

Abstract
The genetic code is established in aminoacylation reactions catalyzed by aminoacyl-tRNA synthetases. Many aminoacyl-tRNA synthetases require an additional domain for editing, to correct errors made by the catalytic domain. A nonfunctional editing domain results in an ambiguous genetic code, where a single codon is not translated as a specific amino acid but rather as a statistical distribution of amino acids. Here, wide-ranging consequences of genetic code ambiguity in Escherichia coli were investigated with an editing-defective isoleucyl-tRNA synthetase. Ambiguity retarded cell growth at most temperatures in rich and minimal media. These growth rate differences were seen regardless of the carbon source. Inclusion of an amino acid analogue that is misactivated (and not cleared) diminished growth rate by up to 100-fold relative to an isogenic strain with normal editing function. Experiments with target-specific antibiotics for ribosomes, DNA replication, and cell wall biosynthesis, in conjunction with measurements of mutation frequencies, were consistent with global changes in protein function caused by errors of translation and not editing-induced mutational errors. Thus, a single defective editing domain caused translationally generated global effects on protein functions that, in turn, provide powerful selective pressures for maintenance of editing by aminoacyl-tRNA synthetases.
Keywords: aminoacylation errors, genetic code ambiguity, amino acid misincorporation
It is thought that the genetic code initially had an ambiguous format, in which codons specified groups of similar amino acids (1, 2). As a consequence, the earliest proteins were not distinct chemical entities but were statistical in nature, because the earliest genes gave rise to families of closely related but diverse sequences. Strong selective pressure in favor of those species with the best activities generated pure chemical entities and forced the code into the precise form that it has today. Here we focus on one aspect of how selection operates to retain the modern genetic code.
The natural function of aminoacyl-tRNA synthetases (AARSs), to match amino acids with nucleotide triplets (as anticodons in tRNAs), is the focal point for understanding the origin and development of the genetic code. Primitive synthetases (3), or possibly ribozymes that had aminoacylation activity (4, 5), are thought to have driven the transition from the RNA world to the theater of proteins. The aminoacylation reaction typically occurs in two steps (6):
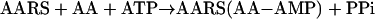 |
[1] |
and
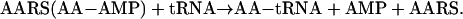 |
[2] |
In reaction 1, the amino acid AA is condensed with ATP to form the highly reactive aminoacyl adenylate. In reaction 2, the activated amino acid is joined through an ester link to one of the hydroxyl groups at the 3′ end of the cognate tRNA. (It is by this reaction that each amino acid is associated with an anticodon, which is embedded in the tRNA.) In addition to the active site for aminoacylation, many of these enzymes have an editing domain, a second, discrete active site that clears errors of amino acid misactivation or misacylation (7). These errors occur as a result of inherent difficulties in obtaining amino acid side chain recognition specificity that matches the accuracy of the code. For example, reaction 1 is not sufficiently accurate for isoleucyl-tRNA synthetase (IleRS) to discriminate between isoleucine vs. valine. In these instances, the misactivated amino acid (AA-AMP), or the mischarged tRNA (AA-tRNA), is cleared by hydrolytic editing (8, 9).
Previous work established that an editing-deficient IleRS generated statistical proteins in vivo (10). A strain of Escherichia coli was constructed that carries a chromosomal ileS allele encoding an editing-deficient IleRS. Under specialized conditions, this strain used Nrv to supplement limiting isoleucine and achieve a growth yield advantage relative to the wild-type strain. However, under most conditions, this strain suffered a growth yield disadvantage in the presence of Nrv. This disadvantage was likely due to the misincorporation of Nrv that, in turn, caused an increased level of misfolded or unfolded proteins in the mutant strain, leading to cell death. These results support the concept that strong selective pressures operate to retain the editing domains of tRNA synthetases.
The full extent of those selective pressures and their outworking are not understood. To explore this issue, we investigated the physiology of an E. coli strain harboring an editing-deficient IleRS encoded by a single chromosomal gene (10). A key objective was to study, under a variety of conditions, the consequences for growth rate, as opposed to just growth yield, of an editing deficiency. The idea was also to link whatever global effects could be measured on cellular proteins to the editing deficiency, per se, and see whether these effects were translation-specific.
