

Abstract
A recent algicidal mode indicates that fungal mycelia can wrap and eliminate almost all co-cultivated algal cells within a short time span. However, the underlying molecular mechanism is rarely understood. We applied proteomic analysis to investigate the algicidal process of Trametes versicolor F21a and identified 3,754 fungal proteins. Of these, 30 fungal enzymes with endo- or exoglycosidase activities such as β-1,3-glucanase, α-galactosidase, α-glucosidase, alginate lyase and chondroitin lyase were significantly up-regulated. These proteins belong to Glycoside Hydrolases, Auxiliary Activities, Carbohydrate Esterases and Polysaccharide Lyases, suggesting that these enzymes may degrade lipopolysaccharides, peptidoglycans and alginic acid of algal cells. Additionally, peptidase, exonuclease, manganese peroxidase and cytochrome c peroxidase, which decompose proteins and DNA or convert other small molecules of algal cells, could be other major decomposition enzymes. Gene Ontology and KEGG pathway enrichment analysis demonstrated that pyruvate metabolism and tricarboxylic acid cycle pathways play a critical role in response to adverse environment via increasing energy production to synthesize lytic enzymes or uptake molecules. Carbon metabolism, selenocompound metabolism, sulfur assimilation and metabolism, as well as several amino acid biosynthesis pathways could play vital roles in the synthesis of nutrients required by fungal mycelia.
Introduction
Blue algae (Blue-green algae or Cyanobacteria) belong to a phylum of bacteria, and can produce energy by photosynthesis1. Most members possess nitrogen fixation capacity and resistance to adverse environments2, 3. Although blue algae produce most of the microbial biomass for primary consumers in waterbodies, the rapid growth of cyanobacteria often results in algal blooms in lakes under eutrophication4, 5. Algal blooms outbreaks have significantly negative impacts on ecosystems as well as on human beings, such as reduced transparency and dissolved oxygen, generation of stench, and threat to aquatic biological safety6–9. Thus, it is extremely urgent to establish an efficient method for controlling the incidence of algal blooms and their toxic metabolites. Currently, three main approaches are used to control algal blooms involving chemical strategies, physical methods and biomanipulation10. Chemical strategies are prone to generate secondary pollution in the environment11. Physical treatments are suitable for short-term solutions, but cannot completely eliminate the blooms11. In contrast, biomanipulation is considered to be a cost-effective and safe approach to eliminate algal blooms7.
The accumulated evidences indicate that several bacteria and fungi can inhibit algal growth or degrade algal cells4. Numerous investigations have mainly focused on algicidal bacteria, while fungi with algicidal potential have been less well studied8. The antagonistic activities of 62 fungal isolates toward cyanobacteria were first observed in 1978. The cephalosporin group of antibiotics was deemed to be important algicidal factors released by Acremonium and Emericellopsis spp.12. In addition, a novel type of algicidal fungus was first identified owing to its ability to lyse live blue algae7. Mycelia of Trichaptum abietinum 1302BG can wrap algal cells and remove most algae within 48 h, resulting in clarification of the algal solution. Similar algicidal properties were also observed in the fungus L. spadicea 8. We had earlier identified Trametes versicolor F21a, Bjerkandera adusta T1, T. hirsuta T24, and Irpex lacteus T2b to have strong algicidal abilities. Of these, T. versicolor F21a showed the strongest algicidal ability by eliminating almost all tested living algal cells within 30 h11.
Several aspects of the underlying algicidal mechanism of these fungi were investigated at the cellular and biochemical levels. The interaction between fungus T. abietinum 1302BG and algal cells was investigated by scanning electron microscopy and transmission electron microscopy7. The hyphae of T. abietinum 1302BG could initially recognize the algal cells, and then wrap and degrade them7. Flow cytometry and fourier transform infrared spectroscopy were also employed to assess and compare the cell and surface morphology of the fungus Phanerochaete chrysosporium 10, revealing that P. chrysosporium primarily destroyed the cell membrane, cell wall and subsequently the pyridine ring of chlorophyll. Preliminary investigation of a few extracellular enzymes of T. versicolor F21a during the algicidal process indicated that the activities of cellulase, β-glucosidase, protease, alkaline phosphatase, laccase and manganese peroxidase were significantly induced in different phases of fungal mycelia13. The numbers and types of enzymes with decomposition abilities are far greater than the number of enzymes tested. For example, comparative analyses of 31 fungal genomes showed that as many as 250 genes are candidates for decomposition of lignin and cellulose14. Until now, it is still not known how many types of decomposition enzymes are involved in the complex algicidal process.
In this study, proteomic analysis was used to detect and quantify the whole proteins during the algicidal process of the fungus T. versicolor F21a. The aims of the present study were: (1) to use an approach combining TMT labeling and LC-MS/MS to reveal the whole proteins of T. versicolor F21a during the algicidal process; (2) to reveal the types of fungal decomposition enzymes and main metabolism pathways during the algicidal process. Our results will provide detailed information on the expressed decomposition enzymes as well as the major metabolism pathways operating during the algicidal process, and will be helpful in further understanding the underlying molecular mechanism of this novel fungi-algae interaction.
Results
Effect of T. versicolor F21a on chlorophyll-a contents of M. aeruginosa PCC7806
Growth of M. aeruginosa PCC7806 was assessed when co-cultivated with mycelia of T. versicolor F21a. The initial chlorophyll-a content of M. aeruginosa PCC7806 was 658.3 μg·L−1. After co-cultivation for 12 h and 24 h, the chlorophyll-a contents decreased to 372.6 μg·L−1 (43.40%) and 142.3 μg·L−1 (78.38%), respectively (Fig. 1). The algal media co-cultivated with fungal mycelia turned transparent after 48 h of treatment.
Figure 1.
