

Abstract
Expensive computation in handling a large number of sequences limits the application of local multiple sequence alignment. We present an Eulerian path approach to local multiple alignment for DNA sequences. The computational time and memory usage of this approach is approximately linear to the total size of sequences analyzed; hence, it can handle thousands of sequences or millions of letters simultaneously. By constructing a De Bruijn graph, most of the conserved segments are amplified as heavy Eulerian paths in the graph, and the original patterns distributed in sequences are recovered even if they do not exist in any single sequence. This approach can accurately detect unknown conserved regions, for both short and long, conserved and degenerate patterns. We further present a Poisson heuristic to estimate the significance of a local multiple alignment. The performance of our method is demonstrated by finding Alu repeats in the human genome. We compare the results with Alus marked by repeatmasker, where the two programs are in good agreement. Our method is robust under various conditions and superior to other methods in terms of efficiency and accuracy.
Keywords: De Bruijn graph, sequence repeats, declumping, repeat finding
As molecular sequence databases are growing rapidly, powerful tools for manipulating the large data sets and extracting useful information become increasingly important. Sequence alignment is one of the commonly used methods because sequence homologies often provide clues to biological function. We have developed a local multiple alignment algorithm that can efficiently find conserved segments among a large number of sequences and in long sequences. In each iteration of our method, the consensus sequence pattern among the sequences is constructed from a De Bruijn graph, and then this consensus is used as a query to locate all instances of the pattern. The numbers and lengths of sequences that might process such a pattern are unknown. We regard a pattern to be an estimate of an ancestral sequence.
The initial motivation for the method arises from the algorithm for fragment assembly in DNA sequencing using the Eulerian superpath approach (1, 2), where a De Bruijn graph of DNA fragments is constructed and the assembled genome is an Eulerian path in the graph. We have developed an Eulerian path approach to global multiple alignment (3), and our approach to local multiple alignment is a further extension. Compared with global alignment that attempts to align the entire sequences, local alignment is more difficult because the locations, sizes, structures, and numbers of conserved regions are completely unknown. By locations we are referring to the large number of choices as to what portions of sequences to align.
Many local multiple alignment methods have been developed in the past decades, such as pima (4, 5), pralign (6), macaw (7), match-box (8), and dialign (9). One subproblem of local alignment is motif finding, which has been widely applied in studying noncoding regions (mostly short regions close to genes) that may contain regulatory motifs. Regulatory motifs are often weakly conserved, and methods developed in motif finding often use statistical techniques that are necessary to capture the weak, but still significant, signals hiding in sequences. The Gibbs motif sampler (10) uses the Gibbs sampling technique to iteratively sample motifs until convergence. meme (11) fits a mixture model by expectation maximization and combines motifs into a hidden Markov model for database searching. Those methods, however, are limited by the size of data they can analyze and the length of motifs they can model. As a result, they are not efficient in finding local alignments of variant lengths, particularly when the total size of data is large.
Another specific problem of local alignment emerged recently as entire genome sequences became available, that is, large-size sequence comparison. Several methods have been proposed to find conserved regions across multiple genome-sized sequences. The methods, such as mlagan (12) and mavid (13), first find locally well conserved regions as anchors, and then chain anchors into larger alignments. tba (14) computes a set of local alignments called blockset and chains blocks according to a reference sequence. Nevertheless, those methods are designed for whole-genome comparison and are different from typical local multiple alignment that seeks all possible combination of similar regions. It is not appropriate to apply those methods for finding less conserved regions, especially when the regions are “randomly” located in multiple sequences.
Few methods have been proposed to find local alignments among a large number of sequences, both accurately and efficiently. Given a large number of sequences, the computation time often is reduced by using pairwise sequence comparison or comparing each sequence with a database. Pairwise sequence comparison is not accurate in multiple alignment, because the pairwise alignment errors may accumulate and ruin the final result when the number of sequences is large, or it may simply miss the local conservation that is insignificant when only two sequences are considered at a time. Similarly, comparisons of each sequence with a database can find only conserved regions that are similar to known sequences or consensuses, and the local alignment result is very likely to be biased. We propose a different approach that is more accurate and efficient when presented with a large number of sequences or long sequences. In addition, our method can locate both similar and degenerately conserved regions of various sizes simultaneously, without increasing computational load.