Materials and Methods
Strain Construction. To ensure the isogenic character of strains used herein, one E. coli strain was used to reconstruct an editing-deficient mutant and wild-type strains. E. coli strain PS6231 [Δtdk::KnR, ileS T241-N250Ala (10)] was transduced with P1vir lysate generated from strain MG1655 (11) and selected on LB + deoxythymidine (dT, 40 μg/ml) + tri-methoprim (trim, 5 μg/ml) to select for tdk revertants. Clones were restreaked several times on the same medium. Selected clones were also streaked on LB + kanamycin (50 μg/ml) to confirm loss of the insertion marker; on minimal medium with 0.2% glucose [MSglc (12)] to confirm continued prototrophy; and on MSglc + Nrv (5 mM) to confirm continued sensitivity to Nrv. Three clones generated by this method were called PS8078, PS8079, and PS8080, and are used throughout this study as the editing-deficient strains. Clone PS8078 was again transduced with P1vir generated from strain MG1655 and selected on MSglc + Nrv + dT + trim. Clones were restreaked several times on the same media, MSglc + Nrv or MSglc + dT + trim. Clones with the wild-type phenotype were finally isolated and called PS8229, PS8231, and PS8233 and were used throughout this study as the wild-type strains.
E. coli strains ΔdnaQ::TcR (renamed here PS8250) and N3835 (ΔmutS::SpecR, renamed here PS8248) were from J. Shapiro (University of Chicago, Chicago) and F. Taddei (Hôpital Necker-Enfants, Paris). P1vir lysates of strain PS8250 were generated and used to transduce strains PS8078, PS8079, PS8080, PS8229, PS8231, and PS8233. Transductants were selected on LB + tetracycline (Tc, 10 μg/ml), thereby generating strains PS8319, PS8321, and PS8323 (all of which are ileSAla, ΔdnaQ::TcR) and PS8287, PS8288, and PS8289 (all of which are ΔdnaQ::TcR). All strains were grown overnight in LB medium and stored at –80°C in 10% DMSO.
Growth Curves. Strains were inoculated into LB media either directly from frozen stocks or from colonies on plates and grown overnight at 37°C. These cultures were diluted 1:1,000 into 250 μl of fresh LB medium in wells of a 96-well plate. To generate growth curves in minimal media, overnight LB cultures were diluted 1:1,000 into MSglc and again grown overnight. These cultures were diluted 1:1,000 into 250 μl of MSglc in 96-well plates. Microplate cultures were grown in LB or MSglc medium at various temperatures as indicated or at 37°C with various concentrations of Nrv in MSglc or at 37°C in minimal media with various concentrations of several carbon sources. Growth was carried out in a PowerWave 200 Microplate reader (Bio-Tek, Winooski, VT), with shaking and incubation for up to 2 days, and growth curves were generated by following the optical density at 595 nm (13). To determine growth rate, growth curves were fitted by nonlinear regression to the logistic growth equation:
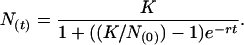 |
[3] |
N(t) is the population at time t (estimated by the optical density of the culture); N(0) is the initial population; K is the carrying capacity, which is the population limit of the specific culture conditions; and r is the intrinsic growth rate of the strains given the specific culture conditions (14). At minimum, growth curves were generated with each of three strains of editing-deficient and wild-type genotypes to ensure replicability; each of these clones was typically repeated in triplicate to internally control the experiments. Each replicate was fitted to Eq. 3, and growth rates were then averaged.
Halo Assays. Overnight LB cultures were diluted 1:500 into fresh LB medium or sterile PBS, and 1 ml was allowed to coat the surface of a measured 25-ml agar plate. Excess culture was removed, and the plates were allowed to dry. Once dry, a 50-μl plug was removed from the center of the plate and then filled with a 50-mg/ml stock solution of antibiotic [except nalidixic (Nal) acid, which was at 5 mg/ml in water at pH 11]. Alternatively, 50 μl of Nrv (100 mM), N-methyl-N′-nitro-N-nitrosoguanidine (1 mg/ml in 0.1 M citrate, pH 5.5) or 2-aminopurine (2.1 mg/ml) was added to the well. For antibiotic and mutagen halo assays, LB plates were used, whereas MSglc plates were used for Nrv halo assays. Solutions were allowed to adsorb into the agar and then were incubated at 37°C overnight. Clearing zone diameters were measured, and then the surface area of the clearing zone was calculated. Clearing zones were never generated by water or citrate buffer controls; the area of the plug was subtracted from the total clearing zone area.