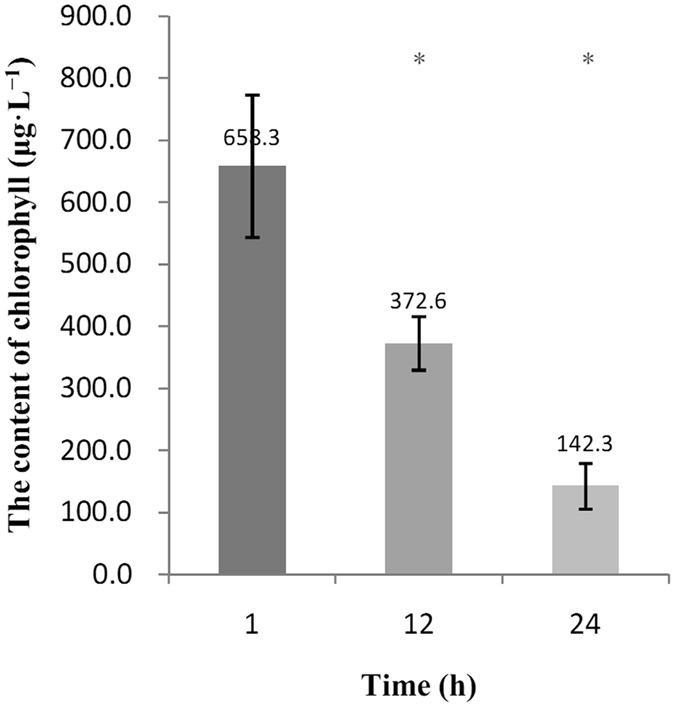
Changes in chlorophyll-a content during the algicidal process of T. versicolor F21. T12, 12 h fungal mycelia co-cultivated with algal cells; T1, 1 h fungal mycelia co-cultivated with algal cells. Significant differences between the control and treatments are indicated by asterisk (p < 0.05).
Identification of proteins related to algicidal activity using TMT LC-MS/MS
The draft genome sequence of T. versicolor FP-101664 SS1 was completed by the Department of Energy Joint Genome Institute (JGI)14, which facilitated systematic analysis of proteins related to the algicidal process of T. versicolor F21. To ensure the quality of proteomic analysis, the generated MS data were assessed. As shown in Fig. 2a, the distribution of mass error was close to zero with most of the values being less than 0.02 Da, indicating mass accuracy of the MS data (Fig. 2a). The length of most peptides was between 8 and 16 (Fig. 2b). Based on the above data, sample preparations were regarded to have reached the standard.
Figure 2.

Proteins identified during the algicidal process of T. versicolor F21a. (a) Mass error distribution of the identified peptides, (b) Peptide length distribution, (c) Distribution of significantly differentially regulated proteins. Q1 (0 < Ratio L/H < 0.67), Q2 (0.67 < Ratio L/H < 0.83), Q3 (1.2 < Ratio L/H < 1.5) and Q4 (Ratio L/H > 1.5), p < 0.05.
A total of 3,754 proteins were identified by TMT Labeling coupled with LC-MS/MS analysis, among which 2,809 proteins could be quantified. Genes encoding 207 proteins were up-regulated and those encoding 140 proteins were down-regulated in the 12 h fungal samples when compared with those at 1 h (Fig. 2c).
Functional enrichment of proteins related to algicidal activity
Four categories of cellular components, 12 categories of molecular functions and ten categories of biological processes were the enriched Gene Ontology (GO) terms15 among all differentially expressed genes (Table 1). Up-regulated proteins in the cellular component category belonged to “cell part” and “intracellular organelle” (Fig. 3a). Up-regulated proteins in the molecular function category belonged to “oxidoreductase activity, acting on peroxide as acceptor”, “oxidoreductase activity acting on the CH-OH group of donors”, “oxygen binding”, “adenylylsulfate kinase activity” and “antioxidant activity” (Fig. 3b). Up-regulated proteins in the biological process category belonged to “sulfur compound metabolic process”, “sulfate assimilation” and “sulfur compound biosynthetic process” (Fig. 3c).
Table 1.
The GO term enrichment of different proteins.
| GO Terms Level 1 | GO Terms Description | Mapping | Background | Fisher’ exact test P value | −log10(P value) |
|---|---|---|---|---|---|
| Cellular Component | ribosome | 10 | 66 | 0.007384 | 2.13 |
| protein complex | 3 | 168 | 0.022873 | 1.64 | |
| intracellular membrane-bounded organelle | 8 | 285 | 0.028706 | 1.54 | |
| membrane-bounded organelle | 8 | 285 | 0.028706 | 1.54 | |
| Molecular Function | oxidoreductase activity | 38 | 361 | 0.000305 | 3.52 |
| structural constituent of ribosome | 10 | 55 | 0.002324 | 2.63 | |
| cofactor binding | 12 | 92 | 0.012495 | 1.90 | |
| adenylylsulfate kinase activity | 2 | 2 | 0.016525 | 1.78 | |
| transferase activity, transferring acyl groups | 2 | 2 | 0.016525 | 1.78 | |
| oxidoreductase activity, acting on a sulfur group of donors | 3 | 9 | 0.024393 | 1.61 | |
| catalytic activity | 104 | 1560 | 0.025367 | 1.60 | |
| structural molecule activity | 10 | 78 | 0.026482 | 1.58 | |
| antioxidant activity | 4 | 18 | 0.028929 | 1.54 | |
| coenzyme binding | 9 | 69 | 0.036116 | 1.44 | |
| oxidoreductase activity, acting on the CH-NH group of donors | 2 | 4 | 0.038428 | 1.42 | |
| oxidoreductase activity, acting on the CH-OH group of donors | 4 | 22 | 0.049980 | 1.30 | |
| Biological Process | dicarboxylic acid metabolic process | 4 | 7 | 0.002086 | 2.68 |
| organic acid biosynthetic process | 9 | 56 | 0.007818 | 2.11 | |
| sulfur compound metabolic process | 5 | 19 | 0.008294 | 2.08 | |
| sulfur compound biosynthetic process | 4 | 12 | 0.009284 | 2.03 | |
| branched-chain amino acid biosynthetic process | 3 | 6 | 0.010509 | 1.98 | |
| sulfate assimilation | 2 | 2 | 0.016525 | 1.78 | |
| dicarboxylic acid biosynthetic process | 2 | 2 | 0.016525 | 1.78 | |
| cellular amino acid biosynthetic process | 7 | 43 | 0.017344 | 1.76 | |
| branched-chain amino acid metabolic process | 3 | 8 | 0.019044 | 1.72 | |
| aspartate family amino acid metabolic process | 3 | 9 | 0.024393 | 1.61 |
Figure 3.