A special form of local multiple alignment is repeat finding, which is to find repeated regions within a single sequence. Repeat structure is abundant in eukaryote genomes, and it provides valuable clues to molecular mechanisms. reputer (15, 16) was developed to efficiently find maximal repeats within genome-sized sequences. Another interesting work (17) found repeated structures directly from short DNA fragments of sequence assembly, before the entire genome sequence was assembled. aba (18) focuses on classifying different classes of repeats or conserved regions by using a graph structure, and the alignments are initially computed by using other software. We compared our method with reputer in the repeat finding problem and found that our method is more sensitive to detecting degenerate repeats. repeatmasker, a program searching for repeats against a repeat database [Repbase Update (19)], is used as the reference to test the program's accuracy. Note that repeat finding is a special form of local multiple alignment, where alignment of multiple sequences can be transformed to repeat finding by concatenating all sequences into one.
Different classes of patterns often coexist in the same data at different locations. Instances of each class are output as one local alignment. An interesting and important idea is to evaluate the significance of each output alignment. As an example, two local multiple alignments of similar sizes are output by our program (Fig. 1), and it is not obvious which one is more significant. We use a Poisson heuristic to estimate their significance under a random model.
Fig. 1.

An example of two local multiple alignments output by our program. Sequence indices and positions are shown on the left. The significance of each alignment can be estimated by a Poisson heuristic.
Method
The pipeline of our algorithm is as follows: (i) construct a De Bruijn graph by overlapping k-tuples; (ii) cut “thin” edges by estimating the statistical significance of each edge with a Poisson heuristic; (iii) resolve cycles in the graph; (vi) extract a heaviest path as the consensus; (v) use fast local pairwise alignment and the declumping algorithm to find all subsequences similar to the consensus; (vi) construct and output a multiple alignment from pairwise alignments; and (vii) declump the De Bruijn graph and return to step iv to find other patterns.
Constructing a De Bruijn Graph. Given a set of DNA sequences, we break each sequence into overlapping k-tuples. Two adjacent k-tuples overlap at (k-1) letters. Each k-tuple is represented by a directed edge in the graph, and identical k-tuples are represented by the same edge. Two edges are connected by a node if they represent at least one pair of adjacent k-tuples in a sequence, where the node represents the (k-1)-tuple overlap. Finally, we record the position and sequence index of each k-tuple in the corresponding edge. An example of a three-tuple De Bruijn graph of three sequences (ATGT, ATGC, and CTGT) is shown in Fig. 2. Under this construction, each sequence is mapped to a path traversing the graph. If a k-tuple appears in multiple sequences, the corresponding sequence paths will intersect at the edge representing this k-tuple. Based on this property, we define the multiplicity of an edge to be the number of sequence paths visiting the edge, i.e., the number of sequences containing the corresponding k-tuple. Intuitively, the larger the multiplicity is, the more likely the edge represents a conserved k-tuple. Vice versa, the conserved subsequences tend to be amplified in the graph by edges with large multiplicities. By traversing the graph, we can find the consensus of a pattern in the resolution of k-tuple even if the original pattern does not exist in any sequence. We demonstrate the fidelity of this idea by both theoretical analysis and its application to simulated and real data.
Fig. 2.

Three DNA sequences are build into a three-tuple De Bruijn graph by “gluing” identical edges and vertices. Each sequence is a path traversing the graph.
Removing Thin Edges. Significant regions of conservation have been found among DNA/protein sequences that overall have little similarity. Dissimilar regions are often uninteresting and make the local alignment problem more difficult to solve. In the De Bruijn graph, they correspond to a huge number of thin edges, whose multiplicities are small. Thin edges therefore are removed from the graph. Without knowing the locations of conserved segments, we remove an edge by estimating the probability that the edge is presented by chance under a random model (see Significance Estimation for details). Fig. 3 shows an example of before and after removing thin edges.
