

Abstract
Myelin basic protein (MBP) is a major candidate autoantigen in multiple sclerosis (MS). Its immunodominant epitope, MBP 85–99, forms a complex with human leukocyte antigen (HLA)-DR2 with which multiple sclerosis is genetically associated. Copolymer 1 (Copaxone), a random amino acid copolymer [poly (Y,E,A,K)n] as well as two modified synthetic copolymers [poly (F,Y,A,K)n and poly (V,W,A,K)n] also form complexes with HLA-DR2 (DRA/DRB1*1501) and compete with MBP 85–99 for binding. Moreover, two high-affinity synthetic peptide 15-mers that could inhibit binding even more effectively were previously designed. Here, we show that further-modified peptide 15-mers inhibited even more strongly (in order J5 > J3 > J2) both the binding of MBP 85–99 to HLA-DR2 and IL-2 production by two MBP 85–99-specific HLA-DR2-restricted T cells. J5, J3, and J2 also suppressed both MBP 85–99-induced experimental autoimmune encephalomyelitis (EAE) in humanized mice and proteolipid protein 139–151-induced EAE in SJL/J mice. Moreover, none of these previously uncharacterized peptide inhibitors crossreacted with MBP 85–99- or proteolipid protein 139–151-specific T cells. In both cases, spleen and lymph node cultures stimulated with these peptides produced large amounts of Th2 cytokines (IL-4 and IL-10), and adoptive transfer of established T cell lines suppressed disease induction. These peptide 15-mers provide specific, nonrandom sequences that appear to be at least as effective as random copolymers in suppressing EAE in several models.
Keywords: Cop1, HLA-DR, multiple sclerosis, T cells
Multiple sclerosis (MS), an autoimmune inflammatory disease of the CNS, is associated with the HLA-DR2 haplotype (DRA*0101, DRB1*1501, DQA1*0102, DQB1*0602) (1–3). Myelin basic protein (MBP) is one of the major candidate autoantigens in the pathogenesis of MS. Particularly, T cell reactivity to the immunodominant MBP 85–99 epitope is found in subjects carrying human leukocyte antigen (HLA)-DR2, a genetic marker for susceptibility to MS. HLA-DR2-restricted MBP-specific T cells are clonally expanded and activated in MS patients (4–9). Furthermore, HLA-DR2/MBP 85–99 complexes have been detected in the CNS plaques of these patients (10). Critical residues for binding to HLA-DR2 and for T cell antigen receptor (TCR) recognition of the MBP 85–99 epitope have been defined (11, 12).
Several therapeutic approaches to MS have been attempted by using cytokines, copolymers, dimers of class II major histocompatibility complex-peptide complexes, peptide antigens that induce anergy, vaccination with TCR, and an altered peptide ligand (13–19). Many of these studies aimed to interfere with the MBP 85–99-specific T cell recognition and/or to deviate the T cell response from the Th1 to the Th2 phenotype. Copolymer 1 [Cop1, Copaxone, Glatiramer Acetate, poly(Y, E, A, K)n], the only approved drug known to reduce MBP-specific T cell responses, reduces the relapse rate by ≈30% in relapsing-remitting forms of MS (20–26). Cop1 binds to several human HLA-DR molecules, including HLA-DR2, and blocks modestly the presentation of MBP 85–99 (27, 28). The effectiveness of amino acid copolymers with in vitro and in vivo assays was greatly improved by modifications that enhanced their binding to HLA-DR2 (29, 30). We hypothesized that by similarly improving the binding of synthetic peptide mimetics (13, 29, 30), their effectiveness might also be greatly enhanced. Thus, T cell recognition of MBP 85–99 might be more effectively inhibited and/or additional immunosuppressive functions of the copolymers might be enhanced by the peptide mimetics. In this paper we report generation of three peptide inhibitors based on the binding motif of MBP 85–99 to HLA-DR2 with improved functions in inhibiting T cell responses and experimental autoimmune encephalomyelitis (EAE). These 15-mers (J2, J3, and J5) competed with MBP 85–99 for binding to HLA-DR2, inhibited IL-2 secretion by MBP 85–99-specific T cell clones and induced production of Th2 cytokines by splenocytes. Additionally they did not crossreact with MBP 85–99- or PLP 139–151-specific T cells. They also suppressed EAE in two different murine disease models in a manner equivalent to that achieved by using the modified random amino acid copolymers.
Materials and Methods
Generation of Peptide-Specific Short-Term T Cell Lines and Proliferative Responses to Peptides. Mice were immunized with proteolipid protein (PLP) 139–151 or peptide 15-mers (100 μg per mouse) emulsified in complete Freund's adjuvant (CFA) (Difco). Ten days later, single-cell suspensions were prepared from spleens and lymph nodes and they were stimulated with corresponding peptides at a concentration of 10 μg/ml in the presence of antigen-presenting cells. After two rounds of restimulation, viable lymphocytes were separated by Ficoll/Hypaque density gradient centrifugation and used to test their specificity at a dose range of 0–50 μg/ml. As a negative control, Nase 101–120 peptide, which also binds I-As, was used. To determine the proliferative response of transgenic (tg) T cells expressing the HLA-DR2-restricted/MBP 85–99-specific TCR (hVβ2+CD4+) and non-tg T cells (hVβ2–CD4+), splenocytes were fractionated from double-tg humanized mice that express both HLA-DR2 and a MBP 85–99-specific hVβ2 T cell receptor from an MS patient (30) by flow cytometry and seeded into a 96-well U-bottom microtiter plate in complete medium at 4 × 105 cells per well. Irradiated splenocytes of humanized mice, also expressing HLA-DR2, were used as antigen-presenting cells. Cultures were stimulated with 10 μg/ml of MBP 85–99 or 100 μg/ml peptides J2, J3, or J5). Proliferative responses were measured as described in refs. 17 and 29.