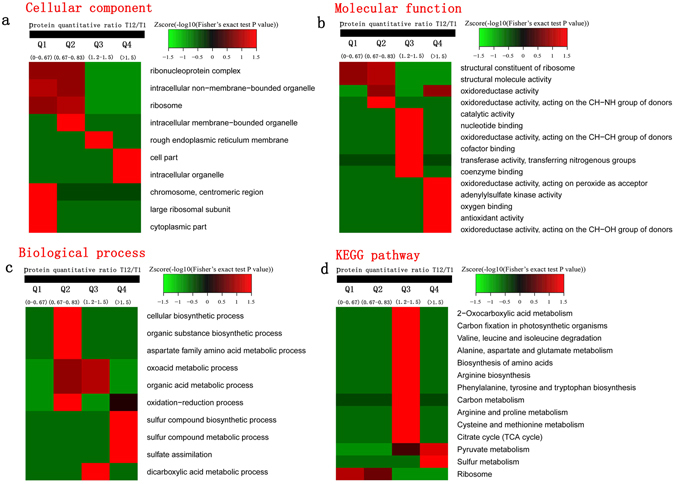
Functional enrichment-based clustering analysis of Quantitative Categories. (a) Cellular component, (b) Molecular function, (c) Biological process, (d) KEGG pathway.
KEGG pathway enrichment analysis revealed that the major enriched pathways were “ribosome”, “carbon metabolism”, “pyruvate metabolism”, “citrate cycle (TCA cycle)”, “glycolysis/gluconeogenesis”, “biosynthesis of amino acids”, etc. (Table 2, Figs S1–S3)16–18. KEGG pathway cluster analysis showed that “pyruvate metabolism” and “sulfur metabolism” were significantly up-regulated (Fig. 3d). Particularly, many genes of carbon metabolism, citrate cycle (TCA cycle) pathways, and biosynthesis of amino acids were up-regulated (Figs S1–S3)16–18.
Table 2.
KEGG pathway-based enrichment analysis of different proteins.
| KEGG pathway | Mapping | Background | Fisher’ exact test p value | −log10(P value) |
|---|---|---|---|---|
| Ribosome | 18 | 104 | 0.000181 | 3.74 |
| Carbon metabolism | 17 | 106 | 0.000494 | 3.31 |
| Pyruvate metabolism | 8 | 38 | 0.008602 | 2.07 |
| Pentose and glucuronate interconversions | 6 | 27 | 0.021394 | 1.67 |
| Selenocompound metabolism | 3 | 6 | 0.021394 | 1.67 |
| Glycolysis/Gluconeogenesis | 8 | 49 | 0.024284 | 1.61 |
| Arginine biosynthesis | 4 | 16 | 0.031222 | 1.51 |
| Alanine, aspartate and glutamate metabolism | 5 | 24 | 0.031222 | 1.51 |
| Carbon fixation in photosynthetic organisms | 4 | 16 | 0.031222 | 1.51 |
| 2-Oxocarboxylic acid metabolism | 6 | 33 | 0.031222 | 1.51 |
| Biosynthesis of amino acids | 13 | 115 | 0.031222 | 1.51 |
| Citrate cycle (TCA cycle) | 5 | 29 | 0.045084 | 1.35 |
| Phenylalanine metabolism | 4 | 19 | 0.045084 | 1.35 |
| Phenylalanine, tyrosine and tryptophan biosynthesis | 4 | 19 | 0.045084 | 1.35 |
| Carbon fixation pathways in prokaryotes | 3 | 10 | 0.045084 | 1.35 |
Classification of subcellular location
The significantly up-regulated proteins were mainly located in the cytosol, mitochondria, nucleus, plasma membrane and extracellular space, while the frequently down-regulated proteins were located in the cytosol, mitochondria, nucleus, plasma membrane, and extracellular space (Fig. 4). Remarkably, extracellular proteins comprised ~10% of all significantly up-regulated proteins.
Figure 4.

The subcellular locations of up-regulated (a) and down-regulated (b) proteins.
Domain enrichment-based clustering analysis
Protein domain enrichment-based clustering analysis of differentially regulated proteins showed that NmrA-like domain and Oxidoreductase domain were the significantly down-regulated fungal proteins during the algicidal process (Fig. 5).
Figure 5.

Protein domain enrichment-based clustering analysis of different proteins.
Biomass degradation enzymes
312 different proteins with biomass degradation capacities were predicted from the T. versicolor FP-101664 SS1 genome (Fig. 6a). They can be classified into four classes, namely Auxiliary Activities, Carbohydrate Esterases, Glycoside Hydrolases, and Polysaccharide Lyases (Table S1). These enzymes can be further classified into 46 subclasses. Majority of the degradation enzymes belong to Glycoside Hydrolases.