Fig. 3.

De Bruijn graph before (a) and after (b) removing thin edges. Corresponding sequences contain copies of two patterns. (a) The bold edges represent the conserved patterns. (b) Conserved patterns remain in the graph, and the bold edges represent the heaviest path (10 sequences, 100 bp each, tuple size of 10).
Resolving Cycles. Because of repeats and random matching, the initial De Bruijn graph is very likely to have cycles. For example, if there exist a three-tandem repeat (a repeat unit consecutively repeated three times) in some sequences, the repeat unit (≥ k letters) will be represented as a cycle in the graph since identical k-tuples are represented by the same edge, and thus any sequence path containing the three-tandem repeat will visit the cycle three times. It is hard and often ambiguous to determine how many times a cycle should be visited when reconstructing consensuses. We applied an idea similar to the superpath solution in ref. 2 to resolve all cycles in the graph, where the cycle representing the three-tandem repeat were transformed to acyclically connected edges (3-fold relative to the size of the cycle) through which we can reconstruct the three-tandem repeat without ambiguity. The key observation is that we know how to traverse the graph after each single sequence, which makes resolving cycles possible. More details can be found in refs. 2 and 3.
Consensus Alignment. After removing thin edges and resolving cycles, we apply a heaviest path algorithm to find the consensus sequence. The heaviest path algorithm is a shortest path algorithm with negative edge weights and costs linear time when the graph is acyclic. The weight function for edge e is defined as W(e) = (me - c) × (le - o), where me is the edge multiplicity, c is a constant to address the random matches, le is the edge length, and o is a constant to avoid overweighting from k-tuple overlaps. More sophisticated weight functions can be easily applied when additional information is available, such as quality scores of base calling.
For each consensus, we apply a banded version of local pairwise alignment (20) and a declumping algorithm (21) to find segments similar to the consensus. The declumping algorithm allows multiple independent segments to be found within one sequence. Segments found from all sequences then are assembled into a local multiple alignment. It is worth mentioning that our method does not require the pattern to appear in all sequences, and/or have specified lengths, as do many statistical and deterministic methods.
Declumping Graph. It is often the case that several different patterns present within the same sequence set. Our program outputs one local alignment at a time, and we ran the program iteratively to find additional patterns. Declumping graph is a procedure that removes information of previously output local alignments from the graph, and thus allows additional patterns to be found. This is done by removing the information of k-tuples within the output alignments (instead of removing the edges of the consensus path) from the graph. As an example, two patterns, XYZ and PYQ, share a subpattern, Y, and their paths intersect at Y in the graph. We first output a local alignment of pattern XYZ and declump the graph so that the path of PYQ appears as the next consensus. Note that we do not remove the edge of Y but only reduce its multiplicity, because Y also appears in PYQ. The steps of finding a consensus-consensus alignment-declumping graph are repeated until no significant local alignments are left.
There arises an issue when multiple patterns are present in the same sequence set. As in the above example, when both patterns XYZ and PYQ exist, it is possible that our heaviest path approach mistakenly chooses XYQ as the consensus if k-tuples in Q are more abundant than in Z. Our current solution to this issue is to analyze the distribution of k-tuples from the heaviest path and choose a subset of edges that is likely to contain one particular pattern and run the heaviest path algorithm again within the subset. This issue, however, could be more complicated where the fundamental repeat units occurring as tandem repeats can result in a consensus of two or more fundamental units concatenated. This situation requires further analysis.
Significance Estimation
We recall that local alignment P values are invaluable in database searches (22, 23). The statistical nature of matchings among random sequences has been studied (21, 24-28). In our problem, we estimate the P value of a local multiple alignment to remove thin edges formed by random matches, as well as to rank multiple outputs by statistical significance. We have presented a large deviation estimation on the minimum multiplicity of mutation-free edges in our previous paper (3). Intuitively, edges with multiplicity much less than the expected minimum are most likely to consist of random matches. The large deviation estimation under local alignment conditions, however, is more complicated than in the global case. This is because now the positions and the orders of conserved regions in each sequence are totally random. Without additional considerations, such an estimation will only provide a lower bound that is too small to be useful. Aldous (29) has formulated the Poisson clumping heuristic that is used instead in our estimation.
