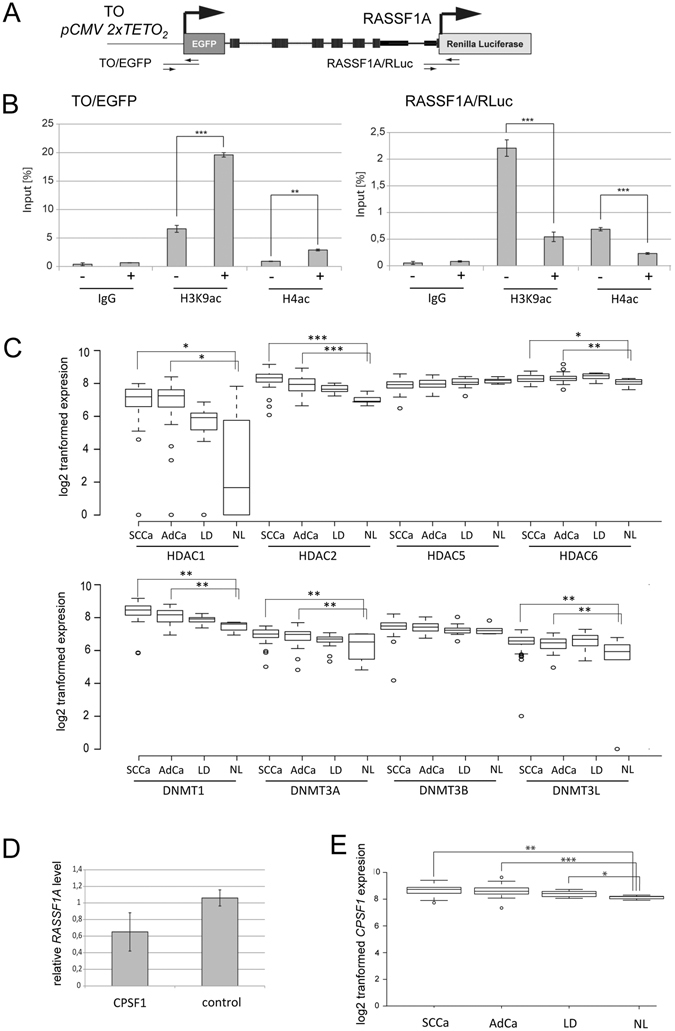
Epigenetic regulation of tandem genes in lung cancer. (A) Outline of the tandem oriented reporter gene construct analyzed by chromatin immuno-precipitation (ChIP). The analyzed TO/EGFP and RASSF1A/RLuc promoter regions are depicted. For details see Fig. 5. (B) TREx293 cells stably transfected with TO-EGFP-2.3-RLuc (clone B7) were induced with 4 mM Dox (+) or uninduced (−) for four days (d) and histone modifications were analyzed by ChIP. Chromatin was precipitated with anti histone H3K9ac, histone H4ac and IgG antibodies (negative control) antibodies. DNA was isolated and analyzed by qPCR. Levels are plotted relative to 1% of input sample. (C) Expression of HDAC1, HDAC2, HDAC5, HDAC6, DNMT1, DNMT3A, DNMT3B and DNMT3L was analyzed by microarray in 49 primary squamous cell lung cancer (SCCa), 41 primary lung adenocarcinoma (AdCa), 15 non malignant lung disease samples (LD) and 6 normal lung tissues (NL)2. Data are depicted as log2 transformed expression. (D) CPSF1 induced repression of RASSF1A. CPSF1 was cloned in pCMV-Tag1 and overexpressed in A427 cells. Endogenous RASSF1A level were analyzed by qRT-PCR and plotted relative to the empty control vector (=1). (E) Expression levels of CPSF1 in SCCa, AdCa, LD and NL. *p < 0.05, **p < 0.01 and ***p < 0.001.
To further dissect the epigenetic deregulation in primary lung cancer, we analyzed the expression levels of HDAC1, HDAC2, HDAC5, HDAC6, DNMT1, DNMT3A, DNMT3B and DNMT3L in 49 SCCa, 41 AdCa, 15 not malignant lung disease samples (LD) and 6 normal lung tissues (NL) (Fig. 6C). Expression levels were obtained a microarray previously generated in our laboratory2. Compared to normal lung samples, we observed significantly higher levels of HDAC1, HDAC2, HDAC6, DNMT1, DNMT3A and DNMT3L in primary SCCa and AdCa (Fig. 6C). Significantly increased levels of UHRF1BP, c-Myc and EZH2 were also observed in cancer samples compared to normal lung (Fig. S4). However, for HDAC5 and DNMT3B this increase was not significant (Fig. 6C).