Mutation Frequency Determination. Overnight LB cultures of editing-deficient, wild-type, as well as mutator strains PS8250, PS8248, PS8319, PS8321, PS8323, PS8287, PS8288, and PS8289, were diluted 1:106 and allowed to grow overnight for a total of ≈20 generations. Dilutions were titered on LB, as well as on LB + Rifampicin (170 μg/ml) and LB + Nal (15 μg/ml) (15). Titers of resistant clones were compared with total titer of colony-forming units to determine the frequency of spontaneously resistant mutants for each strain.
Results
Effects of Ambient Environment on Growth Phenotype of a Strain Bearing an Editing-Defective tRNA Synthetase. The connective polypeptide 1 (CP1) editing domain of a subgroup of class I tRNA synthetases is inserted into and splits the active site Rossmann fold of the catalytic center for aminoacylation (16–18). (This subgroup of closely related synthetases includes those for isoleucine, leucine, and valine.) Amino acids that are misactivated are translocated, in a tRNA-dependent step, from the active site to the center for editing, where they are cleared by hydrolysis [of either the misactivated aminoacyl adenylate or mischarged tRNA (Eq. 1 and 2)] (19, 20). An editing-defective IleRS (designated IleRSAla) was created by replacement of a 10-aa sequence within CP1 with a decameric sequence of oligoalanine (10). As a result, this enzyme cannot clear misactivated amino acids. At the same time, aminoacylation activity of IleRSAla is little affected by the oligoalanine replacement. Consequently, the enzyme mischarges tRNAIle with amino acids, such as valine, whose isopropyl group fits into the binding pocket for the isobutyl side chain of isoleucine.
To test the effect of the disruption of the center for editing on growth rate and the sensitivity of growth rate to environment, a strain harboring the ileSAla allele was compared with that of the isogenic wild-type strain. Growth rates of these strains were monitored over a range of temperatures in rich and minimal media (Fig. 1). Regardless of medium, the editing-deficient strain suffered growth rate inhibition at 37°C. Similarly, the growth rate defect of the editing-deficient strain was increased at the upper end of the temperature range tested (i.e., 42–45°C) and even more pronounced in minimal media, where the editing-deficient strain failed to show any signs of growth after 48 h at temperatures above 40°C. Possibly, a greater degree of protein misfolding at higher temperatures combines with amino acid misincorporation to exacerbate the growth-rate defect (21–23). Consistent with this interpretation, at lower temperatures in both rich (30°C) and minimal (32–35°C) media, growth rates of editing-defective and wild-type strains converged (or nearly converged). These results show that genetic code ambiguity caused by errors from defective editing may be exacerbated or ameliorated by the environment, such as the ambient temperature.
Fig. 1.

Effect of temperature on the growth rate of editing-deficient and wild-type strains. Growth curves of the strains were generated in a microplate reader. Data were fitted to the logistic growth equation (see Materials and Methods) to generate an intrinsic growth rate (a growth rate of 1 h–1 corresponds to a doubling time of ≈42 min). Strains were tested in rich media (LB) (A) and minimal media (MSglc) (B). Only at the lowest temperatures tested do strains have comparable growth rates. Error bars give the standard deviation of the growth rates of three replicates of each of three clones.
In addition, growth on different concentrations (0.25–25 mM) of succinate, lactose, and glucose showed that, as carbon sources are varied between limiting and saturating, growth rate differences are exacerbated on substrates that afforded higher growth rates per mol of sugar (data not shown).
Effect of an Isoleucine Analog on Growth Rates. Nrv extends the valine side chain by one methylene group, with branching at the γ instead of β carbon. Thus, the Nrv side chain has the same number of carbon atoms as isoleucine and, not surprisingly, is activated by IleRS. Although the presence of Nrv can reduce the growth yield in the strain carrying the ileSAla allele (10), the effect on growth rate has not previously been measured. To test the effects of Nrv, growth rates of wild-type and editing-deficient strains were measured over wide concentration ranges (0.0001–1 mM) of Nrv. (Both strains were isoleucine prototrophs.) More than a 100-fold difference in growth rate or susceptibility to Nrv was observed between the editing-deficient and wild-type strains (Fig. 2).