Figure 6.

Summary of detected degradation enzymes during the algicidal process of T. versicolor F21a. (a) Classification of detected degradation enzymes, (b) Number of detected degradation enzymes.
12 h after co-cultivation, 84 of the 312 fungal proteins could be detected by TMT Labeling LC-MS/MS analysis (Fig. 6b, Table S2). The detected proteins were grouped into 31 subclasses (Table S1). 44 out of 187 proteins belonged to Glycoside Hydrolases. More than half of the proteins (18 out of 35, and 6 out of 11) detected consisted of Carbohydrate Esterases and Polysaccharide Lyases. When compared with the 1 h samples, 30 proteins were up-regulated in the fungal mycelia at 12 h (Table 3). Total of 13 up-regulated proteins with quantitative ratios above 1.2, particularly proteins with ID 44897 (manganese peroxidase), 134935 (β-1,3-glucanase), 58033 (α-glucosidase) and 111754 (chondroitin ABC lyase) were up-regulated with quantitative ratios above 2 (Table 3). Approximately one third of up-regulated proteins contain secretary signal peptides, while 7 of 30 proteins contain signal peptides that can guide the transportation of the mature protein to the mitochondrion.
Table 3.
Up-regulated decomposition enzymes of T. versicolor F21a.
| Protein Id | NCBI annotation | CAZymes classification | Location | T12/T1 Ratio | P value | Significant |
|---|---|---|---|---|---|---|
| 26239 | manganese peroxidase | AA2 | S | 1.47 | 0.286007 | |
| 26601 | cytochrome c peroxidase | AA2 | M | 1.329 | 0.005797 | yes |
| 44897 | manganese peroxidase | AA2 | S | 3.702 | 0.004783 | yes |
| 174536 | glucose-methanol-choline (gmc) oxidoreductase family | AA3 | — | 1.168 | 0.289844 | |
| 174721 | pyranose oxidase, alcohol oxidase | AA3 | M | 1.264 | 0.23381 | |
| 57372 | glucose-methanol-choline (gmc) oxidoreductase family | AA3 | S | 1.116 | 0.388914 | |
| 59756 | alcohol oxidase | AA3 | — | 1.011 | 0.895376 | |
| 61229 | copper radical oxidase, galactose oxidase, glyoxal oxidase | AA5 | — | 1.95 | 0.000907 | yes |
| 116106 | phospholipase carboxylesterase | CE1 | M | 1.067 | 0.466033 | |
| 27033 | 3-hydroxyacyl CoA dehydrogenase | CE1 | — | 1.218 | 0.039281 | yes |
| 171192 | β-glucosidase | GH1 | — | 1.187 | 0.424115 | |
| 75497 | α-1,3-glucan synthase | GH13 | S | 1.223 | 0.006005 | yes |
| 156418 | Six-hairpin glycosidase | GH15 | — | 1.167 | 0.015216 | yes |
| 134935 | β-1,3-glucanase | GH16 | — | 2.461 | 0.004073 | yes |
| 137261 | glucosidase | GH16 | M | 1.065 | 0.409968 | |
| 175484 | endo-1,3(4)- β-glucanase | GH16 | S | 1.261 | 0.035876 | yes |
| 159574 | α-galactosidase | GH27 | S | 1.003 | 0.98303 | |
| 146818 | β-glucosidase | GH3 | — | 1.092 | 0.347247 | |
| 173291 | α-glucosidase | GH31 | S | 1.167 | 0.050646 | |
| 58033 | α-glucosidase | GH31 | S | 2.145 | 0.00371 | yes |
| 123722 | Mannosyl-oligosaccharide 1,2-α-mannosidase | GH47 | M | 1.265 | 0.002694 | yes |
| 54985 | Mannosyl-oligosaccharide 1,2-α-mannosidase | GH47 | M | 1.119 | 0.348562 | |
| 172368 | Glucan 1,3-β-glucosidase 3 | GH5 | — | 1.05 | 0.548148 | |
| 32196 | glucocerebrosidase | GH5 | — | 1.131 | 0.312239 | |
| 120979 | exo-β-1,3-glucanase | GH55 | — | 1.016 | 0.937197 | |
| 37162 | cellobiohydrolase | GH74 | S | 1.065 | 0.06072 | |
| 34231 | alginate lyase | PL14 | S | 1.136 | 0.304725 | |
| 138905 | 3-ketoacyl-CoA thiolase 5, peroxisomal | PL15 | — | 1.214 | 0.074484 | |
| 111754 | chondroitin ABC lyase | PL8 | M | 6.881 | 0.001413 | yes |
| 159259 | Chondroitinase-AC | PL8 | S | 1.06 | 0.669082 |
Location, Prediction of localization; C, Chloroplast; M, Mitochondrion; S, Secretory pathway; -, Any other location; GH, Glycoside Hydrolases; AA, Auxiliary Activities; CE, Carbohydrate Esterases; PL, Polysaccharide Lyases; Changes of more than 1.2 and P values less than 0.05 were considered significant.
Additionally, several fungal proteins belonging to peptidase, exonuclease, manganese peroxidase, cytochrome c peroxidase, glucose-methanol-choline oxidoreductase and alcohol oxidase categories were also up-regulated during the algicidal process (Table S3).
Enzymatic activities during the algicidal process
As can be seen in Fig. 7, the activity of α-glucosidase in mycelia was effectively improved in treated samples. Two peaks were presented in the co-cultivated samples of 1 h and 12 h, respectively. No obvious difference was observed in 6 h, 24 h and 48 h. By contrast, the activity of β-1,3-glucanase was significantly increased at the first 6 h and then decreased, but the activity in mycelia with algal cells during the algicidal process was much higher than that in control. Similarly, the chondroitin lyase contents were also marked enhanced in the treated samples, which reached the highest level at 24 h. The expression patterns of these enzymes were consistent with the results of the proteomic data.