Significance of Local Multiple Alignments. To avoid redundant output that essentially represents the same alignment, we define a clump to be a set of alignments that either shares at least one matched column with or is a subset of a reference alignment. The reference alignment can be arbitrarily defined, and only one alignment per clump will be output.
In the exact matching case, an alignment is a set of identical tuples from different sequences. It has been proved that the asymptotic distribution of the number of clumps is Poisson (30). That is, given N sequences of L letters from a finite alphabet set Σ, define the size of a clump to be (n, l) if its reference alignment consists of n identical l-tuples from n different sequences, where the reference alignment is inextensible. Under this definition, the asymptotic distribution of the number of clumps of size at least (n, l) is approximately Poisson when N is bounded and L → ∞.
When allowing gaps under a wide range of penalty scoring, the Poisson approximation is still valid but the parameters are to be estimated by simulation. The Poisson heuristic has been successfully applied in estimating the significance of pairwise alignment (23), where two parameters γ and p(2) are set, such that 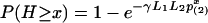 . H is the optimal clump score (let W be the number of clumps that has score ≥ x; then H < x is equivalent to W = 0), p(2) is the probability that two letters are identical, and L1, L2 are the adjusted lengths of two sequences. In fact, the factor
. H is the optimal clump score (let W be the number of clumps that has score ≥ x; then H < x is equivalent to W = 0), p(2) is the probability that two letters are identical, and L1, L2 are the adjusted lengths of two sequences. In fact, the factor 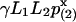 is an approximation to the expected number of clumps with score ≥ x. For multiple alignment, analogously, we approximate the expected number of clumps of size at least
is an approximation to the expected number of clumps with score ≥ x. For multiple alignment, analogously, we approximate the expected number of clumps of size at least 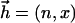 by
by 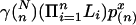 , and hence
, and hence 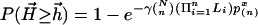 (30), where n is the number of rows and x is the alignment score normalized by n. Parameters γ and p(n) are estimated by simulation. In addition, for the significance of suboptimal alignments, the p value of the rth optimal alignment as an example, can be easily estimated as
(30), where n is the number of rows and x is the alignment score normalized by n. Parameters γ and p(n) are estimated by simulation. In addition, for the significance of suboptimal alignments, the p value of the rth optimal alignment as an example, can be easily estimated as
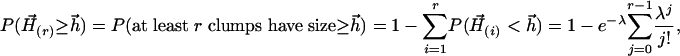 |
and
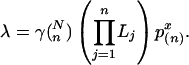 |
The accuracy of the Poisson approximation depends on sequence lengths, where long sequences are required for good approximations. In addition, large variation among sequence lengths reduce the accuracy (23, 29, 30).
Declump Pairwise Alignment. After obtaining a consensus path from the graph, we use the banded pairwise alignment and the declumping algorithm (21) to find segments similar to the consensus. For each pairwise alignment matrix (between the consensus and each sequence), we iteratively record the current optimal alignment followed by declumping the matrix, until the current optimal alignment has the p value above a significance threshold p0. The threshold p0 is chosen in the following way: assume the Poisson distribution of 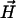 with distribution function F(x), and let
with distribution function F(x), and let 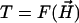 , then T is a random variable distributed as U(0, 1) under the random model. A larger T indicates a more significant alignment. From N sequences, we have N random variables Ti from each sequence (i = 1, 2,..., N). If we choose p0 such that P(max(Ti) < 1 - p0) > 1 - α, we will have (1 - α) × 100% chance to detect significant alignments. By selecting a small α, we can solve p0 by P(max(Ti) < 1 - p0) = (1 - p0)N = 1 - α.