Fig. 2.

Effect of Nrv concentration on growth rate. Growth rates were determined for editing-deficient and wild-type strains over 4 orders of magnitude of Nrv concentration (Left). A 2-order-of-magnitude effect was seen between editing-deficient and wild-type strains. Error bars indicate the standard deviations of the growth rates of three replicates of each of three clones. In addition, halo assays testing for sensitivity to Nrv showed a 2.5-fold difference in clearing zone area (Right). Error bars give the standard deviations of the clearing zones of three clones.
To further test and support these conclusions, the editing-deficient and wild-type strains were tested in a halo assay. In this assay, concentrated Nrv filled a 50-μl plug in the center of a bacteria-coated plate and diffused to create a radial gradient. After overnight growth, a radial clearing zone around the plug monitored the relative susceptibility to Nrv of the wild-type and ileSAla-containing strains. The area of the clearing zone was calculated from its radius. Compared with its wild-type counterpart, the editing-deficient strain had an ≈2.5-fold increased clearing zone (Fig. 2). These results are consistent with the impaired growth rate of the ileSAla-harboring strain when challenged with Nrv. Thus, in an environment where an organism is exposed to noncanonical amino acid variants, powerful selective pressure operates to retain the editing function and preserve the canonical genetic code.
Global Effects on Protein Function Tested with Antibiotics. Cellular functions were assayed in a physiological setting by using antibiotics. Pursuant to this objective, a halo assay was used. Four antibiotics were selected to target the A site on the ribosome: neomycin, paromomycin, streptomycin, and tobramycin. (Neomycin and paromomycin are identical with the exception of a single functional group.) These antibiotics enabled investigation of the effects on ribosome function of an editing-defective IleRS. In addition, by choosing antibiotics that target the same site, we could determine whether any effects seen were idiosyncratic to the antibiotic.
Each of the ribosome-specific antibiotics was significantly more potent when tested against the editing-deficient strain (Fig. 3). Although the greatest percentage difference was seen with neomycin, the enhanced sensitivity of the ileSAla-containing strain was clear-cut with all four drugs. The internal consistency of these results showed clearly that the ribosomal A site was perturbed when proteins were synthesized with an editing-defective IleRS.
Fig. 3.

Growth halo test of antibiotics. Editing-deficient and wild-type strains were tested for sensitivity to antibiotics by diffusion in halo tests (see Materials and Methods). The diameters of the clearing zones caused by the antibiotics were measured, and the area of the clearing zone was calculated. In all cases, the editing-deficient strain was found to be more sensitive to antibiotics. Error bars give the standard deviations of the clearing zones of three clones.
To broaden the study beyond the ribosome, additional antibiotics and targets were chosen. We were especially concerned that the translation apparatus (i.e., ribosomes), per se, might be more susceptible to errors of proteins synthesis caused by the aberrant IleRS. We selected two targets that were not directly connected to protein synthesis, DNA replication, and cell wall biosynthesis. Specifically, Nal is directed to DNA gyrase and thereby inhibits DNA replication. Ampicillin, on the other hand, is an alternate (suicide) substrate for cell wall biosynthesis and prevents cell division. Both antibiotics generated larger clearing zones in the editing-deficient strain (Fig. 3). On a relative basis, the effects seen with Nal and ampicillin were comparable to those observed with the four ribosome-directed drugs. These results support the hypothesis that cellular function is deficient on a global basis in the editing-deficient strain.
Because the ribosomes of the editing-deficient strains are more sensitive to antibiotics than are the isogenic wild-type strains, translational errors might be partly attributable to the ribosome, in addition to IleRSAla. Under this model, ribosomal proteins with translational errors would be more prone to make errors, thereby feeding back into the translation system to increase the rate of translational errors. Frameshifting mutations would not be directly attributed to the isoleucyl-tRNA synthetase, so it was thought that, if these errors could be detected, they could be attributed to the ribosome. However, flow cytometry of editing-deficient and wild-type cells harboring a plasmid that expressed an out-of-frame mutant of green fluorescent protein showed no significant recovery of fluorescence in the editing-deficient strain (data not shown). Thus, any errors of translation in the editing-defective cells were due to amino acid misincorporation, rather than to frameshift errors. Therefore, although the ribosome may be more susceptible to antibiotics, it does not amplify effects seen with IleRSAla and thereby increase translational errors.