Effect of Peptide 15-mers on the Induction of EAE. Female SJL/J or humanized mice were immunized s.c., each with 50 μg of PLP 139–151 or 150 μg MBP 85–99 alone, or coimmunized with these antigens and 150 μg peptide 15-mers emulsified in CFA. Pertussis toxin, 150 ng (List Biological Laboratories, Campbell, CA), was given for humanized mice 8–12 weeks old i.p. on the day of immunization and on day 2 postimmunization and for SJL/J mice i.v. on day 2 only. The mice were monitored for appearance of EAE and scored from 0–5 as described in refs. 17, 29, and 30.
Adoptive Transfer of Peptide-Specific T Cell Lines. SJL/J mice were immunized with either 50 μg of PLP 139–151 peptide or 250 μg of peptides J2, J3, or J5. Cell lines were prepared and used as described in refs. 17 and 29.
Additional Materials or Methods. Additional materials or methods included mice (17, 29, 30), peptide synthesis (13), isolation of empty HLA-DR2 (12, 13), inhibition by peptide 15-mers of MBP 85–99 binding to HLA-DR2 (DRA/DRB1*1501) (13, 17), sorting of tg and endogenous (non-tg) T cells from humanized mice by flow cytometry (30), cytokine measurement by ELISA (30), and detection of PLP 139–151-reactive CD4+ T cells by using I-As/PLP 139–151 tetramers (31, 32).
Results
Design of Synthetic Peptide 15-mers. The design of the peptide 15-mers was based on the following criteria: (i) MBP 85–99 has V89 at P1 and F92 at P4. (ii) Y at the P1 of peptides or copolymers (as found in Copolymer 1) is too large for the P1 pocket. Such peptides (as used in ref. 13) are, therefore, poor binders to HLA-DR2. F at P1 of MBP 85–99 was less effective than V in a T cell stimulation assay (11, 17). (iii) A peptide with the large hydrophobic amino acid W substituted for F at P4 of MBP 85–99 was found to effectively stimulate a HLA-DR2-restricted T cell clone (11, 12). Moreover, in the binding motif of Cop1 to HLA-DR2, Y was eluted from the P4 pocket (28). Here, both the P1 and P4 residues of the previously described 15-mers nos. 100 and 101 (13) (J1 and J11 in Table 1) were therefore substituted with hydrophobic amino acids of different sizes V, F, or Y. (iv) K at P5, the major TCR contact residue in MBP 85–99, was retained in all peptides. (v) Further, the residues at P6 to P11 in MBP 85–99 were substituted by alanine, except that in some peptides, proline was placed at the penultimate position P10 because proline residues are resistant to exopeptidases in serum. For the purpose of in vivo EAE studies, P at P–2 (as found in MBP 85–99) and at P–5 were also included in some peptides for the same reason. These prolines did not change the binding affinity of peptides for HLA-DR2 (Table 1). (vi) The TCR contact site V88 of MBP 85–99 at P1 (12) was substituted by lysine; this modification was hypothesized to enhance binding to HLA-DR2 (13). All of these peptide analogues were designed to achieve a minimum possible homology with MBP 85–99, yet bind to HLA-DR2 with high affinity and retain K at P5.
Table 1. Percent inhibition of binding of MBP 85–99-bio at 0.3 μM by peptide competitor at 1 μM.

The parent compounds J1 and J11 (13) are in italics. Modifications are shown in bold.
Inhibition of Binding of MBP 85–99 to HLA-DR2 (DRA/DRB1*1501) by Synthetic Peptides. To examine whether the peptides competed with the immunodominant epitope MBP 85–99 for binding to HLA-DR2 molecules, competitive binding studies were performed. Several peptides inhibited binding of biotinylated MBP 85–99 to HLA-DR2 molecules very efficiently (Table 1). The most effective peptides were J3, J5, J8, J9, J13, J15, and J17. All of these had V or F at P1 and Y or F at P4. Strikingly, placing E at P1 as a control significantly decreased binding (J4 and J6). Notably, Cop1, which contains E as well as Y, had a lower binding affinity for HLA-DR2 and inhibited binding of biotinylated MBP 85–99 less efficiently than the peptides (Table 1). The data obtained for peptides J2-J10, all based on J1 (no. 100), were very similar to those for J12-J17 based on J11(no. 101). Further evaluation was focused on J1-J5. J3 and J5 showed maximum inhibition of MBP 85–99 binding to HLA-DR2, particularly at lower concentrations (Fig. 1A).
Fig. 1.