, then T is a random variable distributed as U(0, 1) under the random model. A larger T indicates a more significant alignment. From N sequences, we have N random variables Ti from each sequence (i = 1, 2,..., N). If we choose p0 such that P(max(Ti) < 1 - p0) > 1 - α, we will have (1 - α) × 100% chance to detect significant alignments. By selecting a small α, we can solve p0 by P(max(Ti) < 1 - p0) = (1 - p0)N = 1 - α.
It is worth mentioning, however, that the significance of a hit not only depends on the alignment score, but also on the total number of hits found within one sequence. The highest-scoring segment may not be significant, whereas some modest-scoring segments could be significant when we take into account the order statistics.
Computation Efficiency
The computational time for our method is approximately linear with respect to the total size of data. Let k be the tuple size, l be the pattern length found in each iteration, N be the number of sequences, and L be the average sequence length. The computation time for graph construction and transformation is approximately O(kNL), and for pairwise alignment with declumping it is O(NLl). The memory usage is O(kNL + l2), because the graph size is proportional to the data size, and the size of alignment matrix is O(l2).
Application in Repeat Finding
In our method, the multiplicity of an edge is defined to be the number of sequences visiting the edge. By slightly modifying this definition to be the number of k-tuple occurrences, our method can be applied in finding repeats from a single sequence. Alu repeats are the most abundant repeats in the human genome. We thus apply our method in the human genome and expect to find Alus, although the Alu repeat is unspecified to our program. The Alu repeat consists of multilevels of shorter forward and palindromic repeats and thus provides a good example for demonstrating the accuracy and robustness of our method.
Five sequences from the human genome were randomly chosen, with sequence lengths ranging from 22 kb to 1 Mb. The data were downloaded from the National Center for Biotechnology Information (NCBI) (www.ncbi.nlm.nih.gov). Two shorter sequences are complete genes (22 and 38 kb, NCBI accession nos. AF435921 and Z15025), and two longer sequences are bacterial artificial chromosome clones (167 and 199 kb, NCBI accession nos. AC034110 and AC010145). The last 1-Mb sequence (chr22) is randomly chosen from the human chromosome 22, the 20- to 21-Mb region from ensembl (www.ensembl.org). As a reference, we used repeatmasker to find all repeats in the above sequences, with the only mask Alus (and 7SLRNA, SVA, and LTR5) option and slow option (which is more sensitive) checked. Alu repeats marked by repeatmasker are regarded as the reference.
Since our method considers forward and reverse complement repeats as two different patterns, we ran our program for two iterations in each test to recover Alus in both directions. We also ran our programs for additional iterations and found non-Alu repeats. One contiguous region may be marked as multiple repeats by either program. Therefore, we use the total length of repeats to define the sensitivity and specificity instead of using Alu units, although the definition should depend on the purpose of repeat finding. Let lt be the total length of repeats found by our program, lr be the total length of repeats masked by repeatmasker, and lc be the total length of letters masked by both programs, then the sensitivity is lc/lr, and the specificity is lc/lh.
As shown in Table 1, our program achieved high sensitivity and specificity in all tests. Its ability to find Alus is caused by the high abundance of Alu repeats in the human genome. The average similarity between Alus and their closest consensus is 86%, as indicated by repeatmasker. If the divergence among Alus is random, we would expect the pairwise similarity between Alus to be 80% or lower. In addition, the specificity of our result is in general lower than sensitivity, because our program also found other repeats (located near some Alus and hence “adhered” to the Alu consensus path in our graph). We manually checked the false-positive and false-negative regions on each sequence. Most false positives are simple repeats, such as (CTT)n, poly(A/T), and some unknown repeats. False negatives fall into two categories, short Alus and repeats from non-Alu families. Two extreme cases of short Alus marked by repeatmasker are a 12-bp Alu in AC034110 and a 13-bp Alu in chr22, both of which are indistinguishable from random hits without targeting Alus. The repeats from non-Alu families were missed by our method because of their low similarity and relatively small number of instances (six 7SLRNA, two LTR5_Hs, six LTR5B, and four SVA in chr22; one SVA in AC034110; and one 7SLRNA in AC010145). An example of repeats in Z15025 is shown in Fig. 4. repeatmasker masked 53 Alu repeats from 13 Alu subfamilies, where our method found them by using only two consensus sequences. Part of the alignment of Alu repeats is shown in Fig. 5. In addition, the 1-Mb chr22 sequence contains 32 subfamilies of 717 Alu repeats that span in total 185 kb and provides the most complicated data in our tests, as shown in Fig. 6. Nevertheless, our program performed consistently over all tested sequences and achieved high accuracy within seconds.