Editing Deficiency Has No Effect on Mutation Frequency. Although the results presented above support the hypothesis that the hindered growth rate of the editing-defective strain was due to global effects on protein function, genetic mutations in the ileSAla-containing strain could also contribute to the observed results. Under this hypothesis, mutations would have accrued in the ileSAla-containing strains, initiated by DNA replication machinery whose fidelity had been hindered by translational errors. To investigate this possibility, two experiments were carried out. First, halo tests were carried out with N-methyl-N′-nitro-N-nitrosoguanidine and 2-aminopurine, to determine whether either or both of these mutagens (which act by different mechanisms) would be more potent in the ileSAla-harboring strain and thereby retard its growth and result in a greater clearing zone. But with both mutagens, the editing-deficient and wild-type strains showed a similar sensitivity (Fig. 4A). Thus, mutagens, per se, appear not to affect the relative viability of the editing-defective strain.
Fig. 4.

Mutation rate effects on editing-deficient and wild-type strains. (A) Halo tests of N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) and 2-aminopurine (2AP). MNNG may induce slightly greater clearing zone in the wild-type strain. Error bars give the standard deviations of the clearing zones of three clones. (B) Frequencies of spontaneous mutants resistant to rifampicin (RifR) and Nal (NalR). Strains tested included the editing-deficient and wild-type strains; ΔdnaQ, mutator versions of these two strains; and two MG1655-derived strains with, separately, ΔdnaQ and ΔmutS (to induce high-mutation frequencies). Mutator strains have greatly enhanced mutation frequencies, whereas editing-deficient and wild-type strains are highly comparable. Error bars give standard deviations of mutation frequencies of three clones.
Next, the frequency of spontaneous mutants that resulted in resistance to either rifampicin or Nal was checked. These frequencies were not significantly different between the editing-deficient and wild-type strains and were significantly lower than strains with mutator phenotypes (Fig. 4B). These results are consistent with those in Fig. 4A, in that mutational effects seem not to be exacerbated by an editing-defective state. Finally, the editing-deficient and wild-type strains were transduced with the ΔdnaQ mutation. The mutation frequencies of these strains became comparable to that of strain PS8250, which is derived from MG1655 and carries the ΔdnaQ mutator genotype, but not significantly different from one another (Fig. 4B). Thus, an increased mutation frequency does not contribute to the retarded growth rate seen when editing is defective, and an editing deficiency does not contribute to the frequency of spontaneous mutations.
Discussion
Earlier work with amino acid analogues showed that growth defects can occur when organisms are exposed to amino acid analogues. For example, phage Qβ was grown under conditions that allowed the incorporation of ≈65% 6-fluorotryptophan. This incorporation caused a >10-fold growth defect for the phage. In contrast, eight other tryptophan analogues had only a slight effect (24). When E. coli was grown under conditions of tryptophan analogue incorporation, almost no effect on growth rate occurred until the media carried a ratio of >97% analogue relative to the natural amino acid (25). A trifluorinated analogue of isoleucine caused growth rate inhibition in an isoleucine auxotroph (26). (This inhibition may occur because trifluoro-isoleucine bypasses the editing machinery.) In contrast, in this work and in certain related studies (13), growth rate inhibition comes from the misincorporation of natural amino acids due to genetic code ambiguity associated with canonical amino acids. This growth rate inhibition was shown here as likely due to average global effects on protein function.