Peptides J1-J5 inhibit binding of MBP 85–99 to HLA-DR2 (DRA/DRB1*1501) and inhibit proliferation of MBP 85–99 T cells. (A) Competitive binding. Recombinant HLA-DR2 molecules were incubated with 0.13 μM biotinylated MBP 85–99 alone or in the presence of unlabeled MBP 85–99 or peptides J1-J5 (1–100 μM). The reaction mixtures were transferred to the plates coated with mAb LB3.1 that binds HLA-DR2 molecules and incubated for 55 min, followed by removal of the supernatant. Streptavidin-conjugated alkaline phosphatase and its substrate were added and incubated for 55 min. The absorbance was measured at 450 nm by using a microtiter plate reader. (B) Proliferation inhibition. Irradiated MGAR cells (HLA-DR2 homozygous) as antigen-presenting cells were coincubated with MBP 85–99 (12.5 μM) and various concentrations of the peptides J2, J3, or J5 or Cop1 for 2 h. MBP 85–99-specific T cell hybridoma 8073 were then added and the mixtures incubated for 24 h. Culture supernatants were used to test the proliferative response of IL-2-dependent CTLL, and the response was measured as cpm 16 h after pulsing with 3[H]thymidine.
Inhibition by Synthetic Peptide 15-mers of IL-2 Secretion by an HLA-DR2-Restricted, MBP 85–99-Specific T Cell Hybridoma. The fact that the peptides competed with MBP 85–99 in binding to HLA-DR2 provided a basis to test the hypothesis that they also inhibit MBP-specific T cells. The T cell hybridoma 8073 that had been transfected with the MBP 85–99-specific TCR from clone Ob1A.12, derived from a HLA-DR2+ patient with relapsing-remitting MS (33), was used. The parent peptide J1, J2, J3, and J5 completely inhibited MBP 85–99-induced IL-2 secretion (as measured by proliferation of the IL-2-responsive CTLL line) by hybridoma 8073 in a dose-dependant manner (Fig. 1B). This inhibition was significantly greater than the effect of either Cop 1 or J4 (Fig. 1B) (P ≤ 0.05). J5 was the best inhibitor, as well as binder, suggesting that the V at P1 and Y at P4 were critical for enhanced binding of these peptides to HLA-DR2. Inhibition by the peptides of IL-2 production by the 2E12 T cell line obtained from a double-tg humanized mouse (33) was comparable to that obtained with the 8073 T cell hybridoma (data not shown).
Synthetic Peptide 15-mers Are Immunogenic. To examine immunogenicity of the synthetic peptides, humanized mice (30) were immunized with 100 μg each of J2, J3, J5, and MBP 85–99. Spleen and lymph node cells were collected 10 days later and restimulated in vitro with the corresponding peptides to generate cell lines. As expected, MBP 85–99 elicited a strong proliferative response of the line that was generated by using this antigen (Fig. 2A Left). Likewise, in the mice immunized with J3 or J5 alone a lower response was induced suggesting, by analogy to responses to copolymers (30), that the non-tg, endogenous T cells (that represent ≈70% of the T cells in the humanized mice) might have responded to these peptides presented by HLA-DR2. However, a negligible response to J2 (that bound weakly) was seen. Immunogenicity of these peptides was further verified in SJL/J mice (in which I-As in the only expressed class II major histocompatibility complex protein) and compared with PLP 139–151, one of the most immunodominant and encephalitogenic epitopes of PLP in SJL/J mice (34, 35). SJL/J mice immunized with these peptides were able to elicit proliferative response after restimulation by peptide 15-mers J2, J3, and J5 (Fig. 2A Right). Thus, the peptides are able to bind to both HLA-DR2 and I-As and induce peptide-specific T cells.
Fig. 2.

Generation, cross-reactivity, and cytokine production of T cells were examined (A) Peptides J2, J3, and J5 are immunogenic. Humanized (Left) or SJL/J (Right) mice were immunized with MBP 85–99 or PLP 139–151, respectively, or peptides J2, J3, and J5 (100 μg in CFA). After 10 days, splenocytes were restimulated three times at 2-week intervals with the corresponding peptides at 10 μg/ml in the presence of irradiated antigen-presenting cells. Two weeks later, viable cells were stimulated with corresponding peptides for two days. Sixteen hours after pulsing with 3[H]thymidine, proliferative response was measured as cpm. (B) J2, J3, and J5 do not stimulate MBP 85–99- or PLP 139–151-specific T cells. (Left) MBP 85–99-specific tg and endogenous (non-tg) cells from humanized mice were sorted by flow cytometry and cultured with MBP 85–99 or peptides J2, J3, or J5 at 50 μg/ml. Their proliferative response was measured as cpm after pulsing with 3[H]thymidine. (Right) PLP 139–151-specific T cells were incubated with PLP 139–151 or peptides J2, J3, or J5, and the proliferative response was measured as cpm after pulsing with 3[H] thymidine. (C) Peptides J2, J3, and J5 induce T cells that produce Th2, but not Th1, cytokine responses. SJL/J mice were immunized with either PLP 139–151 or peptides J2, J3, or J5 as described in Materials and Methods. After 10 days, single-cell suspensions derived from lymph nodes and spleens were restimulated with the corresponding peptides in the presence of antigen presenting cells for 2 days. Culture supernatants were examined for secretion of IL-2, IL-4, IL-10, and IFN-γ by ELISA.
