

Abstract
Ficolins are a family of pattern recognition molecules that are capable of activating the lectin pathway of complement. A limited number of reports have demonstrated a protective role of ficolins in animal models of infection. In addition, an immune modulatory role of ficolins has been suggested. Yet, the contribution of ficolins to inflammatory disease processes remains elusive. To address this, we investigated ficolin deficient mice during a lipopolysaccharide (LPS)-induced model of systemic inflammation. Although murine serum ficolin was shown to bind LPS in vitro, there was no difference between wildtype and ficolin deficient mice in morbidity and mortality by LPS-induced inflammation. Moreover, there was no difference between wildtype and ficolin deficient mice in the inflammatory cytokine profiles after LPS challenge. These findings were substantiated by microarray analysis revealing an unaltered spleen transcriptome profile in ficolin deficient mice compared to wildtype mice. Collectively, results from this study demonstrate that ficolins are not involved in host response to LPS-induced systemic inflammation.
Introduction
The ficolins are a family of pattern recognition molecules that initiate the lectin pathway of complement1–3. Humans express three ficolins: ficolin-1(M-ficolin)4, 5 in bone marrow and myeloid cells, ficolin-2(L-ficolin)6 in liver and ficolin-3(H-ficolin)7 in liver and lungs. All the human ficolins are present in serum8–10. Mice express two ficolins: ficolin-A11 in liver and spleen and ficolin-B12 in bone marrow. Both proteins are present in serum, although ficolin-B only in very low concentration13. Ficolin-B is the homologue of human ficolin-1, ficolin-A is closely related to human ficolin-2 whereas a third ficolin gene exists as a pseudogene in mice14. The ficolins recognize pathogen-associated molecular patterns on the surface of microorganisms15–19 and DNA on apoptotic and necrotic host cells20, 21. Upon pathogen recognition, the ficolin associated serine proteases, named MASPs, catalyze cleavage of complement components C4 and C2, leading to C3 convertase (C4b2a) formation22. The C3 convertase cleaves C3 into the opsonin C3b and the anaphylatoxin C3a. Activation of C3 also leads to downstream formation of the C5 convertase (C4b2a3b) which cleaves C5 into the anaphylatoxin C5a and the fragment C5b. C5b initiates formation of the lytic terminal membrane attack complex (C5b-9). In this way, effector functions of the complement cascade, i.e. opsonization, lysis and generation of an inflammatory response, result in elimination of the target. Therefore, complement and ficolins generally have a host-protective effect.
In certain situations, however, an exaggerated activation of the complement cascade can have detrimental effects due to activation of inflammatory cells and secretion of pro-inflammatory cytokines23, 24. Sepsis is a systemic overwhelming inflammatory response, resulting from uncontrolled bacterial spread during infection, which can lead to tissue damage, multiple organ failure and death25. Because this uncontrolled production of pro-inflammatory cytokines can be more damaging than the primary infection, it is important to attenuate this excessive reaction. Lipopolysaccharide (LPS) from the outer membrane of Gram-negative bacteria plays a major role in activating the inflammatory response during sepsis, and administration of LPS in animal models has been widely used to study the inflammatory response to Gram-negative bacteria26.
While a protective effect of ficolins has been demonstrated in clinical studies27–29 and in mouse models of infection13, 30–32, the contribution of ficolins to inflammatory processes is not well-characterized. However, we recently observed that ficolin deficient mice showed decreased production of pro-inflammatory cytokines during local fungal lung infection in vivo 33. Others have reported that ficolin-2 promotes production of pro-inflammatory cytokines in mouse macrophages in vitro, and that ficolin-A promotes TNF and IL-17 production in response to LPS in vivo 32.
To elaborate on these previous discoveries and to better understand the role of ficolins during inflammation, we subjected ficolin deficient mice to LPS-induced inflammation and evaluated their inflammatory response and survival.
Results
Ficolin-A binds to LPS in vitro
To determine whether ficolin-A binds to LPS, ELISA plates were coated with LPS (from Escherichia coli 0111:B4) and serial dilutions of wildtype (WT) or ficolin knockout (KO) serum were added and ficolin-A binding to LPS was detected with an anti-ficolin-A antibody. Plates coated with the ficolin-A ligand acetylated bovine serum albumin (AcBSA) served as a positive control while plates coated with BSA served as a negative control. The results showed a dose-dependent binding of ficolin-A from WT serum to AcBSA and LPS (Fig. 1A,B). As expected, no binding from ficolin KO serum to either of the coats was observed. In another experiment, mouse serum (WT or ficolin KO) was diluted 1:50 and pre-incubated with serial dilutions of LPS for 2 h. When the samples were added to plates coated with AcBSA and ficolin-A was detected with anti-ficolin-A antibody, a dose-dependent inhibition by LPS of ficolin-A binding to its known ligand AcBSA was revealed. These results demonstrated that ficolin-A from mouse serum binds to the LPS used for the subsequent in vivo experiments.
Figure 1.
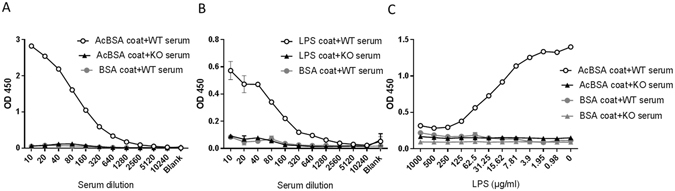
Serum ficolin-A binds to LPS in vitro. ELISA plates were coated with of BSA (negative control), the ficolin-A ligand AcBSA (positive control) or LPS (all 5 µg/ml). In A and B, serial dilutions of mouse serum (WT or ficolin KO) were added to the plates and ficolin-A was detected with an anti-ficolin-A antibody. In C, mouse serum (WT or ficolin KO) was diluted 1:50 and pre-incubated with serial dilutions of LPS for 2 h. Subsequently, the samples were added to plates coated with AcBSA or BSA and ficolin-A was detected with an anti-ficolin-A antibody. All graphs show mean ± SEM of duplicate wells and are representative of three independent experiments.
Ficolin deficiency does not impact morbidity and mortality by LPS-induced inflammation
In order to assess the role of ficolins during LPS-induced inflammation in vivo, groups of WT mice and ficolin KO mice were injected intraperitoneally (i.p.) with four different doses of LPS and survival time and body weight change was monitored for seven days (Fig. 2). The clinical scores of the animals are presented in Supplementary Fig. S1. Given the highest dose of LPS (25 mg/kg), all mice in WT and ficolin KO groups reached the humane endpoint before day 2 and the median survival time of both groups was 1 day (Fig. 2A). Given a dose of 10 mg/kg, survival rate of WT mice was 47% and of ficolin KO mice 33% and their median survival times were 7 and 2 days, respectively. The difference in survival rates was not statistically significant. After receiving a dose of 5 mg/kg, the survival rate of both WT and ficolin KO groups was 87% and the median survival time was undefined. Given a dose of 2.5 mg/kg, all animals in both groups survived and their median survival time was undefined.