To reveal additional factors that might be involved in the epigenetic occlusion of the downstream gene by HDACs and the transcriptional machinery, the published interactome data of the histone deacetylase family was analyzed28. In particular, the physical interaction of HDAC6 with cleavage and polyadenylation specificity factor 1 (CPSF1) has been reported28. CPSF1 is involved in the regulation of transcription termination by RNA polymerase II. Accordingly, we analyzed the effect of CPSF1 overexpression on endogenous RASSF1A expression (Fig. 6D), which demonstrated reduced RASSF1A levels in the CPSF1 transfected cells (Fig. 6D). In addition, we compared CPSF1 levels in primary lung SCCa, AdCa and normal lung (Fig. 6E), from which a significant increase of CPSF1 expression was detected in the lung cancer samples compared to normal lung. These results suggested that CPSF1 could be involved in the epigenetic inactivation of RASSF1A in lung cancer.
Discussion
The epigenetic inactivation of TSG by promoter hypermethylation represents a key event in the pathogenesis of lung cancer. Depending on the genetic and environmental context several mechanisms have been reported that may contribute to the silencing of genes in cancer. Deregulation of epigenetic key factors, including DNMT, HDAC and nucleosome remodelers and modifiers have been reported in lung cancer and may represent interesting therapeutic targets29–34. In the present study, we revealed that for genes localized in tandem orientation the downstream gene could be susceptible to epigenetic promoter occlusion depending on the distance between the TES of the upstream gene and the TSS of the downstream gene (Fig. 4). Notably, the RASSF1A lung tumor suppressor gene is located in such a configuration, suggesting that its promoter occlusion might contribute to its epigenetic silencing which constitutes a frequent event in lung and other cancers2, 4, 35. Here, we confirmed that RASSF1A is frequently hypermethylated in 69 NSCLC; conversely the upstream gene ZMYND10 is relatively hypomethylated in these tumor samples compared to normal lung tissues (Fig. 4E and G). To analyze the mechanism of RASSF1A occlusion in further detail and we generated an inducible tandem oriented reporter system that mimics the genomic organization of RASSF1A and its upstream gene ZMYND10 (Fig. 5). We observed that induction of the upstream gene resulted in a decreased expression of the RASSF1A reporter gene. Furthermore, the effect was dependent on the distance between the polyA site and the transcriptional start site (TSS) of RASSF1A (Fig. 5). In addition, we also observed a distance dependency of the TSS-associated CGI hypermethylation as detected by the 450K array (Fig. 4B and C). Consistent with these observations, epigenetic inactivation of other genes, such as CDKN1C, AOX1 or CPT1C that are located in a similar configuration, has also been reported in cancers25–27. It will be interesting to confirm the mechanism of downstream promoter occlusion in other tandem-oriented genes.
At the level of chromatin, we observed that the reduction of the RASSF1A promoter activity was associated with the deacetylation of histones H3 and H4 (Fig. 6). In lung cancer samples, we also observed a significant overexpression of HDAC1, HDAC2, HDAC6 and also DNMT1 and DNMT3A (Fig. 6). In comparison36, observed an increased expression of HDAC6 in lung AdCa and the aberrant expression correlated negatively with patient prognosis. Recently, it has also been reported that HDAC6 suppresses RASSF1A expression37. The aberrant expression of DNMT1 and DNMT3 has also been documented for lung cancer30, 38. In particular, wild type TP53 regulates the DNMT1 level and overexpression of DNMT1 has been correlated with mutated p53 39. The interaction of HDACs with DNMTs and CPSF1 has also been revealed28, 40, 41. As the upregulation of CPSF4 in cancer has been reported42, we analyzed the expression of CPSF1 and found that it was overexpressed in lung cancer (Fig. 6) and that its, overexpression resulted in reduced RASSF1A levels (Fig. 6D). Our data therefore suggest that the aberrant expression of CPSF1, HDAC6 and DNMT in cancer may cause promoter occlusion of RASSF1A. This is consistent with results from our previous studies, which indicate that histone deacetylation precedes the hypermethylation of the RASSF1A promoter43.