Peptide 15-mer-Reactive T Cell Lines Do Not Crossreact with Those of MBP 85–99. Based on previous work with copolymers (29), tg T cells (≈30% of the T cells) were assumed to be the main responders to MBP 85–99 in humanized mice with a smaller response of the endogenous T cells. Therefore, MBP 85–99-specific tg (hVβ2+CD4+) and endogenous, non-tg (hVβ2–CD4+) T cells were sorted by flow cytometry from spleen cells of double-tg humanized mice. Both sets were stimulated with 10 μg/ml MBP 85–99 or 100 μg/ml of J2, J3, or J5 in the presence of irradiated syngeneic splenocytes. MBP 85–99 efficiently stimulated the tg CD4+ T cells to an extent significantly higher than that obtained from endogenous CD4+ cells (Fig. 2B Left). By contrast, J2, J3, and J5 did not induce proliferation of either MBP 85–99-specific tg or endogenous CD4+ cells, confirming that these peptides do not crossreact with this human MBP 85–99-specific TCR or with the nonclonal endogenous TCR that respond to MBP 85–99 (Fig. 2B). The lack of response to J2, J3, or J5 was not due to cell death because in all of the treatment groups, viable cell numbers were comparable (data not shown).
Lack of cross-reactivity to these peptides was further verified by using a PLP 139–151-specific cell line. This line responded only to PLP 139–151 and neither to the control peptide NASE 101–120 nor to J2, J3, or J5 (Fig. 2B Right). Similarly, T cell lines derived from SJL/J mice immunized with J2, J3, or J5 responded only to the cognate peptide and not to PLP 139–151 (data not shown).
Peptide 15-mer-Specific T Cells Produce Th2 Cytokines Preferentially. The cytokine profiles of short-term lines stimulated with J2, J3, or J5 were compared with those of PLP 139–151-stimulated cultures. SJL/J mice were immunized with PLP 139–151, J2, J3, or J5 in CFA and, after 10 days, spleen and lymph node cells were stimulated with their cognate peptides in the presence of irradiated antigen-presenting cells for 2 days (see Materials and Methods). In cultures stimulated with PLP 139–151, IL-2 and IFN-γ were produced in significant amounts, whereas neither was produced in those stimulated with the peptide 15-mers (Fig. 2C). In contrast, IL-10 production was dominant in cultures stimulated with peptides J3 and J5 in amounts ≈3-fold higher than in those stimulated with PLP 139–151 or peptide J2. Secretion of IL-4 was comparable in all of the cultures regardless of peptide used.
J2, J3, and J5 Ameliorate EAE Induced by MBP 85–99 in Humanized Mice or PLP 139–151 in SJL/J Mice. The ability of J2, J3, and J5 to ameliorate the severity of EAE in vivo was tested in two models of EAE: (i) MBP 85–99-induced EAE in HLA-DR2 and MBP 85–99-specific TCR double-tg humanized mice or (ii) PLP 139–151-induced EAE in SJL/J mice. In the first model, the humanized mice, after immunization with MBP 85–99, all mice in the group developed clinical signs of EAE between days 6 and 9 (Fig. 3A). Although one mouse died, the remaining mice reached a mean maximum score of 2.5 and then entered a chronic phase of the disease with a mean of 2.0. On the other hand, in the mice coimmunized with the J2, J3, or J5 and MBP 85–99, the onset of disease was delayed by 2–5 days, and the disease severity was strikingly reduced during the acute phase of EAE. The maximum mean scores of J2-, J3-, and J5-treated mice were ≈1 (limp tail). Importantly, no mortalities occurred in any of the coimmunized groups. In a previous study, Cop1 reduced the signs of EAE but less effectively (30). In the second model, EAE was induced in SJL/J mice by immunizing with PLP 139–151. The first sign of EAE appeared at day 7 and reached a maximum mean score of 3 by day 16 (Fig. 3B). Three of the eight mice immunized with PLP 139–151 died. In parallel, groups of six mice were coimmunized with Cop 1 or with J2, J3, and J5 together with PLP 139–151. In the mice coimmunized with Cop 1, the severity of EAE was moderately suppressed (mean maximum score 2.5 at about day 22, followed by slow recovery) (see Fig. 5B). By contrast, mice coimmunized with J2, J3, or J5 developed only a mild disease (mean clinical score ≈1). No mortalities occurred in any of these groups. Thus, the severity of EAE in J2, J3, or J5 coimmunized groups was significantly lower than in mice immunized with PLP 139–151 or coimmunized with Cop 1.
Fig. 3.

Peptide 15-mers reduce severity of EAE. EAE was induced in humanized or SJL/J mice by immunizing with MBP 85–99 (150 μg) (A) or PLP 139–151 (50 μg) (B), respectively, with or without peptides J2, J3, or J5 (100 μg) or Cop 1 (150 μg) (see Materials and Methods). Appearance of clinical signs of EAE was monitored daily, and the disease severity was scored. This data represent the average of three experiments. Cop1 was not included in the MBP 85–99 experiment because of a limitation in the availability of humanized mice, but its effect is shown in figure 2 A of ref. 29. P values vs. MBP 85–99 on day 16 (n = 8 per group): J2; <0.001; J3, <0.01; J5, <0.008; vs. PLP 139–151 on day 22 (n = 7 per group): J2, <0.008; J3, <0.005; J5, <0.002; vs. Cop1: J2; <0.11; J3, 0.05; J5, <0.03. Each experiment was performed three times.
Fig. 5.