Figure 2.
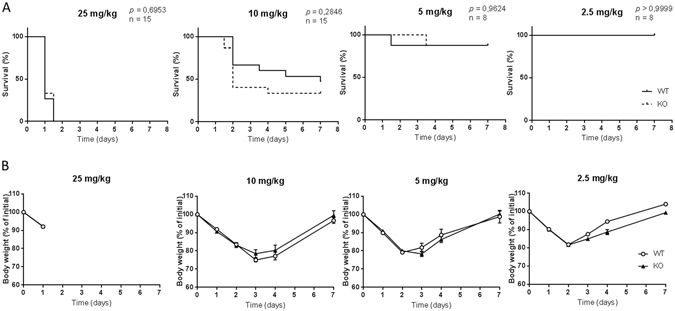
Survival and body weight change is similar in WT and ficolin KO mice after LPS challenge. WT and ficolin KO mice were intraperitoneally injected with LPS of indicated doses and monitored for survival (A) and body weight change (B) for 7 days. Results were obtained from one (5 mg/kg and 2.5 mg/kg; n = 8) or two (25 mg/kg and 10 mg/kg; n = 15) experiments. In A, Kaplan–Meier survival curves were compared using the log-rank test. In B, graphs show mean ± SEM.
Consistent with the survival recordings, there was no difference in weight loss between WT and ficolin KO mice (Fig. 2B). The clinical scores (Supplementary Fig. S1) and the body weight changes (Fig. 2B) revealed that the mice were affected by the two low doses of LPS although most of them survived the LPS challenge. In addition, control groups of WT and ficolin KO mice received 100 µl of PBS i.p.: their survival was 100% and their body weight remained the same during the 7 day period (Supplementary Fig. S2).
Ficolin expression is unaltered by LPS-induced inflammation
Next, we assessed the expression of ficolins 6 h after LPS challenge of a lethal dose (10 mg/kg). qPCR analysis showed a fold-change in gene expression of ficolin-A in liver of 0.9 and in spleen of 0.7 and of ficolin-B in bone marrow of 1.2 after LPS challenge (Fig. 3); none of these changes were statistically significant.
Figure 3.
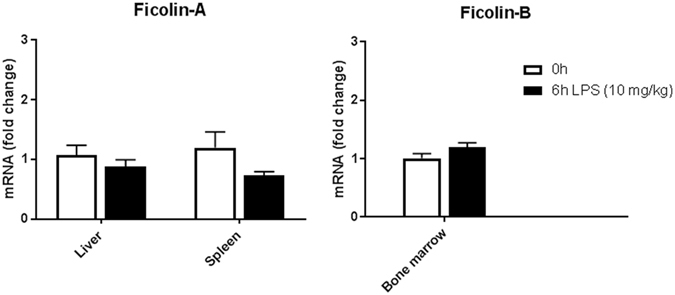
Ficolin expression is unaltered in mice after LPS challenge. Groups of WT mice were untreated or intraperitoneally injected with LPS of a lethal dose (10 mg/kg) and sacrificed after 6 h. Liver, spleen and bone marrow were removed and expression of ficolin-A and ficolin-B in tissue homogenates was measured by qPCR. Data was obtained from one experiment and represent mean ± SEM (n = 5) as the fold change from naive WT mice.
Ficolin deficiency does not impact the cytokine profile in response to LPS-induced inflammation
To evaluate the LPS-induced inflammatory response in the animals, we analyzed the cytokine profiles after LPS challenge (Figs 4–8). First, we determined the gene expression of inflammatory cytokines in spleens of WT and ficolin KO mice (Fig. 4). Groups of mice were either challenged with a lethal dose of LPS (10 mg/kg) and sacrificed after 6 h or challenged with a sublethal dose of LPS (2.5 mg/kg) and sacrificed after 6 h or 24 h, and the mRNA levels of IL-1β, TNF, IL-6 and IL-10 in spleen homogenates were determined. Comparing the expression of the cytokines induced by the two different doses at 6 h, all four cytokines were expressed to a higher extend after the lethal dose. After challenge with a lethal dose of LPS, the expression of all four cytokines were lower at 6 h in ficolin KO mice compared to WT, but only the difference in IL-1β levels was statistically significant. After a sublethal dose of LPS, the pro-inflammatory cytokines (IL-1β, TNF and IL-6) peaked at 6 h, while the anti-inflammatory cytokine IL-10 peaked at 24 h. There was no difference in expression between ficolin KO mice and WT mice challenged with a sublethal dose of LPS.
Figure 4.
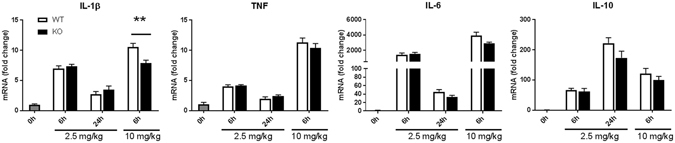
Cyokine expression in spleens of WT and ficolin KO mice after LPS challenge. Groups of WT and ficolin KO mice were intraperitoneally injected with LPS of a sublethal (2.5 mg/kg) or lethal (10 mg/kg) dose and sacrificed at indicated time points. Spleens were removed and expression of inflammatory cytokines in spleen homogenates was measured by qPCR. Data represent mean ± SEM as the fold increase over the control group (naive WT mice); n = 6–8 mice in LPS groups and n = 2 mice in the control group. **p < 0.01 for WT to ficolin KO comparisons (unpaired two-tailed Student’s t test).
Figure 8.
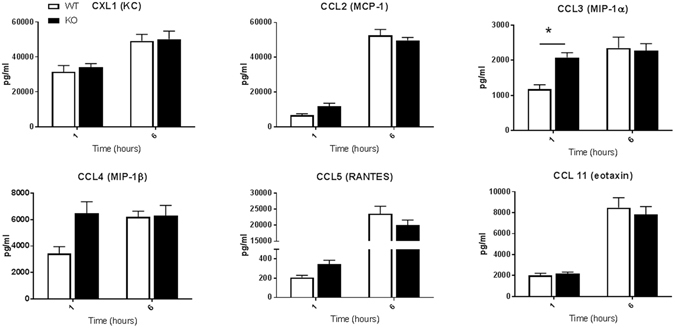
Serum chemokine levels in WT and ficolin KO mice after LPS challenge with a lethal dose. Groups of WT and ficolin KO mice were intraperitoneally injected with LPS of a lethal dose (10 mg/kg). At 1 h or 6 h after challenge, blood was collected before the mouse was sacrificed. The serum concentration of chemokines was measured by a multiplex Luminex assay; the data was obtained from two independent experiments per time point and represent mean ± SEM (n = 11 mice per group per time point). *p < 0.05 for WT to ficolin KO comparisons (unpaired two-tailed Student’s t test).