Conversely, it is also important to note that DNMT and HDAC inhibitors reactivate RASSF1A expression in cancer15, 43, 44. It will be interesting to analyze if suppression of ZMYND10 leads to reactivation of the RASSF1A in cancer cells. Since RNA interference reduces RNA levels by a post-transcriptional mechanism this technique is not useful to reduce ZMYND10 transcription rate. However genomic editing with the CRISPR/Cas9 technology could be utilized to alter regulatory sequences of the ZYMND10 gene. Another factor that might be involved in the reactivation of the RASSF1A tumor suppressor gene is the insulator binding protein CTCF14, 15. Specifically, it has been reported that CTCF separates chromatin boundaries and activates the expression of tumor suppressor genes by epigenetic mechanisms13, 14, 45.
The results from the present study also indicated that intra- and intergenic CGI are methylated to a higher degree in lung cancer cells than in normal lung. Furthermore, CGI regions located at the 3′-UTR exhibited a significant tumor-specific hypermethylation in lung cancer (Fig. 2) indicating that transcriptional termination may correlate with aberrant methylation of CGI at TES. This suggests that TES-associated CGI that are co-localized at TSS (e.g. the RASSF1A promoter) might be susceptible to epigenetic silencing. Additionally, we observed a correlation between the direction of transcription (5′-3′) and the methylation of CGI. 5′-shores of promoter-associated CGI in lung cancer cell lines were significantly more highly methylated than 3′-shores. We verified this result for the RASSF1A CGI promoter and observed that the -400 bp region which harbors the ZMYND10 TES exhibited increased methylation levels compared to the +400 bp region (Fig. S2). This is consistent with previous findings that CGI shores methylation was associated with gene repression46–49.
In addition, the functional annotation of hypermethylated CGI in lung cancer revealed a correlation with bivalent histone H3K27me3 modifications that serve as polycomb target sites (Table S1). It has been suggested that such bivalent chromatin patterns may predispose tumor suppressor genes to epigenetic silencing12, 50, 51. Furthermore, we observed a lung cancer-specific increase of the expression of the polycomb repressive factor EZH2 (Fig. S4). EZH2 overexpression and aberration are frequently observed in cancer, including lung tumors10, 32, 52. Notably, RASSF1A, as well as PAX5 and MEIS2 have also been previously characterized as polycomb target genes53–55. In the present study, we found lung cancer specific hypermethylation of PAX5 and MEIS2 (Table 1). Aberrant methylation of PAX5 and MEIS, which encode transcription factors harboring a homeobox domain56, 57, has already been reported in different cancer entities16, 17, 20. In accordance with this, our data also suggest that homeobox domain containing genes are significantly associated with CGI hypermethylation in lung cancer (Fig. S1).
In summary, we identified a novel putative epigenetic regulatory mechanism that is involved in the inactivation of a lung cancer related gene was identified. Our results suggest that the downstream gene promoter could be susceptible to epigenetic silencing when organized in tandem orientation with a CGI harboring a TES of the upstream gene and a TSS of the downstream gene. As the RASSF1A tumor suppressor gene represents such a downstream tandem gene, the epigenetic mechanism was confirmed for the RASSF1A promoter. HDAC and CPSF1 were identified as factors involved in this repressive effect.
Materials and Methods
Cell culture and tumor cells
The A549, H322 and A427 human cell lines (ATCC) were maintained in DMEM F12 medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (PAA, Austria), 100 units/ml penicillin, 0.1 mg/ml streptomycin, and 2 mM L-glutamine at 37 °C in a 5% CO2, 95% O2. NHBEC were obtained from Clonetics (Belgium) and grown in BEGM. TREx293 cells, which stably express the Tet repressor (Thermo Fisher, USA), were transfected with the expression vector pcDNA4TO and selected with Zeocin (Invitrogen). HEK293T and TREx293 were transfected using polyethylenimine. All patients gave a written consent at initial clinical investigation. Consent has been obtained to publish in an online-access publication, if the information could lead to the identification of a study participant. The study and experimental protocols were approved by the ethical committee of the Medical Faculty of University Halle-Wittenberg, Germany2. All experiments were performed in accordance with relevant guidelines and regulations.