Peptide 15-mers suppress proliferation of PLP 139–151-specific CD4+ T cell responses. SJL/J mice were immunized with PLP 139–151 in CFA. After 10 days, lymphocytes from axillary and inguinal lymph nodes were cultured with PLP 139–151 alone or PLP 139–151 together with J2, J3, or J5 (Left) at 10 μg/ml. Four days later, viable lymphoblasts were incubated with I-As tetramers [PLP 139–151 or TMEV 70–86 (Right)] for 3 h, as described in ref. 32. The I-As/tetramer-stained cells were determined in the live CD4+ T cell subset after eliminating dead cells (7-AAD positive).
Suppression by Adoptive Transfer of Peptide-Specific T Cell Lines. SJL/J mice were immunized s.c. with either 50 μg of PLP 139–151 or 250 μg of J2, J3, or J5. On day 10, splenocytes were restimulated three times in vitro every other week with peptide-pulsed irradiated splenocytes from naïve mice. After the third round of stimulation, >92% of the cells present in all of the cultures were CD3+ T cells. Then, 5 × 106 T cells of the J2, J3, or J5 stimulated cultures were administered i.v. per mouse. EAE was induced the next day by using PLP 139–151 in CFA. Compared with PLP 139–151 controls, all of the mice that received peptide-specific T cells had a delayed onset of a much milder form of EAE (Fig. 4).
Fig. 4.

Suppression of EAE upon adoptive transfer of peptide-specific T cell lines. Peptide-specific T cells (5 × 106) from lines established after immunization with J2, J3, or J5 alone were transferred into naïve SJL/J mice, and then, on the next day, the mice were immunized s.c. with 50 μg of PLP 139–151 in CFA. Appearance of clinical signs of EAE was monitored daily and the disease severity was scored. P values vs. control on day 22 (n = 9 per group): J2, <0.0005; J3, <0.0003; J5, <0.0001. Experiment performed twice.
Peptide 15-mers Inhibit the Expansion of PLP 139–151-Specific T Cells. To determine whether J2, J3, and J5 affect the expansion of antigen-specific T cells, I-As/PLP 139–151 tetramers were used. Lymphocytes from SJL mice either immunized with PLP 139–151 or coimmunized with the peptides and PLP 139–151 were cultured with PLP 139–151 for 4 days. As expected, SJL mice immunized with PLP 139–151 led to the expansion of PLP tetramer reactive cells (1.5%), and the response was specific because there was no response to Theiler's murine encephalomyelitis virus (TMEV) control tetramers (Fig. 5). In striking contrast, expansion of PLP 139–151-specific T cells was reduced markedly in mice coimmunized with peptides J5 or J2 and PLP 139–151.
Discussion
Peptides of 15 amino acids in length were previously designed based on the crude binding motif of Copolymer 1 to HLA-DR2 (13). About 120 peptides were synthesized and assayed for their ability to compete with MBP 85–99 for binding to HLA-DR2 and for inhibition of proliferation of two T cell hybridomas that carry the TCR derived from two different MS patients (13). Among the peptides synthesized, two peptides (nos. 100 and 101 in ref. 13, J1 and J11 in this article) were unusually active in both assays. These peptides were identical to MBP 85–99 at only two residues, but notably they both retained K at P5, the dominant T cell stimulatory residue (11, 12) and had a large hydrophobic amino acid Y at both of the putative anchor residues P1 and P4.
This study is an effort to improve the binding affinity of these peptides for HLA-DR2 based on the knowledge of how peptides bind to this protein (11, 12) while retaining their ability to inhibit proliferation of encephalitogenic T cell clones in vitro, but more importantly, to test these peptides in in vivo assays of amelioration of EAE in mice and to study their mechanisms. The following modifications have been made. Peptides J1 and J11 have Y at P4, which is well accommodated in the large hydrophobic P4 pocket of HLA-DR2 (Table 1). However, they also have Y at the putative P1 position, which is too large for the P1 pocket. This small dimorphic pocket is formed in part by β86V (rather than β86G) of HLA-DR2, although Y can be forced into it at high concentrations. Thus, V, which is found at P1 in MBP 85–99, and F, which may just fill the P1 pocket, were systematically substituted at this position. Two peptides, J4 and J6, contained E at the putative P1 position; the negative charge of E precluded binding to HLA-DR2, and they served as controls. Finally, P found as P87 (P–2) in MBP 85–99 was added at this position in some peptides or at the very beginning of the peptide (P–5) as well as at the penultimate 14th residue (P10), in the hope that it might prevent degradation by aminopeptidases and carboxypeptidases found in serum. In some peptides, Y at the putative P4 position was substituted by F; the very large hydrophobic P4 pocket of HLA-DR2 can accommodate F, Y, or even W (11).
Most of the peptides inhibited binding of biotinylated MBP 85–99 to some extent as did Cop1 (Fig. 1A and Table 1). However, several stood out as inhibiting binding much better than the others, >25% at the lowest concentration tested, and were far more effective in competing with biotinylated MBP than either MBP 85–99 itself or particularly Cop1. These same peptides were also the best at inhibiting proliferation of the T cell hybridomas (Fig. 1B). However, this assay was compromised by the high value (presumably a blank value) shown by both peptides J4 and J6. These peptides served as negative controls and did not bind at all to HLA-DR2 at the highest concentrations tested.