To further assess the inflammatory response in mice after LPS challenge, we measured the serum levels of 22 inflammatory cytokines and chemokines at various time points after challenge with a sublethal or a lethal dose of LPS (Figs 5–8).
Figure 5.
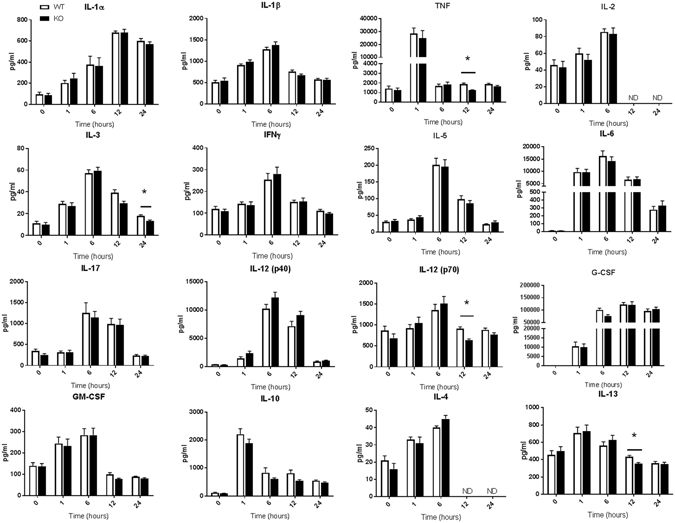
Serum cytokine levels in WT and ficolin KO mice after LPS challenge with a sublethal dose. Groups of WT and ficolin KO mice were intraperitoneally injected with LPS of a sublethal dose (2.5 mg/kg). At indicated time points, blood was collected before the mouse was sacrificed. The serum concentration of inflammatory cytokines was measured by a multiplex Luminex assay; the data was obtained from two independent experiments per time point and represent mean ± SEM (n = 11 mice per group per time point). *p < 0.05 for WT to ficolin KO comparisons (unpaired two-tailed Student’s t test). ND = not determined.
Groups of WT and ficolin KO mice given a sublethal dose of 2.5 mg/kg of LPS i.p. were bled and sacrificed at 0 h, 1 h, 6 h, 12 h or 24 h after LPS challenge (Figs 5,6). Most of the cytokines peaked at 6 h and about half of the cytokines returned to basal level at 24 h. TNF and IL-10 peaked at 1 h whereas IL-1α peaked at 12 h (Fig. 5). Three of the six chemokines peaked early at1h, whereas the other three peaked at 6 h (Fig. 6). The most increased cytokines after LPS challenge included TNF, IL-6, IL-12(p40), G-CSF and the chemokines. Overall, levels of the 22 cytokines and chemokines were similar in WT and ficolin KO mice challenged with a sublethal LPS dose and only levels of a few cytokines were significantly lower in KO mice; TNF at 12 h, IL-3 at 24 h, IL-12(p70) at 12 h and the anti-inflammatory IL-13 at 12 h.
Figure 6.

Serum chemokine levels in WT and ficolin KO mice after LPS challenge with a sublethal dose. Groups of WT and ficolin KO mice were intraperitoneally injected with LPS of a sublethal dose (2.5 mg/kg). At indicated time points, blood was collected before the mouse was sacrificed. The serum concentration of chemokines was measured by a multiplex Luminex assay; the data was obtained from two independent experiments per time point and represent mean ± SEM (n = 11 mice per group per time point).
In another set of experiments, groups of WT and ficolin KO mice were given a lethal dose of 10 mg/kg of LPS i.p. and sacrificed at 1 h or 6 h to assess the serum cytokine and chemokine levels (Figs 7, 8). The general cytokine profiles resembled those of mice given a sublethal dose; most of the cytokines peaked at 6 h, whereas TNF and IL-10 peaked at 1 h, and the most increased cytokines were TNF-α, IL-6, IL-12(p40), G-CSF and the chemokines. In accordance to mice given the sublethal dose, the chemokines that peaked after 6 hours were CCL2, CCL5 and CCL11. However, regarding CCL3 and CCL4, the levels clearly declined from 1 h to 6 h in both WT and ficolin KO mice given the sublethal dose (Fig. 6), whereas the levels increased in WT mice and remained the same in KO mice given a lethal dose (Fig. 8). Except for a statistically significant higher level of G-CSF and CCL3 in ficolin KO mice at 1 h, there was no difference in the general pattern of cytokine and chemokine levels between WT and ficolin KO mice given a lethal dose of LPS.
Figure 7.
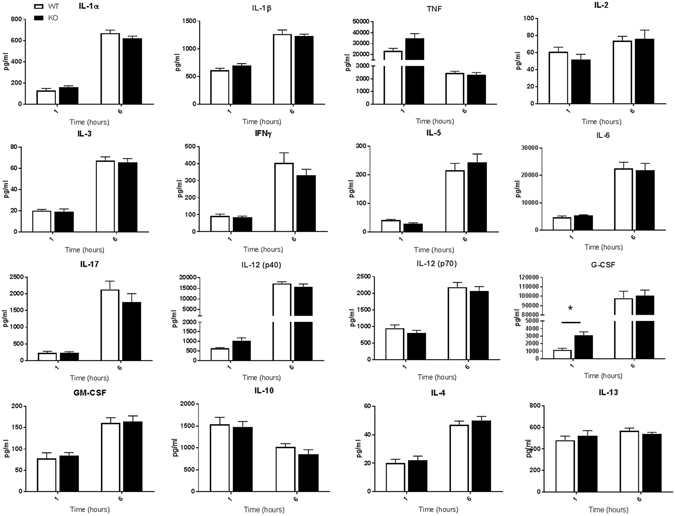
Serum cytokine levels in WT and ficolin KO mice after LPS challenge with a lethal dose. Groups of WT and ficolin KO mice were intraperitoneally injected with LPS of a lethal dose (10 mg/kg). At 1 h or 6 h after challenge, blood was collected before the mouse was sacrificed. The serum concentration of inflammatory cytokines was measured by a multiplex Luminex assay; the data was obtained from two independent experiments per time point and represent mean ± SEM (n = 11 mice per group per time point). *p < 0.05 for WT to ficolin KO comparisons (unpaired two-tailed Student’s t test).