Infinium HumanMethylation450K BeadChip
For the bead chip array 500 ng of genomic DNA was treated with bisulfite and Infinium Beadchip (Illumina) analysis was performed by Life & Brain GmbH (Bonn). The Illumina HumanMethylation450 K panel targets CpG sites located within the proximal promoter regions of transcription start sites of consensus coding sequences in the NCBI Database (Genome Build 36; www.ncbi.nlm.nih.gov). This bead chip technology allows the assessment of 482,421 highly informative CpG sites per sample at single-nucleotide resolution58. Relative methylation levels and differential methylation were calculated using the default settings in GenomeStudio software (Illumina). All subsequent calculations were based on average beta values and DiffScores as extracted from the GenomeStudio analysis. Beadchip methylation data has been deposited at the Gene Expression Omnibus (GEO) repository: GSE92843.
Methylation analysis
DNA was isolated by phenol-chloroform extraction and then bisulfite treated prior to pyrosequencing59. A total of 200 ng was used for PCR with the primers listed in Table S2. Methylation status was quantified utilizing a sequencing primer with PyroMark Q24 utilizing a sequencing primer (Qiagen). M.SssI methylase (NEB) was used for the in vitro methylation of genomic DNA.
Expression analysis
RNA was isolated using the Isol-RNA lysis procedure (5´Prime) from cell lines. RNA was digested with DNase (Fermentas) and then reverse transcribed as previously described60. RT-PCR was performed with the primers listed in Table S2. qRT-PCR was performed in triplicates with the SYBR Master Mix (Life Technologies) using a Rotor-Gene 3000 (Qiagen). Expression analysis of 49 primary lung SCCa, 41 lung AdCa, 15 non-malignant lung disease samples and 6 normal lung tissues was performed using a micro array generated and were previously described2.
Cloning of the tandem reporter system
A 2.3 kb fragment of the ZYMND10-RASSF1A locus was amplified from fibroblast DNA with primers RF1AXHOASEU1: 5′-CTCGAGATTAATACTGTGGAGGGCTGGAAGACCGG and PSL1: 5′-GAATTCACCGGTTCAGGCTCCCCCGACATGGC and cloned into the pRLnull vector (Promega). EGFP was obtained form pEGFP-C1 and cloned upstream in the 2.3-kb RASSF1A pRLnull construct. The stop codon of EGFP was modified by in vitro mutagenesis (Primer StopEGFPMutL1: 5′-GACTCAGATCTCGAGATCCTGCCTCTACTCAC and the reverse complement primer) to allow expression of the EGFP-ZYMND10 fusion protein. The EGFP-2.3-RLuc cassette was then cloned into pcDNA4/TO/myc-His-C and the resulting construct was termed TO-EGFP-2,3-RLuc. To generate TO-EGFPpA-2,3-RLuc the 312 bp polyA sequence of SV40 was amplified from the vector pEGFP-C1 (EGFPBsiWIU1: 5′-ACCGTACGCGAGCTCAAGCT and EGFPBsiWIL1: TGCGTACGTAAGATACATTG) and cloned in a novel BsiWI site 3′ of EGFP (generated by in vitro mutagenesis with primer BsiwIMutU1: 5′-AGGCAGGATCTCGAGATCTGAGCGTACGCTTGTACAGCTCGTCCATGCCG and the reverse complement). Finally, in the construct TO-EGFP-2,3-RLuc a 1.8 kb XhoI fragment of the ZYMND10 locus was deleted and the backbone was re-ligated to generate the TO-EGFP-0,5-RLuc construct.
Constructs
The following vectors and plasmids were used: pEGFP-C1 (Clontech); pcDNA4/TO/myc-His-C (Invitrogen, USA); pCMV-Tag1 (Stratagene), pRL-Null (Promega) and pGL3.1 (Promega). The cDNA of CPSF1 was obtained in pOTB7 (IRAUp969E0360D; Source BioScience, UK). By in vitro mutagenesis a BamHI (BamEcoMutU1: 5′-GTCGGCTCCAACTGCCAGGATCCGAATTCGCCCGGGTT and reverse complement primer) and EcoRV (EcoRVMutU1: 5′-CGTCACCGCCCACTTCTAGATATCTGGATGCCGTCACCACCAG and the reverse complement primer) restriction sites were generated flanking the cDNA and the ORF of CPSF1 was cloned in frame in pCMV-Tag1. HDAC1, HDAC2, HDAC3-FLAG, HDAC4-FLAG, HDAC5-FLAG, HDAC6, HDAC8, HDAC10-FLAG and Myc-Sirtuin 1 were obtained from Lienhard Schmitz (Justus-Liebig-University, Germany). The constructs were confirmed through conventional sequencing.