Because the group of peptides based on peptide J1 and J11 gave similar data, only the first group was evaluated in vivo. J2 (the proline substituted derivative of J1) and both J3 and J5 (also with proline substitutions and with F or V at P1, respectively) were preferentially examined. Two different mouse strains were used. SJL/J mice bear I-As as the only class II major histocompatibility complex protein, whereas in the humanized mouse, all murine class II genes have been knocked out and replaced by DRA and DRB1*1501 that encode HLA-DR2. In addition, this mouse bears as a transgene the TCR from an MS patient (the same TCR that is present in the 8073 T cell hybridoma). These mice were immunized with peptide MBP 85–99 (humanized mouse) or PLP 139–151 (SJL/J mouse). Splenocyte lines were established by stimulation with these same peptides or with peptides J2, J3, or J5. Proliferative responses of these lines to each of the immunogens were obtained (except in the case of the line established from the humanized mouse with the J2 peptide, in which case proliferation was very weak) (Fig. 2A). Notably, the T cell lines that responded to PLP 139–151 or sorted tg cells that responded to MBP 85–99 did not crossreact with any of the peptides J2, J3, and J5 or with Cop1 (Fig. 2B). The lines established after immunization with J2, J3, or J5 produced the Th2 cytokines IL-4 and IL-10 but notably not the Th1 cytokines IFN-γ and IL-2 that were produced by PLP 139–151-specific lines (Fig. 2C). The peptides were also remarkably effective in the amelioration of EAE in both mouse models in comparison with Cop1 (Fig. 3), and adoptive transfer of peptide specific cell lines strongly suppressed the subsequent induction of EAE by PLP 139–151 with effectiveness in order of J5 > J3 > J2-specific lines (Fig. 4). Additionally, in SJL/J mice, the synthetic peptides, particularly J2 and J5, prevented the expansion of PLP 139–151-specific CD4 cells in vitro (Fig. 5). These effects essentially reproduce those previously found by using random amino acid copolymers (29, 30).
Previously, an altered MBP 85–99 was designed by the substitutions E85A(D-Ala), N86K, F91L, and K93A in MBP 85–99 (using the numbering as in Table 1) (36), after an earlier study of single alanine substitution (37). This altered peptide ligand was shown to be effective in binding to HLA-DR2 molecules and also therapeutically proved beneficial in an animal model of MS (38). However, contrary to expectation, the altered peptide ligand exacerbated the clinical signs of MS, an effect attributed to cross-reactivity with MBP 85–99 (39).
Induction of EAE is mediated by Th1 cells, and IFN-γ is an important mediator of encephalomyelitis (40, 41). On the other hand, Th2 cytokines (IL-4 and IL-10) have been shown to possess antiinflammatory properties, neutralizing the effects of 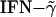 . Mice tg for IL-10 are highly resistant to myelin oligodendrocyte glycoprotein 35–55-induced EAE (42, 43). Because these synthetic peptides induced dominant Th2 cytokines, these Th2 cytokines presumably contributed to the reduction of disease severity in both EAE models. Induction of cell death of antigen-specific T cells was not a possibility because no nonspecific cell death was seen in cultures treated with the synthetic peptides alone.
. Mice tg for IL-10 are highly resistant to myelin oligodendrocyte glycoprotein 35–55-induced EAE (42, 43). Because these synthetic peptides induced dominant Th2 cytokines, these Th2 cytokines presumably contributed to the reduction of disease severity in both EAE models. Induction of cell death of antigen-specific T cells was not a possibility because no nonspecific cell death was seen in cultures treated with the synthetic peptides alone.
Thus, peptide 15-mers of defined sequence have been synthesized that can substitute for random amino acid copolymers in the amelioration of EAE. Three such copolymers have been described as follows: viz., Copolymer 1 [YEAK, poly(Y,E,A,K)n]; FYAK, [poly(F,Y,A,K)n]; and VWAK, [poly(V,W,A,K)n] (29, 30). Cop1 is made in solution by polymerization of N-carboxy amino acid anhydrides, whereas FYAK and VWAK were made by the more recent solid phase chemistry. In either case, the precise constituents of these random copolymer mixtures are not reproducible from batch to batch. The length of the copolymers produced varies from 50 to 80 amino acids. Copolymers of 50 amino acids in length or larger are biologically active. Considerable reduction in efficacy occurs with copolymers of 35 amino acids in length and related random peptides of 15 amino acids (slightly larger than the groove in HLA-DR2 to which peptides and copolymers bind) are essentially inactive (17). Based on the data presented here, a size of 50 or larger may be required to generate within the random copolymers a specific sequence or sequences in sufficient quantity to produce the desired effect. The present peptides and the three copolymers appear to share the ability to induce the antiinflammatory cytokines IL-4 and IL-10. Among these, FYAK and peptides J3 and J5 are the most effective in this respect. The minimum similarity to MBP 85–99 and the lack of cross-reactivity with cell lines specific for either MBP 85–99 or PLP 139–151 suggest that the synthetic peptides described here may be suitable for clinical trials in human subjects with MS.
Acknowledgments
We thank M. L. Wong for expert technical assistance; N. Reppas for helpful discussions; T. Aldridge and the Bauer Center for Genomics Research for use of equipment and facilities; and Robert McGilp and Deneen Kozoriz for flow cytometry sorting. This work was supported by National Institutes of Health Research Grants R01 AI 49524 (to J.L.S.), R01 NS30843 (to V.K.K.), R01 NS38037 (to H. L. Weiner), and National Multiple Sclerosis Society Grant RG2571-D-9 (to V.K.K.).