Altogether, these results showed no overall difference between WT and ficolin KO mice in the inflammatory cytokine response after LPS challenge, regardless of the dose given and the time point investigated.
Ficolin deficiency does not alter the spleen transcriptome profile
Next, we performed microarray analysis to gain insight to the genome-wide transcriptional changes accompanying ficolin deficiency in spleens from naive or LPS challenged (10 mg/kg for 6 h) mice. Using fold-change > 1.5 and p < 0.05 as criteria for significance (Fig. 9A), we identified 24 differentially expressed genes (DEGs) between ficolin KO and WT mice in the naive state (Supplementary Table S1) and 59 DEGs between ficolin KO mice and WT mice in the LPS challenged state (Supplementary Table S2). If setting the significance border to q < 0.05 only Fcna (ficolin-A) would be considered as differentially expressed between KO and WT groups. In general, most of the DEGs did not appear to be related to LPS-induced inflammation. Of note is, however, that Reg2 (regenerating islet-derived 2) was upregulated in ficolin KO mice (fold-change 7.0) in the naive state, whereas the same gene was downregulated in ficolin KO mice (fold-change 0.1) in the LPS challenged state. We also compared the number of changed genes in ficolin KO mice after LPS challenge to the number of changed genes in WT mice after LPS challenge (Fig. 9B). Using fold-change > 2.0 and q < 0.05 as criteria for significance, we identified 1470 genes that were altered in both WT and ficolin KO mice after LPS challenge. In addition, 434 genes were changed only in WT mice, whereas other 335 genes were changed only in ficolin KO mice. Among the mostly LPS-responsive genes common for ficolin KO and WT mice were chemokines, cytokines and other pro-inflammatory mediators, including Ccl2, Ccl3, Ifng, Il6, Mmp13 (matrix metallopeptidase 13), Irg1 (Immune Responsive Gene 1), Saa3 (serum amyloid A 3) and Ptgs2 (prostaglandin-endoperoxide synthase 2) (Fig. 9C).
Figure 9.
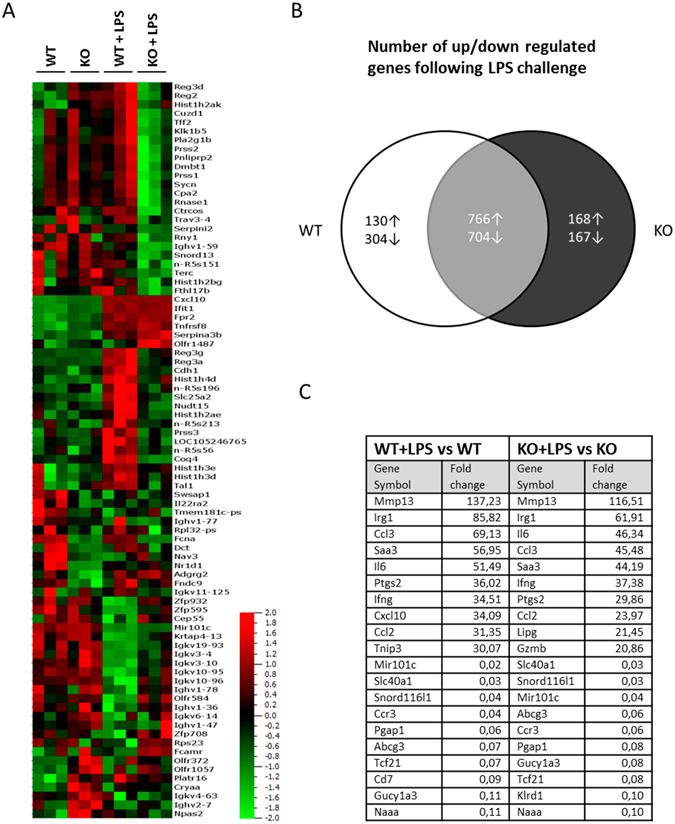
Microarray analysis of differentially expressed genes in WT and ficolin KO mice after LPS challenge. Groups of WT and ficolin KO mice were untreated or intraperitoneally injected with LPS of a lethal dose (10 mg/kg) and sacrificed after 6 h (n = 3 mice per group). Total RNA extracted from spleens was hybridized to the Affymetrix GeneChip Mouse Genome 2.0 ST arrays chip. (A) Heatmap showing all experimental samples and 85 genes found as differentially expressed between WT and KO mice in either naive or LPS-challenged state (cut-off: fold-change > 1.5 and p < 0.05). Samples are ordered by the four experimental groups and variables order by hierarchical clustering. (B) Venn diagram showing the number of differentially expressed genes induced by LPS challenge (cut-off: fold-change > 2.0 and q < 0.05). The white area contains the number of genes that were only affected in WT mice upon LPS challenge; the black area contains the number of genes that were only affected in ficolin KO mice upon LPS challenge; the grey area contains the number of overlapping genes between WT and ficolin KO mice that were affected upon LPS challenge. (C) Table showing the top 20 most changed genes (10 up and 10 downregulated) after LPS challenge in WT and ficolin KO mice (cut-off: fold-change > 2.0 and q < 0.05).
Overall, the results show that many genes change in response to LPS in both ficolin KO and WT mice; however, lack of ficolins does not change the transcriptional program in spleens, neither in the naive state nor after LPS challenge. These gene expression data therefore substantiate that ficolin deficiency does not alter the host response to LPS-induced inflammation in mice.
Discussion
In the present study we show for the first time that ficolins do not contribute to the LPS-induced inflammatory response in vivo in mice. The data are consistent using a broad panel of readouts; microarrays and selected qPCRs in tissues in addition to a number of single inflammatory mediators measured in serum. Convincingly, there were no overall differences between ficolin KO mice and WT mice.
Few studies involving ficolin KO mice are found in the literature to date. Endo et al. found that survival rates of ficolin-A KO, ficolin-B KO and double KO mice were significantly reduced compared to WT mice after intranasal infection with a Streptococcus pneumoniae strain13. Reconstitution of ficolin-A improved the survival rate of ficolin-A KO mice, but not of double KO mice, revealing the requirement of both ficolin-A and ficolin-B in the defense against S. pneumoniae. Another study also reported a reduced survival rate in ficolin-A KO mice using the same S. pneumoniae strain31. Reduced survival rates of ficolin-A KO mice compared to WT mice after infection with influenza A virus30 and Mycobacterium tuberculosis32 have also been reported. In addition, we showed that ficolin KO mice showed decreased fungal clearance in the early phase of A. fumigatus infection33. A common feature of these studies is that they employ lung infection models.