Figure 7.

Changes of enzymatic activities of β-1,3-glucanase, α-glucosidase, and chondroitin lyase during the algicidal process. Treatment, the fungal mycelia co-cultured with algal cells; Control, the pure fungal mycelia.
Discussion
A small number of studies have shown that majority of the pure cultured blue algae or mixed algal cells collected from Taihu Lake (China) could be eliminated by mycelia of T. abietinum 1302BG, T. versicolor F21, B. adusta T1, P. chrysosporium, etc. Among the previously tested 60 fungal isolates, T. versicolor F21a showed the strongest algicidal ability11. Till date, all tested fungi with relatively strong algicidal ability belonged to saprophytic fungi4, 7, 11. In the T. versicolor genome, we identified 312 genes responsible for degradation polysaccharoses such as lignin and cellulose. Of these, 84 degradation enzymes were detected by proteomic analysis in this paper, with 30 of them being up-regulated in the 12 h-treated samples when compared with the 1 h samples, indicating that numerous degradation enzymes may be involved in breaking algal cells into small molecules13, 19, 20.
Previous studies had showed that elimination of algal cells was not attributed to antibiotics or other small chemicals. To eliminate algae by algicidal fungi, the algal cell envelop must be first broken down by enzyme(s)13. Lipopolysaccharides, proteins, lipids and carotenoids are the main constituents of the outer membrane of cyanobacteria21, and peptidoglycan constitutes the underlying layer21, 22. Du et al.13 believed that extracellular enzymes such as cellulase, β-glucosidase, protease, laccase and manganese peroxidase play important roles during the algicidal process of T. versicolor F21a13. In the present study, dozens of enzymes belonging to the Glycoside Hydrolases and Carbohydrate Esterases subfamilies, which can be classified as endoglycosidase and exoglycosidase, were detected during the algicidal process. Endoglycosidase may play a vital role in attacking and breaking the algal cell envelop. Protein with ID 175484 is an endo-1,3(4)-β-glucanase, which can cleave macromolecules such as peptidoglycan, suggesting its role in cell wall cleavage. Proteomics analysis in our study not only detected more types of extracellular enzymes (isoenzymes), but also observed intracellular enzymes that can potentially accelerate the algicidal process. Many up-regulated fungal proteins contain α-galactosidase (Protein ID: 159574), β-1,3-glucanase (Protein ID: 134935, 172368, 120979), α-glucosidase (Protein ID: 58033), mannosyl-oligosaccharide 1,2-α-mannosidase (Protein ID: 123722, 54985) and cellobiohydrolase (Protein ID: 37162), which can hydrolyze other cell wall components. Peptidase and exonuclease can degrade algal proteins and DNA respectively. Up-regulated manganese peroxidase (2 isoenzymes), cytochrome c peroxidase, glucose-methanol-choline oxidoreductase, alcohol oxidase are helpful to degrade the algal debris into fungal components (Tables S1, S3). Compared with the study of Du et al.13, additional types of degradation enzymes were detected in our investigation. It is worth noting that four up-regulated enzymes (Protein ID: 34231, 138905, 111754, 159259) belong to the Polysaccharide Lyases group. Protein with ID: 111754 is a chondroitin ABC lyase, which cleaves (1–4)-β-galactosaminic bonds between N-acetylgalactosamine and either D-glucuronic acid or L-iduronic acid, indicating that it can degrade peptidoglycans23. Alginate lyase (Protein with ID: 34231) was slightly up-regulated in the 12 h samples as compared to the 1 h samples (Tables S2, S3). Alginate or alginic acid is an anionic polysaccharide that is widely distributed in the cell walls of algae. Alginate lyase catalyzes the degradation of alginate into small molecules by a β-elimination reaction23. To our knowledge, chondroitin ABC lyase and alginate lyase have been identified for the first time in the present study by proteomics techniques as components of key decomposition enzymes during the algicidal process.
Algal cells can be decomposed into small molecules such as N-acetylglucosamine, N-acetylmuramic acid, glucose, amino acids, purines and pyrimidines. To utilize the small molecules from the degraded cells, the molecules must be assimilated into fungal cells by transporters via energy consumption24–26. Some of the molecules can be directly used as substrates to synthesize macromolecules, while others may be converted into other molecules for further utilization by fungal mycelia. Carbon metabolism, selenocompound metabolism, sulfur assimilation and metabolism, and several amino acid biosynthesis pathways were enriched, indicating that they play vital roles in converting small molecules into fungal components (Table 1, Fig. 3d). In particular, pyruvate decarboxylase (EC 4.1.1.1), alcohol dehydrogenase (EC 1.1.1.1), aldehyde dehydrogenase (EC 1.2.1.3), acetyl-CoA synthetase (EC 6.2.1.1) and thioltransacetylase A (EC 2.3.1.12) of the pyruvate metabolism pathway were up-regulated (Fig. S1)18. Proteins related to the tricarboxylic acid cycle were also obviously up-regulated (Table S3, Fig. S2)16–18. Pyruvate carboxylase (EC 6.4.1.1) catalyzes pyruvate, ATP and HCO3− to generate oxaloacetate, which is a component of TCA. Succinate dehydrogenase (EC 1.3.5.1) and succinate-CoA ligase (EC 6.2.1.4 and EC 6.2.1.5) of TCA were up-regulated, suggesting that a large amount of ATP may be produced. As the “molecular unit of currency”27, ATP can be utilized in the transportation of small molecules or participates in the synthesis of macromolecules. Our results indicated that pyruvate metabolism and TCA play a critical role in response to a nutrient poor environment by increasing energy production in the early stages of degradation. Valine, leucine, isoleucine, aspartate and cysteine may be further synthesized (Fig. S3) owing to the difference in amino acid composition between blue algae and fungal mycelia17, 18. The amounts of these amino acids are not sufficient for direct utilization by the fungus, and may be synthesized further in the fungal mycelia.