Abbreviations: CFA, complete Freund's adjuvant; EAE, experimental autoimmune encephalomyelitis; HLA, human leukocyte antigen; MBP, myelin basic protein; MS, multiple sclerosis; PLP, proteolipid protein; TCR, T cell antigen receptor; tg, transgenic.
References
- 1.Spielman, R. S. & Nathanson, N. (1982) Epidemiol. Rev. 4, 45–65. [DOI] [PubMed] [Google Scholar]
- 2.Hillert, J., Kall, T., Vrethem, M., Fredrikson, S., Ohlson, M. & Olerup, O. (1994) J. Neuroimmunol. 50, 95–100. [DOI] [PubMed] [Google Scholar]
- 3.Oksenberg, J. R., Begovich, A. B., Erlich, H. A. & Steinman, L. (1993) J. Am. Med. Assoc. 270, 2362–2369. [PubMed] [Google Scholar]
- 4.Wucherpfennig, K. W., Weiner, H. L. & Hafler, D. A. (1991) Immunol. Today 12, 277–282. [DOI] [PubMed] [Google Scholar]
- 5.Markovic-Plese, S., Fukaura, H., Zhang, J., al-Sabbagh, A., Southwood, S., Sette, A., Kuchroo, V. K. & Hafler, D. A. (1995) J. Immunol. 155, 982–992. [PubMed] [Google Scholar]
- 6.Kerlero de Rosbo, N., Hoffman, M., Mendel, I., Yust, I., Kaye, J., Bakimer, R., Flechter, S., Abramsky, O., Milo, R., Karni, A. & Ben-Nun, A. (1997) Eur. J. Immunol. 27, 3059–3069. [DOI] [PubMed] [Google Scholar]
- 7.Tsuchida, T., Parker, K. C., Turner, R. V., McFarland, H. F., Coligan, J. E. & Biddison, W. E. (1994) Proc. Natl. Acad. Sci. USA 91, 10859–10863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Illes, Z., Kondo, T., Yokoyama, K., Ohashi, T., Tabira, T. & Yamamura, T. (1999) J. Immunol. 162, 1811–1817. [PubMed] [Google Scholar]
- 9.Allegretta, M., Nicklas, J. A., Sriram, S. & Albertini, R. J. (1990) Science 247, 718–721. [DOI] [PubMed] [Google Scholar]
- 10.Krogsgaard, M., Wucherpfennig, K. W., Cannella, B., Hansen, B. E., Svejgaard, A., Pyrdol, J., Ditzel, H., Raine, C., Engberg, J., Fugger, L. & Canella, B. (2000) J. Exp. Med. 191, 1395–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wucherpfennig, K. W., Sette, A., Southwood, S., Oseroff, C., Matsui, M., Strominger, J. L. & Hafler, D. A. (1994) J. Exp. Med. 179, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith, K. J., Pyrdol, J., Gauthier, L., Wiley, D. C. & Wucherpfennig, K. W. (1998) J. Exp. Med. 188, 1511–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fridkis-Hareli, M., Stern, J. N., Fugger, L. & Strominger, J. L. (2001) Hum. Immunol. 62, 753–763. [DOI] [PubMed] [Google Scholar]
- 14.Gaur, A., Wiers, B., Liu, A., Rothbard, J. & Fathman, C. G. (1992) Science 258, 1491–1494. [DOI] [PubMed] [Google Scholar]
- 15.Leonard, J. P., Waldburger, K. E. & Goldman, S. J. (1996) Ann. N.Y. Acad. Sci. 795, 216–226. [DOI] [PubMed] [Google Scholar]
- 16.Nicholson, L. B., Greer, J. M., Sobel, R. A., Lees, M. B. & Kuchroo, V. K. (1995) Immunity 3, 397–405. [DOI] [PubMed] [Google Scholar]
- 17.Fridkis-Hareli, M., Santambrogio, L., Stern, J. N., Fugger, L., Brosnan, C. & Strominger, J. L. (2002) J. Clin. Invest. 109, 1635–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruiz, P. J., DeVoss, J. J., Nguyen, L. V., Fontoura, P. P., Hirschberg, D. L., Mitchell, D. J., Garcia, K. C. & Steinman, L. (2001) J. Immunol. 167, 2688–2693. [DOI] [PubMed] [Google Scholar]
- 19.Goodkin, D. E., Shulman, M., Winkelhake, J., Waubant, E., Andersson, P., Stewart, T., Nelson, S., Fischbein, N., Coyle, P. K., Frohman, E., et al. (2000) Neurology 54, 1414–1420. [DOI] [PubMed] [Google Scholar]
- 20.Teitelbaum, D., Meshorer, A., Hirshfeld, T., Arnon, R. & Sela, M. (1971) Eur. J. Immunol. 1, 242–248. [DOI] [PubMed] [Google Scholar]
- 21.Teitelbaum, D., Webb, C., Meshorer, A., Arnon, R. & Sela, M. (1973) Eur. J. Immunol. 3, 273–279. [DOI] [PubMed] [Google Scholar]
- 22.Teitelbaum, D., Webb, C., Bree, M., Meshorer, A., Arnon, R. & Sela, M. (1974) Clin. Immunol. Immunopathol. 3, 256–262. [DOI] [PubMed] [Google Scholar]