General environmental stressors (high salt, acid, base, calcium chloride, guanidine hydrochloride, and dimethylformamide) generally had little effect on growth rate of the editing-deficient vs. the wild-type strains (data not shown). [Genetic code ambiguity can even enhance resistance to environmental stress in yeast, potentially because of enhanced induction of heat shock and stress response proteins (27). Possibly, when subjected to a general environmental stressor such as acid, enhanced functions that are idiosyncratic to certain proteins and not others facilitate adaptation and compensate for the general inhibitory effects of genetic code ambiguity.] These results suggest that selective pressure for maintenance of the editing function is derived mostly from conditions that enhance genetic code ambiguity, whether they are conditions that allow for synthetase-directed ambiguous incorporation of a natural vs. a nonnatural amino acid (such as isoleucine versus Nrv) or conditions that promote ribosome ambiguity (i.e., antibiotics) that further enhance ambiguity beyond that caused by an editing deficiency.
One natural example of an intermediate step in the recursive splitting of codon blocks that led to the modern genetic code is seen in Candida spp., where Ser-tRNACAG is ambiguously interpreted as encoding serine and leucine (22, 28, 29). In this work, codons for Ile, in the editing-deficient strain, doubtless also allow incorporation not only of Val but also of a few other amino acids that fit into the Ile-binding pocket, such as Cys and Ala. Like E. coli, when confronted with an ambiguous code of the sort generated here, the yeast Saccharomyces cerevisiae suffered growth rate inhibition when it carried the heterologous, ambiguous Candida spp. Ser-tRNACAG. (27).
Similarly, genetic code ambiguity was shown to cause growth defects when the Bacillus subtilis GluRS was overexpressed in E. coli (30). This protein is known to mischarge the E. coli 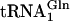 with Glu. Growth defects were ameliorated by coexpression of
with Glu. Growth defects were ameliorated by coexpression of 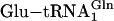 amidotransferase, which converts the mischarged tRNA to the correct species.
amidotransferase, which converts the mischarged tRNA to the correct species.
The DNA replication machinery is most likely affected by the presence of an editing-defective tRNA synthetase. And yet the presence of the ileSAla allele did not lead to an increased mutation frequency. A possible explanation is the editing activity of nucleic acid polymerases. This activity enforces incorporation of complementary bases into a nascent nucleic acid chain, rejecting transient mismatches (31). In addition, mismatch repair systems exist to identify and correct errors that are incorporated into the new DNA chain (31). Thus, were amino acid substitutions to occur in DNA polymerase, errors caused by these substitutions could still be corrected at the editing step.
Acknowledgments
We thank Professors Jeffrey Wong and Tamara Hendrickson for helpful comments on the manuscript. We also thank Dr. E. Westhof for suggesting the use of several antibiotics and Drs. J. Shapiro and F. Taddei for the gift of mutator strains. This work was supported by National Institutes of Health Grant GM23562 and National Science Foundation Grant MCB-0128901.
Abbreviations: AARS, aminoacyl-tRNA synthetase; Nrv, norvaline; IleRS, isoleucyl-tRNA synthetase; MSglc, minimal medium with 0.2% glucose; Nal, nalidixic acid.
See Commentary on page 1273.
References
- 1.Jimenez-Sanchez, A. (1995) J. Mol. Evol. 41, 712–716. [DOI] [PubMed] [Google Scholar]
- 2.Schimmel, P. & Ribas de Pouplana, L. (2001) Cold Spring Harbor Symp. Quant. Biol. 66, 161–166. [DOI] [PubMed] [Google Scholar]
- 3.Ribas de Pouplana, L. & Schimmel, P. (2000) Cell Mol. Life Sci. 57, 865–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee, N., Bessho, Y., Wei, K., Szostak, J. W. & Suga, H. (2000) Nat. Struct. Biol. 7, 28–33. [DOI] [PubMed] [Google Scholar]
- 5.Saito, H., Kourouklis, D. & Suga, H. (2001) EMBO J. 20, 1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ibba, M. & Soll, D. (2000) Annu. Rev. Biochem. 69, 617–650. [DOI] [PubMed] [Google Scholar]
- 7.Hendrickson, T. L. & Schimmel, P. (2003) in Translation Mechanisms, eds. Lapointe, J. & Brakier-Gingras, L. (Kluwer/Plenum, Dordrecht, The Netherlands), pp. 34–64.