Luciferase assays
Luciferase promoter assays were performed using the Dual-Luciferase Reporter System (Promega) and assessed using the microplatte illuminometer ORIONL (Berthold). The transfection efficiency was controlled using the corresponding empty control vector (pGL3.1: 300 ng firefly luciferase respectively). The obtained data were normalized to the corresponding control vectors.
Western blot and antibodies
For western blot analysis, 20–30 µg protein lysates were separated using 12% PAGE-SDS gels and blotted on a PVDF membrane (Amersham). All antibodies were obtained from Santa Cruz Biotechnologies (Dallas, USA): anti GAPDH FL-335 (1:10000), anti Myc-Tag (1:2000) and anti GFP rabbit polyclonal serum (1:1000). For detection a goat HRP-coupled anti rabbit antibody (sc-2004) was utilized and detected with an enhanced chemiluminescence reagent (WCI-HRP-Substrate, Millipore) using a VersaDoc Imager.
Chromatin immunoprecipitation (ChIP) and antibodies
ChIPs was performed as described previously13. DNA was recovered by using QIAquick PCR Purification Kit (Qiagen) and PCR amplification with the following primers: EGFPRTF1: 5′-ACGTAAACGGCCACAAGTTC, EGFPRTR1: 5′-AAGTCGTGCTGCTTCATGTG, RF1ALucChIPU1: 5′-GCTCTCCTCAGCTCCTTCC, RF1ALucChIPL1: 5′-GTGCCTCACGACCAACTTCT. The TO/EGFP and RASSF1A promoters were amplified using the primers F1/R1 (187 bp), and U1/L1 (195 bp) respectively. qPCR was performed in triplicate using the SYBR Select Master Mix (Life Technologies) using Rotor-Gene 3000 (Qiagen).
Statistical analysis
Statistical and correlations analyses were performed using R version 3.1.3 (R Foundation). The data are presented as the means of biological triplicates ±S.D. The p-values were quantified by Student’s unpaired t-test, by Chi square test or by Wilcoxon rank-sum test. The differences are considered significant if: *p < 0.05; **p < 0.01; ***p < 0.001.
Electronic supplementary material
Acknowledgements
This work was supported by Deutsches Zentrum für Lungenforschung.
Author Contributions
R.H.D., W.S. and R.S. have created the study. S.K., M.B. and R.H.D. participated in the design of the study. S.K., T.Z. and M.B. acquired data. S.K., T.Z., R.S., M.B. and R.H.D. controlled analyzed and interpreted data. S.S.P. contributed materials. R.H.D. and S.K. prepared the manuscript. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-04248-w
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dammann R, et al. CpG island methylation and expression of tumour-associated genes in lung carcinoma. Eur J Cancer. 2005;41:1223–1236. doi: 10.1016/j.ejca.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 3.Takai D, Gonzales FA, Tsai YC, Thayer MJ, Jones PA. Large scale mapping of methylcytosines in CTCF-binding sites in the human H19 promoter and aberrant hypomethylation in human bladder cancer. Hum Mol Genet. 2001;10:2619–2626. doi: 10.1093/hmg/10.23.2619. [DOI] [PubMed] [Google Scholar]
- 4.Dammann R, et al. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nature genetics. 2000;25:315–319. doi: 10.1038/77083. [DOI] [PubMed] [Google Scholar]
- 5.Dammann R, Takahashi T, Pfeifer GP. The CpG island of the novel tumor suppressor gene RASSF1A is intensely methylated in primary small cell lung carcinomas. Oncogene. 2001;20:3563–3567. doi: 10.1038/sj.onc.1204469. [DOI] [PubMed] [Google Scholar]
- 6.Helmbold P, Lahtz C, Herpel E, Schnabel PA, Dammann RH. Frequent hypermethylation of RASSF1A tumour suppressor gene promoter and presence of Merkel cell polyomavirus in small cell lung cancer. Eur J Cancer. 2009;45:2207–2211. doi: 10.1016/j.ejca.2009.04.038. [DOI] [PubMed] [Google Scholar]