Besides these infection models, ficolin-B KO mice have been investigated in a model of Type1 diabetes related kidney desease34, ficolin-A KO mice have been investigated in a model of collagen antibody-induced arthritis35 and ficolin-A KO as well as ficolin-B KO mice have been subjected to intravenous injection of mitochondria36. There was no difference between ficolin KO mice and WT mice in any of these experimental models. In the present study we found that although serum ficolin-A recognize LPS in vitro, ficolin KO mice did not show an altered response to four different doses of LPS in vivo compared to WT mice. Hence our model of endotoxemia is featured on the list of non-infectious mouse models in which ficolins seem redundant.
Mannose-binding lectin (MBL) is the most widely studied family member of the lectin pathway complement initiators. In 2003 our research group published that MBL gene polymorphisms, giving rise to low levels of MBL, were associated with the development of sepsis and increased risk of fatal outcome from the disease37. Mice express two forms of the molecule: MBL-A and MBL-C. The MBL-A KO mouse has been examined in the cecal ligation and puncture (CLP) model of acute septic peritonitis and interestingly it was revealed that MBL-A KO mice showed increased survival compared to WT mice and to C3 KO mice38. While the CLP model induces a polymicrobial sepsis that mimics the later phase of human sepsis, the LPS model, which is used in the present study, induces systemic inflammation that mimics many of the initial clinical features of sepsis, but without bacteremia26. It would be interesting to test whether ficolin KO mice have a survival advantage in the CLP model like the MBL-A KO mice. All things considered, it is conceivable that an effect of the ficolins require the presence of a (whole) microbe. Recent observations revealed that consumption of ficolin-2 in patients with necrotizing soft tissue infection was associated with increased risk of fatal outcome39. However, necrotizing soft tissue infection is predominantly caused by Gram-positive polymicrobial pathogens such as Group A Streptococcus. Nonetheless, it may underscore the importance of the ficolins in clinical situations involving polymicrobial infections.
An early study found that serum MBL-A levels in mice were increased approximately 2-fold, peaking 32 h after the mice received an LPS-injection, while the MBL-C levels did not increase40. A later study found that serum MBL-A and MBL-C levels were reduced for 1 week after CLP, while the mRNA levels where unchanged, indicating consumption rather than decreased protein expression41. Another study found that ficolin-B mRNA expression was higher in neutrophils isolated from mice subjected to CLP compared to naive mice42. In the present study, we did not detect a change in gene expression of ficolin-A or ficolin-B after LPS challenge. In humans, endobronchial administration of LPS was associated with increased levels of alveolar ficolin-3 and MBL, and intravenous administration of LPS was associated with increased levels of plasma ficolin-143. No change in gene expression of ficolin-1 or ficolin-3 was observed. Interestingly, homozygosity for minor alleles resulting in increased ficolin-1 levels was associated with fatal outcome in patients with systemic inflammation/sepsis44. These studies suggest that, at least in humans, the ficolins play a role in the local and systemic host defense in response to LPS and sepsis. It is important to note that although mouse models are regarded as acceptable models of sepsis in humans, there are multiple factors that differ between the two species, including differences in the ficolin molecules and the host response to LPS.
Prior research has demonstrated the ability of ficolins to modulate the cytokine response in vitro and in vivo. In vitro, one study showed that ficolin-2 promoted production of IFN-γ, IL-17A, TNF and IL-6 by murine macrophages32, whereas another study showed that ficolin-A (or MBL-A) suppressed the LPS-mediated IL-6 and TNF production by murine mast cells45. In vivo, we found that mRNA and proteins levels IL-1β, IL-6 and TNF were reduced by approximately 30% in lungs of ficolin KO mice compared to WT mice after A. fumigatus infection33, whereas others found that mRNA levels of TNF-α, IFN-γ and CLCX2 (MIP-2) was increased in lungs from ficolin-A KO mice compared to WT mice after S. pneumonia infection31. In another study, TNF and IL-17A serum levels were measured in ficolin-A KO mice and WT mice after intravenous LPS challenge: TNF levels were decreased in the KO mice at 3 h and 6 h after LPS challenge (but not at 10 h) and IL-17A levels were decreased in the KO mice at 3 h (but not at 6 h) after LPS challenge32. In the present study there were only a few significantly changed cytokines in ficolin KO mice compared to WT mice, including decreased serum TNF at 12 h after sublethal LPS challenge. However, since none of these cytokines were significantly changed in more than one of the three analyses employed (qPCR, Luminex and microarray), we conclude that there was no overall difference in the cytokine response between ficolin KO mice and WT mice after challenge with a sublethal or a lethal dose of LPS. Collectively, these diverse results from different studies indicate that the ability of the ficolins to modulate cytokine responses is dependent on the underlying disease.
In the present study, whole genome transcriptome analysis confirmed that ficolins were not implicated in LPS-induced inflammation in the animals, and perhaps equally important that there was no significant difference between naive ficolin KO mice and WT mice in spleen expression profiles except, not surprisingly, expression of ficolin-A. Hence, ficolin deficiency per se does not affect transcription in naive mice. Furthermore, the possibility of flanking-allele effects in ficolin KO mice can be ruled out. To our knowledge, this is the first study to provide whole genome transcription profiles of ficolin KO mice.
Despite a growing number of reports on ficolin properties in various in vitro and in vivo model systems, a clear understanding of the actual biological functions of these conserved and phylogenetically ancient molecules remains elusive. In conclusion this study provided important new information about the ficolins. We examined the inflammatory response to LPS in ficolin KO mice in detail; we assessed the cytokine profiles and performed whole genome transcriptome profiling. The results unequivocally documented that ficolins are not involved in the LPS-induced inflammatory response in mice.
Methods
Reagents
LPS (E. coli 0111:B4, product no. L 2630 from Sigma-Aldrich) was suspended at a concentration of 5 mg/ml in sterile PBS and stored in aliquots at −20 °C. LPS was further diluted to the working concentration in sterile PBS on the day of use. Maxisorp polystyrene microtiter plates were purchased from NUNC. PBS-buffer (10 mM Na2HPO4, 1.47 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, pH 7.4) and Barbital- buffer (4 mM C8H11N2NaO3, 145 mM NaCl, 2.6 mM CaCl2, 2.1 mM MgCl2, pH 7.4) and sulphuric acid were all acquired from the hospital pharmacy (RegionH Apoteket, Rigshospitalet, Copenhagen, Denmark). Tween-20 was purchased from Merck. Bovine serum albumin (BSA) (A3803) was purchased from Sigma-Aldrich and acetylated (AcBSA) as described previously46. Horseradish-peroxidase (HRP) conjugated rabbit-anti-rat IgG was from Dako, Streptavidin-HRP was from GE Healthcare and TMB ONE was from Kem-En-Tec Diagnostics.