Proteomic evidence in this study suggested that the disappearance of living algal cells and the increase in fungal biomass could be attributed to a combination of roles of different degradation enzymes, transporters and several key metabolic pathways. The molecular mechanism for the elimination of complex algal cells by fungal mycelia was far more elaborate than the degradation of a single chemical compound. Previous investigation has shown that T. versicolor F21a can quickly and completely degrade co-cultivated algal cells. To eliminate the algal cells, T. versicolor F21a utilizes hundreds of significantly differentially regulated proteins that degrade, transport and convert different algal components into components of the fungal mycelia. Further work is needed to reveal changes and functions of different degradation enzymes during the entire algicidal process of fungi in this new mode.
Materials and Methods
Fungal strain and its cultivation
The fungus T. versicolor F21a was isolated from the soil of Zijin maintain (Nanjing, China) as described by Han et al.11. A piece of round fungal mycelium (7 mm) from the edges of a plate was inoculated in a 9 cm plate containing 15 mL of potato medium (4 g·L−1 potato starch, 20 g·L−1 dextrose, and 15 g·L−1 agar) under static conditions (28 °C). After 5 d of cultivation, the fungal mycelia were used for inoculation in algicidal experiments.
Algal strain and its cultivation
The strain Microcystis aeruginosa PCC7806 was provided by the Freshwater Algae Culture Collection of the Chinese Academy of Sciences (Wuhan, China) and was propagated in BG11 medium11. The strain was kept at 25 °C in an illumination incubator under a 12:12 h (light/dark) cycle at approximately 90 μmol photons m−2 s−1 provided by cool white fluorescent lamps.
Co-cultivation of fungus and algal cells
Batch liquid tests were conducted in 250 mL Erlenmeyer flasks containing 100 mL of algal medium and a fungal inoculum as described by Han et al.11. Briefly, the prepared fungal mycelia were transferred into a flask and co-cultivated in a shaking incubator (25 ± 2 °C, 125 rpm). Co-cultivated fungal mycelia were sampled after 1 h and 12 h. With the consideration of the fungal mycelia need a short time to adjust in the new environment, 1 h samples were used as the initial samples. All experiments were performed in triplicate.
Total chlorophyll-a was extracted with 90% acetone and estimated according to the Standard Methods for the Examination of Water and Wastewater (1998).
Protein extraction, digestion and TMT labeling
Co-cultivation samples of fungal mycelia and algal cells were ground to a powder in liquid nitrogen and three biological replicates of each were prepared for proteomic analyses. The cell powder was resuspended in lysis buffer (8 M urea, 10 mM DTT, 2 mM EDTA and 1% protease inhibitor Cocktail VI) and sonicated thrice on ice. The debris was removed by centrifugation at 20,000 g for 10 min and the remaining protein was precipitated with pre-cooled 15% TCA for 2 h at −20 °C. After centrifugation, the precipitate was washed thrice with cold acetone and redissolved in buffer (8 M urea, 100 mM TEAB, pH 8.0). A 2-D Quant kit (GE Healthcare, USA) was used to determine protein concentrations. The protein solution was digested with trypsin (Promega) at 1:50 trypsin-to-protein mass ratio for the first overnight digestion and 1:100 trypsin-to-protein mass ratio for a second 4 h-digestion. Approximately 100 μg protein from each sample was digested with trypsin for further experiments. After trypsin digestion, the peptide mix was desalted by Strata X C18 SPE column (Phenomenex) and vacuum-dried. The dried peptide mix was reconstituted in 0.5 M TEAB and processed as per the manufacturer’s protocol by the 6-plex TMT kit. Briefly, one unit of TMT reagent (defined as the amount of reagent required to label 100 μg of protein) was thawed and reconstituted in 24 μl ACN. The peptide mixtures were then incubated for 2 h at room temperature and pooled, desalted and dried by vacuum centrifugation.
HPLC fractionation
The labeled peptides were fractionated by high pH reverse-phase HPLC using Agilent 300 Extend C18 columns (5 μm particles, 4.6 mm ID, 250 mm length). Briefly, the peptides were initially separated on a 2% to 60% acetonitrile gradient in 10 mM ammonium bicarbonate at pH 10 over 80 min into 80 fractions. The peptides were then combined as per the manufacturer’s protocol for LC-MS/MS analysis.
Quantitative proteomic analysis by LC-MS/MS
LC-MS/MS Analysis
The labeled peptides were dissolved in 0.1% FA (Formic acid) and directly loaded onto a reversed-phase pre-column (Acclaim PepMap 100, Thermo Fisher Scientific). Peptide separation was performed in a reversed-phase analytical column (Acclaim PepMap RSLC, Thermo Scientific). The gradient was comprised of an increase from 6% to 22% of solvent B (0.1% FA in 98% acetonitrile) over 26 min, then 22% to 35% in 8 min and increasing to 80% in 3 min, then holding at 80% for the last 3 min, at a constant flow rate of 350 nl min−1 on an EASY-nLC 1000 UPLC system. The resulting peptides were analyzed on a Q ExactiveTM hybrid quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific).