- 7.Lou-Qian Z, et al. The prognostic value of epigenetic silencing of p16 gene in NSCLC patients: a systematic review and meta-analysis. PloS one. 2013;8:e54970. doi: 10.1371/journal.pone.0054970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bogdanovic O, Veenstra GJ. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma. 2009;118:549–565. doi: 10.1007/s00412-009-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Torchy MP, Hamiche A, Klaholz BP. Structure and function insights into the NuRD chromatin remodeling complex. Cellular and molecular life sciences: CMLS. 2015;72:2491–2507. doi: 10.1007/s00018-015-1880-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kazanets A, Shorstova T, Hilmi K, Marques M, Witcher M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. Biochim Biophys Acta. 2016;1865:275–288. doi: 10.1016/j.bbcan.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 11.Stirzaker C, Song JZ, Davidson B, Clark SJ. Transcriptional gene silencing promotes DNA hypermethylation through a sequential change in chromatin modifications in cancer cells. Cancer research. 2004;64:3871–3877. doi: 10.1158/0008-5472.CAN-03-3690. [DOI] [PubMed] [Google Scholar]
- 12.Ohm JE, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nature genetics. 2007;39:237–242. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haag T, Richter AM, Schneider MB, Jimenez AP, Dammann RH. The dual specificity phosphatase 2 gene is hypermethylated in human cancer and regulated by epigenetic mechanisms. BMC cancer. 2016;16:49. doi: 10.1186/s12885-016-2087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Witcher M, Emerson BM. Epigenetic silencing of the p16(INK4a) tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Mol Cell. 2009;34:271–284. doi: 10.1016/j.molcel.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang JW, et al. Distinct epigenetic domains separated by a CTCF bound insulator between the tandem genes, BLU and RASSF1A. PloS one. 2010;5:e12847. doi: 10.1371/journal.pone.0012847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lasa A, et al. MEIS 1 expression is downregulated through promoter hypermethylation in AML1-ETO acute myeloid leukemias. Leukemia. 2004;18:1231–1237. doi: 10.1038/sj.leu.2403377. [DOI] [PubMed] [Google Scholar]
- 17.Moelans CB, Verschuur-Maes AH, van Diest PJ. Frequent promoter hypermethylation of BRCA2, CDH13, MSH6, PAX5, PAX6 and WT1 in ductal carcinoma in situ and invasive breast cancer. The Journal of pathology. 2011;225:222–231. doi: 10.1002/path.2930. [DOI] [PubMed] [Google Scholar]
- 18.Shridhar K, et al. DNA methylation markers for oral pre-cancer progression: A critical review. Oral oncology. 2016;53:1–9. doi: 10.1016/j.oraloncology.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eftang LL, et al. GFRA3 promoter methylation may be associated with decreased postoperative survival in gastric cancer. BMC cancer. 2016;16:225. doi: 10.1186/s12885-016-2247-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palmisano WA, et al. Aberrant promoter methylation of the transcription factor genes PAX5 alpha and beta in human cancers. Cancer research. 2003;63:4620–4625. [PubMed] [Google Scholar]
- 21.Shi J, et al. Characterizing the genetic basis of methylome diversity in histologically normal human lung tissue. Nature communications. 2014;5:3365. doi: 10.1038/ncomms4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walter K, et al. DNA methylation profiling defines clinically relevant biological subsets of non-small cell lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:2360–2373. doi: 10.1158/1078-0432.CCR-11-2635-T. [DOI] [PubMed] [Google Scholar]
- 23.Coley HM, et al. The cyclin-dependent kinase inhibitor p57(Kip2) is epigenetically regulated in carboplatin resistance and results in collateral sensitivity to the CDK inhibitor seliciclib in ovarian cancer. British journal of cancer. 2012;106:482–489. doi: 10.1038/bjc.2011.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kobatake T, et al. Aberrant methylation of p57KIP2 gene in lung and breast cancers and malignant mesotheliomas. Oncology reports. 2004;12:1087–1092. [PubMed] [Google Scholar]
- 25.Sato N, Matsubayashi H, Abe T, Fukushima N, Goggins M. Epigenetic down-regulation of CDKN1C/p57KIP2 in pancreatic ductal neoplasms identified by gene expression profiling. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11:4681–4688. doi: 10.1158/1078-0432.CCR-04-2471. [DOI] [PubMed] [Google Scholar]