- 23.Aharoni, R., Teitelbaum, D. & Arnon, R. (1993) Eur. J. Immunol. 23, 17–25. [DOI] [PubMed] [Google Scholar]
- 24.Bornstein, M. B., Miller, A., Slagle, S., Weitzman, M., Crystal, H., Drexler, E., Keilson, M., Merriam, A., Wassertheil-Smoller, S., Spada, V., et al. (1987) N. Engl. J. Med. 317, 408–414. [DOI] [PubMed] [Google Scholar]
- 25.Johnson, K. P., Brooks, B. R., Cohen, J. A., Ford, C. C., Goldstein, J., Lisak, R. P., Myers, L. W., Panitch, H. S., Rose, J. W. & Schiffer, R. B. (1995) Neurology 45, 1268–1276. [DOI] [PubMed] [Google Scholar]
- 26.Johnson, K. P., Brooks, B. R., Cohen, J. A., Ford, C. C., Goldstein, J., Lisak, R. P., Myers, L. W., Panitch, H. S., Rose, J. W., Schiffer, R. B., et al. (1998) Neurology 50, 701–708. [DOI] [PubMed] [Google Scholar]
- 27.Fridkis-Hareli, M. & Strominger, J. L. (1998) J. Immunol. 160, 4386–4397. [PubMed] [Google Scholar]
- 28.Fridkis-Hareli, M., Neveu, J. M., Robinson, R. A., Lane, W. S., Gauthier, L., Wucherpfennig, K. W., Sela, M. & Strominger, J. L. (1999) J. Immunol. 162, 4697–4704. [PubMed] [Google Scholar]
- 29.Stern, J. N., Illes, Z., Reddy, J., Keskin, D. B., Sheu, E., Fridkis-Hareli, M., Nishimura, H., Brosnan, C. F., Santambrogio, L., Kuchroo, V. K. & Strominger, J. L. (2004) Proc. Natl. Acad. Sci. USA 101, 11743–11748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Illes, Z., Stern, J. N., Reddy, J., Waldner, H., Mycko, M. P., Brosnan, C. F., Ellmerich, S., Altmann, D. M., Santambrogio, L., Strominger, J. L. & Kuchroo, V. K. (2004) Proc. Natl. Acad. Sci. USA 101, 11749–11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reddy, J., Bettelli, E., Nicholson, L., Waldner, H., Jang, M. H., Wucherpfennig, K. W. & Kuchroo, V. K. (2003) J. Immunol. 170, 870–877. [DOI] [PubMed] [Google Scholar]
- 32.Reddy, J., Illes, Z., Zhang, X., Encinas, J., Pyrdol, J., Nicholson, L., Sobel, R. A., Wucherpfennig, K. W. & Kuchroo, V. K. (2004) Proc. Natl. Acad. Sci. USA 101, 15434–15439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madsen, L. S., Andersson, E. C., Jansson, L., krogsgaard, M., Andersen, C. B., Engberg, J., Strominger, J. L., Svejgaard, A., Hjorth, J. P., Holmdahl, R., et al. (1999) Nat. Genet. 23, 343–347. [DOI] [PubMed] [Google Scholar]
- 34.Tuohy, V. K., Lu, Z., Sobel, R. A., Laursen, R. A. & Lees, M. B. (1989) J. Immunol. 142, 1523–1527. [PubMed] [Google Scholar]
- 35.Kuchroo, V. K., Sobel, R. A., Yamamura, T., Greenfield, E., Dorf, M. E. & Lees, M. B. (1991) Pathobiology 59, 305–312. [DOI] [PubMed] [Google Scholar]
- 36.Kappos, L., Comi, G., Panitch, H., Oger, J., Antel, J., Conlon, P. & Steinman, L. (2000) Nat. Med. 6, 1176–1182. [DOI] [PubMed] [Google Scholar]
- 37.Brocke, S., Gijbels, K., Allegretta, M., Ferber, I., Piercy, C., Blankenstein, T., Martin, R., Utz, U., Karin, N., Mitchell, D., et al. (1996) Nature 379, 343–346. [DOI] [PubMed] [Google Scholar]
- 38.Karin, N., Mitchell, D. J., Brocke, S., Ling, N. & Steinman, L. (1994) J. Exp. Med. 180, 2227–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bielekova, B., Goodwin, B., Richert, N., Cortese, I., Kondo, T., Afshar, G., Gran, B., Eaton, J., Antel, J., Frank, J. A., et al. (2000) Nat. Med. 6, 1167–1175. [DOI] [PubMed] [Google Scholar]
- 40.Nagelkerken, L. (1998) Braz. J. Med. Biol. Res. 31, 55–60. [DOI] [PubMed] [Google Scholar]
- 41.Druet, P., Sheela, R. & Pelletier, L. (1995) Clin. Exp. Immunol. 101, Suppl. 1, 9–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bettelli, E., Das, M. P., Howard, E. D., Weiner, H. L., Sobel, R. A. & Kuchroo, V. K. (1998) J. Immunol. 161, 3299–3306. [PubMed] [Google Scholar]
- 43.Bettelli, E., Nicholson, L. B. & Kuchroo, V. K. (2003) J. Autoimmun. 20, 265–267. [DOI] [PubMed] [Google Scholar]