ELISA binding assays
For the ELISA binding assay microtiter plates were coated overnight with 100 µl LPS in1M NaCl, AcBSA (positive control47) or BSA (negative control), all 5 µg/ml in PBS. Barbital-Tween buffer was used for all subsequent washing and dilutions. Serial dilutions of mouse serum (WT or KO) were added to the wells and plates were incubated for 2 h at 37 °C. After washing, binding of ficolin-A to the coat was detected with a biotinylated in-house produced rat anti-mouse ficolin-A (1 µg/ml, overnight at 4 °C) followed by washing and Streptavidin-HRP (1:2000, 1 h shaking at room temperature). For the ELISA inhibition assay, mouse serum (WT or KO) was diluted 1:50 in barb-T buffer and pre-incubated with serial dilutions of LPS for 2 h at 37 °C before adding the samples to plates coated with AcBSA or BSA as above. Plates were incubated for 2 h at 37 °C, washed, and binding of ficolin-A to the coat was detected with in-house produced rat anti-mouse ficolin-A (1 µg/ml, overnight at 4 °C) followed by washing and rabbit-anti-rat HRP (1:2000, 1 h shaking at room temperature). All plates were washed and developed for 10–15 min with 100 µl/well TMB-one substrate and the reaction was stopped with 100 µl/well 0.2 M sulphuric acid. The optical density was measured at 450 nm.
Mice
Mice double deficient of ficolin-A and ficolin-B were generated as previously described13, 33. Heterozygous mice were crossed and the resulting offspring were used for homozygous breeding of WT and double KO mice; mice used in this study belong to the F4 generations. Female mice were used at 10–12 weeks of age. The mice were housed in pathogen-free, temperature-controlled and air-conditioned facilities with a 12 h light/dark cycle. Breeding as well as all animal experiments were conducted at the Department of Experimental Medicine, University of Copenhagen, and Rigshospitalet University Hospital, Copenhagen, Denmark. All animal experiments were approved by the Danish Animal Experiments Inspectorate (permission No. 2016-15-0201-00896) and conducted according to the national Animal Experimentation Act (LBK No. 475 from May 15, 2014). The review board at the Faculty of Health and Medical Sciences, University of Copenhagen, approved this study (P16-247).
LPS challenge
Weight-matched mice (20 ± 2 g) received 100 µl i.p. of the indicated doses of LPS. Control mice in the survival study received 100 µl i.p. of PBS. For the survival studies, the mice were assessed every 2-8 hours (based on severity of illness), weighted each morning, and survival/clinical score was recorded twice a day (8 a.m. and 8 p.m.) for 7days. The severity of the clinical appearance was scored as follows: 0, not affected; 1, slightly affected (mild piloerection and slightly hunched posture, normal activity); 2, affected (hunched posture, mild piloerection, less active); 3, severely affected (very hunched posture and severe piloerection, inactive). Animals were euthanized if they reached a score of 3. For the collection of serum and organs for further analysis, the mice were challenged with indicated doses of LPS and sacrificed by cervical dislocation at indicated time points.
Quantitative PCR
Mice were challenged with a lethal dose (10 mg/kg) of LPS and sacrificed after 6 h. In other experiments mice were challenged with a sublethal dose (2.5 mg/kg) and sacrificed after 6 h or 24 h. Naive mice served as controls. Organs were removed and stored in RNAlater (Applied Biosystems). Tissue samples were homogenized with a mechanical homogenizer (TissueRuptor, Qiagen) and RNA was extracted using the Maxwell 16 tissue LEV Total RNA Purification Kit (Promega) following the manufacturer’s recommendations. Total RNA was quantified using a Qubit Fluorometer (Invitrogen) and 1 µg of total RNA was reverse transcribed with the MuLV Reverse transcriptase (Applied Biosystems #N808-0018). Cytokine gene expression in spleens was assessed by quantitative real-time PCR with TaqMan probes specific for mouse TBP (mM00446971_m1), IL-1β (Mm0434228_m1), TNF(Mm00443258_m1), IL-6 (Mm00446190_m1) and IL-10 (Mm00439614_m1), obtained from Applied Biosystems. Quantitative Real-time PCR was performed in triplicate loading 50 ng (IL-1 and TNF) or 100 ng (IL-6 and IL-10) of cDNA per reaction and carried out on the Stratagene Mx3005 P Real-Time PCR system (Agilent Technologies). Reaction conditions included incubation at 50 °C for 2 min. and 95 °C for 10 min. followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. PCR runs included no-template controls and no-reverse transcriptase controls. The target genes were normalized to the housekeeping gene, TBP, and results were expressed as fold changes from naive WT mice (there was no difference in gene expression between naive WT and KO mice, data not shown) using the delta-delta Ct method. Ficolin-A gene expression in liver and spleen and ficolin-B gene expression in bone marrow was assessed under the same conditions as above with TaqMan probes specific for mouse TBP (mM00446971_m1), ficolin-A (Mm00484287_m1) and ficolin-B (Mm01332438_m1) obtained from Applied Biosystems.
Luminex assay
After the administration of a lethal (10 mg/kg) or sublethal (2.5 mg/kg) dose of LPS, serum was obtained from submandibular blood collection before the animal was sacrificed. Blood was also collected from naive mice. The serum was aliquoted and kept at −80 °C until further analysis. In experiments in which a lethal dose of LPS was given, blood was collected at 1 h or 6 h after LPS challenge in separate experiments. In experiments in which a sublethal dose of LPS was given, blood was collected at 1 h, 6 h, 12 h or 24 h after LPS challenge. Blood was also collected from naive mice (0 h). All serum samples were assayed in duplicate with a 23-plex mouse cytokine kit and read using the Bio-Plex 200 system and the Bio-Plex Manager software version 6.0 according to manufacturer’s protocol (Bio-Rad Laboratories). The profile of the cytokines analyzed was IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12 p40, IL-12 p70, IL-13, IL-17, eotaxin (CCL11), G-CSF, GM-CSF, IFN-γ, KC, MCP-1, MIP-1α, MIP-1β, RANTES and TNF. IL-9 was excluded from the analysis because it was undetectable in all samples. On one plate, containing the 12 h and 24 h samples, IL-2 and IL-4 was also undetectable, for which reason these were excluded.
Microarray analysis
RNA from spleens of naive mice and mice challenged with LPS was isolated as described above for qPCR. Subsequently, RNA was reverse transcribed and used for cRNA synthesis, labelling and hybridization with GeneChip Mouse Genome 2.0 ST arrays (Affymetrix) according to the manufacturer standard protocol available at www.affymetrix.com. The arrays were washed and stained with phycoerytrin conjugated streptavidin using the Affymetrix Fluidics Station 450, and the arrays were scanned in the Affymetrix GeneArray 3000 7 G scanner to generate fluorescent images. Cell intensity files (.CEL files) were generated in the GeneChip Command Console Software (AGCC; Affymetrix). The microarray data are deposited to Gene Expression Omnibus (accession number GSE95125).