- 26.Furuta J, et al. Silencing of Peroxiredoxin 2 and aberrant methylation of 33 CpG islands in putative promoter regions in human malignant melanomas. Cancer research. 2006;66:6080–6086. doi: 10.1158/0008-5472.CAN-06-0157. [DOI] [PubMed] [Google Scholar]
- 27.Oster B, et al. Identification and validation of highly frequent CpG island hypermethylation in colorectal adenomas and carcinomas. International journal of cancer. 2011;129:2855–2866. doi: 10.1002/ijc.25951. [DOI] [PubMed] [Google Scholar]
- 28.Joshi P, et al. The functional interactome landscape of the human histone deacetylase family. Molecular systems biology. 2013;9:672. doi: 10.1038/msb.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim H, et al. Elevated mRNA levels of DNA methyltransferase-1 as an independent prognostic factor in primary nonsmall cell lung cancer. Cancer. 2006;107:1042–1049. doi: 10.1002/cncr.22087. [DOI] [PubMed] [Google Scholar]
- 30.Sato M, et al. The expression of DNA methyltransferases and methyl-CpG-binding proteins is not associated with the methylation status of p14(ARF), p16(INK4a) and RASSF1A in human lung cancer cell lines. Oncogene. 2002;21:4822–4829. doi: 10.1038/sj.onc.1205581. [DOI] [PubMed] [Google Scholar]
- 31.Hayami S, et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. International journal of cancer. 2011;128:574–586. doi: 10.1002/ijc.25349. [DOI] [PubMed] [Google Scholar]
- 32.Lv Y, et al. The expression and significance of the enhancer of zeste homolog 2 in lung adenocarcinoma. Oncology reports. 2012;28:147–154. doi: 10.3892/or.2012.1787. [DOI] [PubMed] [Google Scholar]
- 33.Petta V, Gkiozos I, Strimpakos A, Syrigos K. Histones and lung cancer: Are the histone deacetylases a promising therapeutic target? Cancer chemotherapy and pharmacology. 2013;72:935–952. doi: 10.1007/s00280-013-2223-9. [DOI] [PubMed] [Google Scholar]
- 34.Chakravarthi BV, Nepal S, Varambally S. Genomic and Epigenomic Alterations in Cancer. The American journal of pathology. 2016;186:1724–1735. doi: 10.1016/j.ajpath.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richter AM, Pfeifer GP, Dammann RH. The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim Biophys Acta. 2009;1796:114–128. doi: 10.1016/j.bbcan.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 36.Wang Z, et al. HDAC6 promotes cell proliferation and confers resistance to gefitinib in lung adenocarcinoma. Oncology reports. 2016;36:589–597. doi: 10.3892/or.2016.4811. [DOI] [PubMed] [Google Scholar]
- 37.Tao H, Yang JJ, Hu W, Shi KH, Li J. HDAC6 Promotes Cardiac Fibrosis Progression through Suppressing RASSF1A Expression. Cardiology. 2016;133:18–26. doi: 10.1159/000438781. [DOI] [PubMed] [Google Scholar]
- 38.Lin RK, et al. Alteration of DNA methyltransferases contributes to 5′CpG methylation and poor prognosis in lung cancer. Lung Cancer. 2007;55:205–213. doi: 10.1016/j.lungcan.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 39.Lin RK, et al. Dysregulation of p53/Sp1 control leads to DNA methyltransferase-1 overexpression in lung cancer. Cancer research. 2010;70:5807–5817. doi: 10.1158/0008-5472.CAN-09-4161. [DOI] [PubMed] [Google Scholar]
- 40.Geiman TM, et al. DNMT3B interacts with hSNF2H chromatin remodeling enzyme, HDACs 1 and 2, and components of the histone methylation system. Biochem Biophys Res Commun. 2004;318:544–555. doi: 10.1016/j.bbrc.2004.04.058. [DOI] [PubMed] [Google Scholar]
- 41.Brodie SA, et al. Class I HDACs are mediators of smoke carcinogen-induced stabilization of DNMT1 and serve as promising targets for chemoprevention of lung cancer. Cancer Prev Res (Phila) 2014;7:351–361. doi: 10.1158/1940-6207.CAPR-13-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen W, et al. Upregulation of cleavage and polyadenylation specific factor 4 in lung adenocarcinoma and its critical role for cancer cell survival and proliferation. PloS one. 2013;8:e82728. doi: 10.1371/journal.pone.0082728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strunnikova M, et al. Chromatin inactivation precedes de novo DNA methylation during the progressive epigenetic silencing of the RASSF1A promoter. Mol Cell Biol. 2005;25:3923–3933. doi: 10.1128/MCB.25.10.3923-3933.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dammann R, Yang G, Pfeifer GP. Hypermethylation of the cpG island of Ras association domain family 1A (RASSF1A), a putative tumor suppressor gene from the 3p21.3 locus, occurs in a large percentage of human breast cancers. Cancer research. 2001;61:3105–3109. [PubMed] [Google Scholar]