Table 1. Finding Alu repeats in five sequences.
| Seq | Rep, kb | Family | Alu, bp | Div, % | Sn, % | Sp, % | T, s | Sn,* % | Sp,* % | T,* s |
|---|---|---|---|---|---|---|---|---|---|---|
| 22 Kb | 8 | 10 | 261 (69) | 15.0 (6.4) | 98.4 | 98.9 | 3 | 96.3 | 99.4 | 1 |
| 38 Kb | 13 | 13 | 245 (85) | 15.7 (5.7) | 98.8 | 96.4 | 8 | 98.6 | 96.7 | 4 |
| 167 Kb | 25 | 18 | 261 (72) | 12.2 (5.9) | 95.2 | 93.0 | 44 | 93.5 | 95.2 | 14 |
| 199 Kb | 33 | 13 | 277 (55) | 15.0 (5.6) | 99.3 | 92.6 | 62 | 85.2 | 93.7 | 32 |
| 1 Mb | 85 | 32 | 252 (79) | 15.2 (6.1) | 95.3 | 98.9 | 293 | 72.4 | 99.4 | 85 |
Seq, sequence length; Rep, total length of Alus marked by repeatmasker; Family, no. of Alu subfamilies; Alu, average Alu size with SD shown in parentheses; Div, average divergence from the closest subfamily's consensus with SD shown in parentheses. All SDs were computed by excluding repeats from LTRs, 7 SLRNA, and SVA subfamilies. Sn, sensitivity; Sp, specificity; T, computation time.
Indicates results by a fast version of our program. The computation time was greatly reduced, but the last two cases also show reduced sensitivities
Fig. 4.

A 38-kb sequence of a Homo sapiens Bat2 gene. Bars below the genome line are Alu repeats marked by repeatmasker (10 Alu subfamilies are indicated in gray scale). Bars above the genome line are our result, including both forward and reverse complement repeats. One false positive and one false negative are indicated by circles.
Fig. 5.

Partial alignment of Alus (the reverse complement) found in a H. sapiens Bat2 gene. Each row is an aligned Alu.
Fig. 6.

A 1-Mb region of human chromosome 22. Bars above the line are repeats found by our method, and bars below the line are the reference Alus. Forward and backward Alu repeats are shown in different rows. One false-negative example (belonging to the LTR5_Hs subfamily) is indicated by an arrow.
As observed in Table 1, the computation time of our program increases linearly as the size of data increases, and the time also depends on the length of repeats. The banded local pairwise alignment and declumping step is the most time-consuming step, which depends on both the length of consensus and the number of repeats. We used 13-tuples for the first four tests and 22-tuples for the last test because of its large data size and high density of repeats. All tests were performed on a 1.7-GHz and 512-Mb computer.
Alternatively, we can search a region only if it is indicated by our graph, i.e., we only search the region if it has at least one k-tuple in our consensus path in the graph. This strategy will greatly reduce the computation time, but also may reduce the sensitivity. We ran our program again on the same set of sequences by using the same tuple sizes under this strategy. As shown in Table 1, the computation time for all tests was significantly reduced, but the sensitivities of the last two tests were reduced as well. The missing repeats are probably more degenerate than the others so that they are missed when using a large tuple size. We reduced the tuple size to 12 for AC010145 and 20 for chr22 and observed that the sensitivity for AC010145 increased from 85.2% to 98.6% and for chr22 increased from 72.4% to 86.8%. This finding indicates that our method can directly locate a large portion of repeats only by checking the consensus path through the graph, even when the pairwise similarity of repeats is 80% or lower.