- 8.Baldwin, A. N. & Berg, P. (1966) J. Biol. Chem. 241, 839–845. [PubMed] [Google Scholar]
- 9.Fersht, A. R. (1977) Biochemistry 16, 1025–1030. [DOI] [PubMed] [Google Scholar]
- 10.Pezo, V., Metzgar, D., Hendrickson, T. L., Waas, W. F., Hazebrouck, S., Döring, V., Marlière, P., Schimmel, P. & De Crécy-Lagard, V. (2004) Proc. Natl. Acad. Sci. USA 101, 8593–8597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller, J. H. (1992) A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria (Cold Spring Harbor Lab. Press, Plainview, NY).
- 12.Richaud, C., Mengin-Lecreulx, D., Pochet, S., Johnson, E. J., Cohen, G. N. & Marliere, P. (1993) J. Biol. Chem. 268, 26827–26835. [PubMed] [Google Scholar]
- 13.Hendrickson, T. L., Nomanbhoy, T. K., de Crecy-Lagard, V., Fukai, S., Nureki, O., Yokoyama, S. & Schimmel, P. (2002) Mol. Cell 9, 353–362. [DOI] [PubMed] [Google Scholar]
- 14.Steinhaus, H. (1999) Mathematical Snapshots (Dover, Mineola, NY).
- 15.Ausubel, F. M. (1997) Short Protocols in Molecular Biology: A Compendium of Methods from Current Protocols in Molecular Biology (Wiley, New York).
- 16.Starzyk, R. M., Webster, T. A. & Schimmel, P. (1987) Science 237, 1614–1618. [DOI] [PubMed] [Google Scholar]
- 17.Schimmel, P. & Ribas de Pouplana, L. (1995) Cell 81, 983–986. [DOI] [PubMed] [Google Scholar]
- 18.Lin, L., Hale, S. P. & Schimmel, P. (1996) Nature 384, 33–34. [DOI] [PubMed] [Google Scholar]
- 19.Baldwin, A. N. & Berg, P. (1966) J. Biol. Chem. 241, 831–838. [PubMed] [Google Scholar]
- 20.Eldred, E. W. & Schimmel, P. R. (1972) J. Biol. Chem. 247, 2961–2964. [PubMed] [Google Scholar]
- 21.Santer, U. V., Cekleniak, J., Kansil, S., Santer, M., O'Connor, M. & Dahlberg, A. E. (1995) RNA 1, 89–94. [PMC free article] [PubMed] [Google Scholar]
- 22.Santos, M. A., Perreau, V. M. & Tuite, M. F. (1996) EMBO J. 15, 5060–5068. [PMC free article] [PubMed] [Google Scholar]
- 23.Balashov, S. & Humayun, M. Z. (2003) J. Bacteriol. 185, 5015–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bacher, J. M., Bull, J. J. & Ellington, A. D. (2003) BMC Evol. Biol. 3, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bacher, J. M. & Ellington, A. D. (2001) J. Bacteriol. 183, 5414–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang, P., Tang, Y. & Tirrell, D. A. (2003) J. Am. Chem. Soc. 125, 6900–6906. [DOI] [PubMed] [Google Scholar]
- 27.Santos, M. A., Cheesman, C., Costa, V., Moradas-Ferreira, P. & Tuite, M. F. (1999) Mol. Microbiol. 31, 937–947. [DOI] [PubMed] [Google Scholar]
- 28.Santos, M. A. & Tuite, M. F. (1995) Nucleic Acids Res. 23, 1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Sullivan, J. M., Mihr, M. J., Santos, M. A. & Tuite, M. F. (2001) Yeast 18, 313–322. [DOI] [PubMed] [Google Scholar]
- 30.Baick, J. W., Yoon, J. H., Namgoong, S., Soll, D., Kim, S. I., Eom, S. H. & Hong, K. W. (2004) J. Microbiol. 42, 111–116. [PubMed] [Google Scholar]
- 31.Rupp, W. D. (1996) in Escherichia coli and Salmonella: Cellular and Molecular Biology, eds. Neidhardt, F. C., Curtiss, R., Ingraham, J. L., Lin, E. C. C., Low, K. B., Magasanik, B., Reznikoff, W. S., Riley, M., Schaechter, M. & Umbarger, H. E. (Am. Soc. Microbiol., Washington, DC), Vol. 2, pp. 2277–2294. [Google Scholar]