Database search
Data were deposited in the PRIDE database28 with the identifier PXD005568. The Mascot search engine (v.2.3.0) was employed to process MS/MS data. Tandem mass spectra were searched against 14296 predicted protein sequences T. versicolor FP-101664 SS1 that were downloaded from the fungal genomics resource at JGI (v1.0, 2011-01-05). For peptide search, Trypsin/P was specified as the cleavage enzyme allowing up to 2 missing cleavages. Mass error was set to 10 ppm for precursor ions and 0.02 Da for fragment ions. Carbamidomethyl on Cys, TMT-6Plex on peptides N-term and Lys were specified as the fixed modification. Oxidation on Met was specified as the variable modification. For protein identification, only peptides with scores significant at the 99% confidence level and peptides ion score was set as 20 were considered to be reliable. Only proteins with significant quantitative ratios between the two treatments (p < 0.05) and with fold changes >1.2 or <0.83 were considered to be differentially expressed15.
Protein annotation and enrichment analysis
All identified proteins were annotated based on the UniPort-GOA database, Gene Ontology database, InterProScan database, dbCAN and KEGG Pathway database. The GO function of the proteins was classified into the following three categories: biological process, cellular component and molecular function. The InterPro domain database was used to annotate protein domain function based on protein sequence alignment method. WoLF PSORT was used to predict subcellular localizations of proteins in cells. Fisher’s exact test was used to obtain the enriched functional terms. Categories with P value < 0.05 were considered as enriched.
Statistical analysis
The proteins quantified in this study were divided into four quantitative categories based on the quantification ratio: Q1 (0 < Ratio L/H < 0.67), Q2 (0.67 < Ratio L/H < 0.83), Q3 (1.2 < Ratio L/H < 1.5) and Q4 (Ratio L/H > 1.5)15, 16. Quantitative category based clustering was performed. Q1, Q2, Q3 and Q4 indicated different L/H ratios in the quantification results. Geometric mean of three biological replicates was calculated for each sample and then the ratio was calculated accordingly; P value was calculated from 2-sample t-test.
Clustering Method: All substrate categories obtained after enrichment were collated along with their P values, and then filtered for those categories that were enriched in at least one of the clusters with P value < 0.05. This filtered P value matrix was transformed by the function x = −log10(P value). The x values obtained were z-transformed for each category. These z scores were then clustered by one-way hierarchical clustering (Euclidean distance, average linkage clustering) in Genesis. Cluster membership was visualized by a heat map using the “heatmap.2” function from the “gplots” R-package.
Assay of enzymatic activities
Three significantly up-regulated decomposition enzymes were selected to test their activities. Fungal mycelia with or without algal cells during the entire algicidal process (0 h, 1 h, 6 h, 12 h, 24 h and 48 h) were used to prepare crude enzymes according to Du et al.13. Enzymatic activity of β-1,3-glucanase was determined spectrophotometrically according to Jin et al.29 and Vázquez-Garcidueñas et al.30. Laminarin (Sigma) was used as the substrate and one unit of β-1,3-glucanase activity was defined as the amount of enzyme that catalyzed the release of 1 µg of glucose equivalents in one minute. The activity of α-glucosidase was performed following the modified method of Mohamed et al.31. 3,4-Nitrophenyl α-D-glucopyranoside (Sigma) was used as the substrate. One unit of α-glucosidase activity was defined as the amount of enzyme that catalyzed the release of 1 µmol of pNP equivalents in one minute. Chondroitin lyase was assayed as per the modified method described previously32, 33. Chondroitin sulfate (sigma) was used as the substrate to demonstrate differences between the treatments and controls.
Conclusions
In this study, 3,754 proteins of T. versicolor F21a were identified during the algicidal process. Among them, 2,809 unique proteins could be quantified. 30 proteins with biomass degradation capacities were significantly up-regulated during the process. ~10% of the up-regulated proteins were extracellular enzymes. The major metabolic pathways identified were carbon metabolism, selenocompound metabolism, sulfur assimilation and metabolism, along with several amino acid biosynthesis pathways.
Electronic supplementary material
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (Project No. 51309003 and 31301756), and the Doctoral Fund of Ministry of Education of China (Project No. 20133418120003).
Author Contributions
Xueyan Gao and Congyan Wang designed and carried out majority of the bioinformatics studies. Wei Dai and Shenrong Ren carried out software analyses and performed the experiments. Xueyan Gao and Congyan Wang wrote the manuscript. Fang Tao and Xingbing He designed the study and helped draft the manuscript. Guomin Han and Wei Wang conceived and directed the study. All authors read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Xueyan Gao and Congyan Wang contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-04251-1
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Guomin Han, Email: guominhan@ahau.edu.cn.
Wei Wang, Email: wangweisys@ahau.edu.cn.