- 45.Haag T, Herkt CE, Walesch SK, Richter AM, Dammann RH. The apoptosis associated tyrosine kinase gene is frequently hypermethylated in human cancer and is regulated by epigenetic mechanisms. Genes & cancer. 2014;5:365–374. doi: 10.18632/genesandcancer.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Irizarry RA, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature genetics. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vanderkraats ND, Hiken JF, Decker KF, Edwards JR. Discovering high-resolution patterns of differential DNA methylation that correlate with gene expression changes. Nucleic acids research. 2013;41:6816–6827. doi: 10.1093/nar/gkt482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee CJ, Evans J, Kim K, Chae H, Kim S. Determining the effect of DNA methylation on gene expression in cancer cells. Methods Mol Biol. 2014;1101:161–178. doi: 10.1007/978-1-62703-721-1_9. [DOI] [PubMed] [Google Scholar]
- 49.Rao X, et al. CpG island shore methylation regulates caveolin-1 expression in breast cancer. Oncogene. 2013;32:4519–4528. doi: 10.1038/onc.2012.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hussain M, et al. Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer research. 2009;69:3570–3578. doi: 10.1158/0008-5472.CAN-08-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kondo Y, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nature genetics. 2008;40:741–750. doi: 10.1038/ng.159. [DOI] [PubMed] [Google Scholar]
- 52.Chase A, Cross NC. Aberrations of EZH2 in cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:2613–2618. doi: 10.1158/1078-0432.CCR-10-2156. [DOI] [PubMed] [Google Scholar]
- 53.Beckedorff FC, et al. The intronic long noncoding RNA ANRASSF1 recruits PRC2 to the RASSF1A promoter, reducing the expression of RASSF1A and increasing cell proliferation. PLoS Genet. 2013;9:e1003705. doi: 10.1371/journal.pgen.1003705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ray D, et al. Lineage-inappropriate PAX5 expression in t(8;21) acute myeloid leukemia requires signaling-mediated abrogation of polycomb repression. Blood. 2013;122:759–769. doi: 10.1182/blood-2013-02-482497. [DOI] [PubMed] [Google Scholar]
- 55.Kondo T, et al. Polycomb potentiates meis2 activation in midbrain by mediating interaction of the promoter with a tissue-specific enhancer. Dev Cell. 2014;28:94–101. doi: 10.1016/j.devcel.2013.11.021. [DOI] [PubMed] [Google Scholar]
- 56.Oulad-Abdelghani, M. et al. Meis2, a novel mouse Pbx-related homeobox gene induced by retinoic acid during differentiation of P19 embryonal carcinoma cells. Developmental dynamics: an official publication of the American Association of Anatomists210, 173–183, doi:10.1002/(SICI)1097-0177(199710)210:2<173::AID-AJA9>3.0.CO;2-D (1997). [DOI] [PubMed]
- 57.Eberhard, D. & Busslinger, M. The partial homeodomain of the transcription factor Pax-5 (BSAP) is an interaction motif for the retinoblastoma and TATA-binding proteins. Cancer research59, 1716s–1724s, discussion 1724s–1725s (1999). [PubMed]
- 58.Bibikova M, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 59.Dammann G, et al. Increased DNA methylation of neuropsychiatric genes occurs in borderline personality disorder. Epigenetics: official journal of the DNA Methylation Society. 2011;6:1454–1462. doi: 10.4161/epi.6.12.18363. [DOI] [PubMed] [Google Scholar]
- 60.Richter AM, Walesch SK, Wurl P, Taubert H, Dammann RH. The tumor suppressor RASSF10 is upregulated upon contact inhibition and frequently epigenetically silenced in cancer. Oncogenesis. 2012;1:e18. doi: 10.1038/oncsis.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.