reputer (15, 16) is a program for finding maximum degenerate repeats in genome-sized sequences. We ran reputer on the above sequence sets and compared its performance with our method. Since reputer outputs pairwise relationship between instances of repeats, we merged all repeat pairs together and checked the sensitivity, that is, the proportion of reference Alus covered by reputer's result. reputer requires the user to specify the minimum repeat length and the maximum difference allowed between repeats. We tried several parameters and observed that reputer consistently missed almost half of the reference Alus in each test. We increased the difference allowed between repeats because Alus may be <80% identical and observed no significant improvement in Alu finding, but a sharp increase in computation time. We believe it is because reputer uses exact short matches as seeds and finds repeats by extending each seed. To find degenerate repeats, the length of seeds must be short. Therefore, real seeds will be overwhelmed by random seeds in genome-sized sequences, which greatly increases the computational cost and decreases the accuracy of results. Furthermore, the specification of maximum difference allowed between repeats also limits reputer's accuracy, because repeats of various lengths are restricted by the same number of differences allowed. In our test, however, reputer did find several non-Alu repeats, which are well conserved. As a comparison, our method uses a large tuple size to reduce the number of random matches, so that only a small number of candidates remain. Instead of finding conserved regions directly, our method first constructs a consensus sequence from the De Bruijn graph, and then uses a fast, but exact, version of local consensus alignment to detect conserved regions. Although a large tuple size is often used, our consensus sequence will be accurate with a sufficient number of instances of a pattern in the data. More details about the fidelity of the consensus sequence are presented in ref. 3. Results of simulations testing our method can be found in Supporting Text and Table 2, which are published as supporting information on the PNAS web site.
Discussion
Our Eulerian path approach can accurately find both well conserved and degenerate patterns in a large sequence set. Multiple patterns of lengths in a wide range can be detected simultaneously, and it costs approximately linear time with respect to the size of data. Our method can be applied in analyzing large DNA sequence sets, such as finding unknown locally conserved patterns. As shown by our examples, this method is accurate and robust under various settings, including the size of data, the length of patterns, and the divergence of patterns. The only parameter specified before running our program is the tuple size, which is often arbitrarily chosen. In our experience, the performance of our program is relatively stable when the tuple size is chosen within a certain range (e.g., from 10 to 20). In cases where the tuple size is too small, we do occasionally observe reduced performance, which is probably caused by the graph transformation step that resolves cycles with a local method. Furthermore, how to detect true patterns other than concatenation of different patterns remains an open question. The software and source code are freely available for research purposes on request to yuzhang@stat.harvard.edu.
Most available multiple alignment algorithms are designed for specific purposes and lack the flexibility to be applied in different situations. Few methods have addressed the accuracy issue when presented with a large number of sequences, which is important for multiple alignment. The Eulerian path approach attempts to achieve both accuracy and efficiency through a novel, but general, framework, and it can be extended for studying many different problems. The current version of our method focuses on DNAs only, but it can be further extended to analyze protein sequences. For proteins, however, sequence identity could be 30% and lower, which hinders the direct application of this Eulerian path approach.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant R01 HG02360-01, and Y.Z. was partially supported by a Graduate Merit Award when he was a Ph.D. student at the University of Southern California.