References
- 1.Elliott JA. The seasonal sensitivity of Cyanobacteria and other phytoplankton to changes in flushing rate and water temperature. Global Change Biology. 2010;16:864–876. doi: 10.1111/j.1365-2486.2009.01998.x. [DOI] [Google Scholar]
- 2.Paerl HW, et al. Controlling cyanobacterial blooms in hypertrophic Lake Taihu, China: will nitrogen reductions cause replacement of non-N2 fixing by N2 fixing taxa? PloS one. 2014;9:e113123. doi: 10.1371/journal.pone.0113123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kadam PD, Chuan HH. Rectocutaneous fistula with transmigration of the suture: a rare delayed complication of vault fixation with the sacrospinous ligament. International urogynecology journal. 2016;27:155. doi: 10.1007/s00192-015-2823-5. [DOI] [PubMed] [Google Scholar]
- 4.Mohamed ZA, Hashem M, Alamri SA. Growth inhibition of the cyanobacterium Microcystis aeruginosa and degradation of its microcystin toxins by the fungus Trichoderma citrinoviride. Toxicon. 2014;86:51–58. doi: 10.1016/j.toxicon.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Heisler J, et al. Eutrophication and harmful algal blooms: A scientific consensus. Harmful Algae. 2008;8:3–13. doi: 10.1016/j.hal.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jia Y, Du J, Song F, Zhao G, Tian X. A fungus capable of degrading microcystin-lr in the algal culture of Microcystis aeruginosa PCC7806. Appl Biochem Biotechnol. 2012;166:987–996. doi: 10.1007/s12010-011-9486-6. [DOI] [PubMed] [Google Scholar]
- 7.Jia Y, et al. The efficacy and mechanisms of fungal suppression of freshwater harmful algal bloom species. J Hazard Mater. 2010;183:176–181. doi: 10.1016/j.jhazmat.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 8.Wang Q, et al. Growth inhibition of Microcystis aeruginosa by white-rot fungus Lopharia spadicea. Water Sci Technol. 2010;62:317–323. doi: 10.2166/wst.2010.214. [DOI] [PubMed] [Google Scholar]
- 9.Lance E, Neffling MR, Gerard C, Meriluoto J, Bormans M. Accumulation of free and covalently bound microcystins in tissues of Lymnaea stagnalis (Gastropoda) following toxic cyanobacteria or dissolved microcystin-LR exposure. Environmental pollution. 2010;158:674–680. doi: 10.1016/j.envpol.2009.10.025. [DOI] [PubMed] [Google Scholar]
- 10.Zeng G, Wang P, Wang Y. Algicidal efficiency and mechanism of Phanerochaete chrysosporium against harmful algal bloom species. Algal. Research. 2015;12:182–190. [Google Scholar]
- 11.Han G, et al. Isolation and evaluation of terrestrial fungi with algicidal ability from Zijin Mountain, Nanjing, China. Journal of microbiology. 2011;49:562–567. doi: 10.1007/s12275-011-0496-4. [DOI] [PubMed] [Google Scholar]
- 12.Redhead K, Wright SJ. Isolation and properties of fungi that lyse blue-green algae. Applied & Environmental Microbiology. 1978;35(35):962–969. doi: 10.1128/aem.35.5.962-969.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du J, et al. Potential of extracellular enzymes from Trametes versicolor F21a in Microcystis spp. degradation. Mater Sci Eng C Mater Biol Appl. 2015;48:138–144. doi: 10.1016/j.msec.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Floudas D, et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science. 2012;336:1715–1719. doi: 10.1126/science.1221748. [DOI] [PubMed] [Google Scholar]
- 15.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai Y, et al. Integrative analyses reveal transcriptome-proteome correlation in biological pathways and secondary metabolism clusters in A. flavus in response to temperature. Scientific reports. 2015;5:14582. doi: 10.1038/srep14582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu, B. B., Qin, F. L., Liu, W. W. & Wang, X. F. Differential proteomics profiling of the ova between healthy and Rice stripe virus-infected female insects of Laodelphax striatellus. Scientific reports6, doi:10.1038/srep27216 (2016). [DOI] [PMC free article] [PubMed]
- 21.Fiore M, Trevors J. Cell composition and metal tolerance in cyanobacteria. BioMetals. 1994;7(2):83–103. doi: 10.1007/BF00140478. [DOI] [Google Scholar]
- 22.Hoiczyk E, Hansel A. Cyanobacterial cell walls: news from an unusual prokaryotic envelope. Journal of bacteriology. 2000;182(185):1191–1199. doi: 10.1128/JB.182.5.1191-1199.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic acids research. 2014;42:D490–495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jun, H., Kieselbach, T. & Jonsson, L. J. Enzyme production by filamentous fungi: analysis of the secretome of Trichoderma reesei grown on unconventional carbon source. Microbial cell factories10, doi:10.1186/1475-2859-10-68 (2011). [DOI] [PMC free article] [PubMed]
- 25.Roitsch T, Balibrea ME, Hofmann M, Proels R, Sinha AK. Extracellular invertase: key metabolic enzyme and PR protein. Journal of Experimental Botany. 2003;54:513–524. doi: 10.1093/jxb/erg050. [DOI] [PubMed] [Google Scholar]
- 26.Santulli G, Xie W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:11389–11394. doi: 10.1073/pnas.1513047112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stricker AR, Grosstessner-Hain K, Wurleitner E, Mach RL. Xyr1 (xylanase regulator 1) regulates both the hydrolytic enzyme system and D-xylose metabolism in Hypocrea jecorina. Eukaryotic cell. 2006;5:2128–2137. doi: 10.1128/EC.00211-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vizcaíno JA, et al. 2016 update of the PRIDE database and related tools. Nucleic Acids Res. 2016;44:D447–D456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin X, Feng D, Wang H, Wang J. A novel tissue-specific plantain beta-1,3-glucanase gene that is regulated in response to infection by Fusarium oxysporum fsp. cubense. Biotechnol Lett. 2007;29:1431–1437. doi: 10.1007/s10529-007-9403-9. [DOI] [PubMed] [Google Scholar]
- 30.Vazquez-Garciduenas S, Leal-Morales CA, Herrera-Estrella A. Analysis of the beta-1,3-Glucanolytic System of the Biocontrol Agent Trichoderma harzianum. Applied and environmental microbiology. 1998;64:1442–1446. doi: 10.1128/aem.64.4.1442-1446.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohamed SSH, Hansi PD, Thirumurugan K. Cinnamon extract inhibits α-glucosidase activity and dampens postprandial glucose excursion in diabetic rats. Nutrition & metabolism. 2011;8(1):46–46. doi: 10.1186/1743-7075-8-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saito H, Yamagata T, Suzuki S. Enzymatic methods for the determination of small quantities of isometric chondroition sulfate. Journal of Biological Chemistry. 1968;243:1536–1542. [PubMed] [Google Scholar]
- 33.Sato N, et al. Cloning and expression in Escherichia coli of the gene encoding the Proteus vulgaris chondroitin ABC lyase. Applied microbiology and biotechnology. 1994;41:39–46. doi: 10.1007/BF00166079. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.