Bioinformatics analysis was performed using Qlucore Omics Explorer 3.2. Raw intensity.CEL files were preprocessed by quantile normalization, and gene summaries were extracted via robust multi-array average (RMA)48. By comparison of groups with LPS with groups without LPS (i.e. KO + LPS versus KO-LPS and WT + LPS versus WT-LPS), q < 0.05 was considered as statistically significant. By comparisons of groups KO versus WT (i.e. KO-LPS vs. WT-LPS and KO + LPS vs. WT + LPS), differentially expressed genes passing p < 0.05 were considered as statistically significant. If setting the significance border to q < 0.05 only Fcna would be considered as differentially expressed between KO and WT groups. Hierarchical cluster analysis was performed and visualized using the Qlucore Omics Explorer™ software. All hierarchical clusters are build using average linkage and heat map was generated based on mean m = 0, variance 1 normalization.
Statistics
All data, except microarray data, was analyzed using GraphPad Prism Software, version 6. Kaplan–Meier survival curves were compared using the log-rank test. Comparisons between groups of WT and KO mice were calculated using the unpaired Student’s t-test and p < 0.05 was considered statistically significant. Comparisons of serum cytokine levels were subjected to post hoc Bonferroni correction for multiple testing (22 comparisons).
Electronic supplementary material
Acknowledgements
We greatly appreciate the technical assistance provided by Mr. Jesper Andresen and the scientific advice provided by Dr. Elisabeth Cramer and Dr. Mikkel-Ole Skjødt (Copenhagen). This study was supported by grants from the Danish Research Council of Independent Research, The Novo Nordisk Research Foundation, The Svend Andersen Research Foundation, The research Foundation of Capital Region of Denmark and The University of Copenhagen (to P.G.) and from The Norwegian Council on Cardiovascular Disease, The Odd Fellow Foundation, and the European Community’s Seventh Framework Programme under grant agreement no. 602699 (DIREKT) (to T.E.M.).
Author Contributions
N.G., J.B.C. and P.G. designed the experiments, N.G., O.Ø., C.S. and T.E.M. performed and analyzed the experiments, J.B.C. and P.G. supervised the study, N.G. wrote the manuscript, P.G. revised the manuscript, all authors reviewed and approved the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-04121-w
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Matsushita M, Endo Y, Fujita T. Cutting edge: complement-activating complex of ficolin and mannose-binding lectin-associated serine protease. J. Immunol. 2000;164:2281–4. doi: 10.4049/jimmunol.164.5.2281. [DOI] [PubMed] [Google Scholar]
- 2.Matsushita M, et al. Activation of the lectin complement pathway by H-ficolin (Hakata antigen) J. Immunol. 2002;168:3502–6. doi: 10.4049/jimmunol.168.7.3502. [DOI] [PubMed] [Google Scholar]
- 3.Liu Y, et al. Human M-ficolin is a secretory protein that activates the lectin complement pathway. J. Immunol. 2005;175:3150–6. doi: 10.4049/jimmunol.175.5.3150. [DOI] [PubMed] [Google Scholar]
- 4.Endo Y, Sato Y, Matsushita M, Fujita T. Cloning and characterization of the human lectin P35 gene and its related gene. Genomics. 1996;36:515–21. doi: 10.1006/geno.1996.0497. [DOI] [PubMed] [Google Scholar]
- 5.Lu J, Tay PN, Kon OL, Reid KB. Human ficolin: cDNA cloning, demonstration of peripheral blood leucocytes as the major site of synthesis and assignment of the gene to chromosome 9. Biochem. J. 1996;313(Pt 2):473–8. doi: 10.1042/bj3130473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsushita M, et al. A novel human serum lectin with collagen- and fibrinogen-like domains that functions as an opsonin. J. Biol. Chem. 1996;271:2448–54. doi: 10.1074/jbc.271.5.2448. [DOI] [PubMed] [Google Scholar]
- 7.Sugimoto R, et al. Cloning and characterization of the Hakata antigen, a member of the ficolin/opsonin p35 lectin family. J. Biol. Chem. 1998;273:20721–7. doi: 10.1074/jbc.273.33.20721. [DOI] [PubMed] [Google Scholar]
- 8.Honoré C, et al. The innate pattern recognition molecule Ficolin-1 is secreted by monocytes/macrophages and is circulating in human plasma. Mol. Immunol. 2008;45:2782–9. doi: 10.1016/j.molimm.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Munthe-Fog L, et al. The impact of FCN2 polymorphisms and haplotypes on the Ficolin-2 serum levels. Scand. J. Immunol. 2007;65:383–92. doi: 10.1111/j.1365-3083.2007.01915.x. [DOI] [PubMed] [Google Scholar]
- 10.Munthe-Fog L, et al. Characterization of a polymorphism in the coding sequence of FCN3 resulting in a Ficolin-3 (Hakata antigen) deficiency state. Mol. Immunol. 2008;45:2660–6. doi: 10.1016/j.molimm.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 11.Fujimori Y, et al. Molecular cloning and characterization of mouse ficolin-A. Biochem. Biophys. Res. Commun. 1998;244:796–800. doi: 10.1006/bbrc.1998.8344. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, et al. Ficolin A and ficolin B are expressed in distinct ontogenic patterns and cell types in the mouse. Mol. Immunol. 2005;42:1265–73. doi: 10.1016/j.molimm.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 13.Endo Y, et al. Mice deficient in ficolin, a lectin complement pathway recognition molecule, are susceptible to Streptococcus pneumoniae infection. J. Immunol. 2012;189:5860–6. doi: 10.4049/jimmunol.1200836. [DOI] [PubMed] [Google Scholar]
- 14.Endo Y, et al. Identification of the mouse H-ficolin gene as a pseudogene and orthology between mouse ficolins A/B and human L-/M-ficolins. Genomics. 2004;84:737–44. doi: 10.1016/j.ygeno.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 15.Kjaer TR, et al. M-ficolin binds selectively to the capsular polysaccharides of Streptococcus pneumoniae serotypes 19B and 19C and of a Streptococcus mitis strain. Infect. Immun. 2013;81:452–9. doi: 10.1128/IAI.01148-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Favier A-L, et al. Enhancement of Ebola virus infection via ficolin-1 interaction with the mucin domain of GP glycoprotein. J. Virol. 2016 doi: 10.1128/JVI.00232-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krarup A, Sørensen UBS, Matsushita M, Jensenius JC, Thiel S. Effect of capsulation of opportunistic pathogenic bacteria on binding of the pattern recognition molecules mannan-binding lectin, L-ficolin, and H-ficolin. Infect. Immun. 2005;73:1052–60. doi: 10.1128/IAI.73.2.1052-1060.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cestari I, et al. Role of early lectin pathway activation in the complement-mediated killing of Trypanosoma cruzi. Mol. Immunol. 2009;47:426–37. doi: 10.1016/j.molimm.2009.08.030. [DOI] [PubMed] [Google Scholar]