References
- 1.Idury, R. & Waterman, M. S. (1995) J. Comp. Biol. 2, 291-306. [DOI] [PubMed] [Google Scholar]
- 2.Pevzner, P. A., Tang, H. & Waterman, M. S. (2001) Proc. Natl. Acad. Sci. USA 98, 9748-9753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang, Y. & Waterman, M. S. (2003) J. Comp. Biol. 10, 803-819. [DOI] [PubMed] [Google Scholar]
- 4.Smith, R. F. & Smith, T. F. (1990) Proc. Natl. Acad. Sci. USA 87, 118-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith, R. F. & Smith, T. F. (1992) Protein Eng. 5, 35-41. [DOI] [PubMed] [Google Scholar]
- 6.Waterman, M. S. & Jones, R. (1990) Methods Enzymol. 183, 221-237. [DOI] [PubMed] [Google Scholar]
- 7.Schuler, G. D., Altschul, S. F. & Lipman, D. J. (1991) Proteins 9, 180-190. [DOI] [PubMed] [Google Scholar]
- 8.Depiereux, E., Baudoux, G., Briffeuil, P., Reginster, I., De Bolle, X., Vinals, C. & Feytmans, E. (1997) Comput. Appl. Biosci. 13, 249-256. [DOI] [PubMed] [Google Scholar]
- 9.Morgenstern, B. (1999) Bioinformatics 15, 211-218. [DOI] [PubMed] [Google Scholar]
- 10.Lawrence, C. E., Altschul, S. F., Boguski, M. S., Liu, J. S., Neuwald, A. F. & Wootton, J. C. (1993) Science 262, 208-214. [DOI] [PubMed] [Google Scholar]
- 11.Bailey, T. L. & Elkan, C. (1994) in Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology, eds. Altman, R., Brutlag, D., Karp, P., Lathrop, R. & Searls, D. (American Association for Artificial Intelligence, Menlo Park, CA), pp. 28-36.
- 12.Brudno, M., Do, C. B., Cooper, G. M., Kim, M. F., Davydov, E., Green, E. D., Sidow, A. & Batzoglou, S. (2003) Genome Res. 13, 721-731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bray, N. & Pachter, L. (2003) Nucleic Acids Res. 31, 3525-3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blanchette, M., Kent, W. J., Riemer, C., Elnitski, L., Smit, A. F. A., Roskin, K. M., Baertsch, R., Rosenbloom, K., Clawson, H., Green, E. D., et al. (2004) Genome Res. 14, 708-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurtz, S. & Schleiermacher, C. (1999) Bioinformatics 15, 426-427. [DOI] [PubMed] [Google Scholar]
- 16.Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J. & Giegerich, R. (2001) Nucleic Acids Res. 29, 4633-4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li, X. & Waterman, M. S. (2003) Genome Res. 13, 1916-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pevzner, P. A., Tang, H. & Tesler, G. (2004) in Proceedings of the Eighth Annual International Conference on Computational Molecular Biology, eds. Bourne, P. E. & Gusfield, D. (Association for Computing Machinery, New York), pp. 213-222.
- 19.Jurka, J. (2000) Trends Genet. 16, 418-420. [DOI] [PubMed] [Google Scholar]
- 20.Smith, T. F. & Waterman, M. S. (1981) J. Mol. Biol. 147, 195-197. [DOI] [PubMed] [Google Scholar]
- 21.Waterman, M. S. & Eggert, M. (1987) J. Mol. Biol. 197, 723-728. [DOI] [PubMed] [Google Scholar]
- 22.Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990) J. Mol. Biol. 215, 403-410. [DOI] [PubMed] [Google Scholar]
- 23.Waterman, M. S. & Vingron, M. (1994) Proc. Natl. Acad. Sci. USA 91, 4625-4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith, T. F., Waterman, M. S. & Burks, C. (1985) Nucleic Acids Res. 13, 645-656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arratia, R., Gordon, L. & Waterman, M. S. (1986) Ann. Stat. 14, 971-993. [Google Scholar]
- 26.Waterman, M. S., Gordon, L. & Arratia, R. (1987) Proc. Natl. Acad. Sci. USA 84, 1239-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karlin, S. & Altschul, S. F. (1990) Proc. Natl. Acad. Sci. USA 87, 2264-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lippert, R. A., Zhao, X., Florea, L., Mobarry, C. & Istrail, S. (2004) in Proceedings of the Eighth Annual International Conference on Computational Molecular Biology, eds. Bourne, P. E. & Gusfield, D. (Association for Computing Machinery, New York), pp. 233-241.
- 29.Aldous, D. (1989) Probability Approximations via the Poisson Clumping Heuristic (Springer, New York).
- 30.Waterman, M. S. (1995) Introduction to Computational Biology: Maps, Sequences, and Genomes (Chapman and Hall, London), pp. 253-304.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.