- 19.Verma A, et al. Human H-ficolin inhibits replication of seasonal and pandemic influenza A viruses. J. Immunol. 2012;189:2478–87. doi: 10.4049/jimmunol.1103786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jensen ML, et al. Ficolin-2 recognizes DNA and participates in the clearance of dying host cells. Mol. Immunol. 2007;44:856–65. doi: 10.1016/j.molimm.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Honoré C, et al. The innate immune component ficolin 3 (Hakata antigen) mediates the clearance of late apoptotic cells. Arthritis Rheum. 2007;56:1598–607. doi: 10.1002/art.22564. [DOI] [PubMed] [Google Scholar]
- 22.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 2010;11:785–97. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Markiewski MM, DeAngelis RA, Lambris JD. Complexity of complement activation in sepsis. J. Cell. Mol. Med. 2008;12:2245–54. doi: 10.1111/j.1582-4934.2008.00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ward PA. The dark side of C5a in sepsis. Nat. Rev. Immunol. 2004;4:133–42. doi: 10.1038/nri1269. [DOI] [PubMed] [Google Scholar]
- 25.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 26.Doi K, Leelahavanichkul A, Yuen PST, Star RA. Animal models of sepsis and sepsis-induced kidney injury. J. Clin. Invest. 2009;119:2868–78. doi: 10.1172/JCI39421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boldt ABW, et al. Susceptibility to leprosy is associated with M-ficolin polymorphisms. J. Clin. Immunol. 2013;33:210–9. doi: 10.1007/s10875-012-9770-4. [DOI] [PubMed] [Google Scholar]
- 28.Luz PR, et al. Association of L-ficolin levels and FCN2 genotypes with chronic Chagas disease. PLoS One. 2013;8:e60237. doi: 10.1371/journal.pone.0060237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munthe-Fog L, et al. Immunodeficiency associated with FCN3 mutation and ficolin-3 deficiency. N. Engl. J. Med. 2009;360:2637–44. doi: 10.1056/NEJMoa0900381. [DOI] [PubMed] [Google Scholar]
- 30.Pan Q, et al. L-ficolin binds to the glycoproteins hemagglutinin and neuraminidase and inhibits influenza A virus infection both in vitro and in vivo. J. Innate Immun. 2012;4:312–24. doi: 10.1159/000335670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ali YM, et al. The lectin pathway of complement activation is a critical component of the innate immune response to pneumococcal infection. PLoS Pathog. 2012;8:e1002793. doi: 10.1371/journal.ppat.1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo F, et al. Ficolin-2 defends against virulent Mycobacteria tuberculosis infection in vivo, and its insufficiency is associated with infection in humans. PLoS One. 2013;8:e73859. doi: 10.1371/journal.pone.0073859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Genster N, et al. Ficolins Promote Fungal Clearance in vivo and Modulate the Inflammatory Cytokine Response in Host Defense against Aspergillus fumigatus. J. Innate Immun. 2016;8:579–588. doi: 10.1159/000447714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holt CB, et al. Ficolin B in Diabetic Kidney Disease in a Mouse Model of Type 1 Diabetes. Mediators Inflamm. 2015;2015:653260. doi: 10.1155/2015/653260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Banda NK, et al. Mechanisms of Mannose-Binding Lectin-Associated Serine Proteases-1/3 Activation of the Alternative Pathway of Complement. Mol Immunol. Mol Immunol. 2011;49:281–289. doi: 10.1016/j.molimm.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brinkmann CR, et al. Mitochondria and the lectin pathway of complement. J. Biol. Chem. 2013;288:8016–8027. doi: 10.1074/jbc.M112.430249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garred P, et al. Association of Mannose-Binding Lectin Polymorphisms with Sepsis and Fatal Outcome, in Patients with Systemic Inflammatory Response Syndrome. J. Infect. Dis. 2003;188:1394–1403. doi: 10.1086/379044. [DOI] [PubMed] [Google Scholar]
- 38.Takahashi K, et al. Lack of mannose-binding lectin-A enhances survival in a mouse model of acute septic peritonitis. Microbes Infect. 2002;4:773–784. doi: 10.1016/S1286-4579(02)01597-6. [DOI] [PubMed] [Google Scholar]
- 39.Hansen MB, et al. The Lectin Complement Pathway in Patients with Necrotizing Soft Tissue Infection. J. Innate Immun. 2016;8:507–16. doi: 10.1159/000447327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu H, et al. Characterization and quantification of mouse mannan-binding lectins (MBL-A and MBL-C) and study of acute phase responses. Scand. J. Immunol. 2001;53:489–497. doi: 10.1046/j.1365-3083.2001.00908.x. [DOI] [PubMed] [Google Scholar]
- 41.Windbichler M, et al. Involvement of the lectin pathway of complement activation in antimicrobial immune defense during experimental septic peritonitis. Infect. Immun. 2004;72:5247–52. doi: 10.1128/IAI.72.9.5247-5252.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weber-Steffens D, et al. Immature mouse granulocytic myeloid cells are characterized by production of ficolin-B. Mol. Immunol. 2013;56:488–496. doi: 10.1016/j.molimm.2013.06.015. [DOI] [PubMed] [Google Scholar]
- 43.Plovsing, R. R. et al. Alveolar recruitment of ficolin-3 in response to acute pulmonary inflammation in humans. Immunobiology doi:10.1016/j.imbio.2015.11.015 (2016). [DOI] [PubMed]
- 44.Munthe-Fog L, et al. Variation in FCN1 affects biosynthesis of ficolin-1 and is associated with outcome of systemic inflammation. Genes Immun. 2012;13:515–522. doi: 10.1038/gene.2012.27. [DOI] [PubMed] [Google Scholar]
- 45.Ma YJ, et al. Mouse mannose-binding lectin-A and ficolin-A inhibit lipopolysaccharide-mediated pro-inflammatory responses on mast cells. BMB Rep. 2013;46:376–81. doi: 10.5483/BMBRep.2013.46.7.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hein E, et al. Functional analysis of Ficolin-3 mediated complement activation. PLoS One. 2010;5:e15443. doi: 10.1371/journal.pone.0015443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hummelshøj T, et al. The interaction pattern of murine serum ficolin-A with microorganisms. PLoS One. 2012;7:e38196. doi: 10.1371/journal.pone.0038196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–93. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.