

Summary
The retinoblastoma tumor suppressor protein pRb is a master regulator of cellular proliferation, principally through interaction with E2F and regulation of E2F target genes. Here, we describe the H1.2 linker histone as a major pRb interaction partner. We establish that H1.2 and pRb are found in a chromatin-bound complex on diverse E2F target genes. Interrogating the global influence of H1.2 on the genome-wide distribution of pRb indicated that the E2F target genes affected by H1.2 are functionally linked to cell-cycle control, consistent with the ability of H1.2 to hinder cell proliferation and the elevated levels of chromatin-bound H1-pRb complex, which occur in growth-arrested cells. Our results define a network of E2F target genes as susceptible to the regulatory influence of H1.2, where H1.2 augments global association of pRb with chromatin, enhances transcriptional repression by pRb, and facilitates pRb-dependent cell-cycle arrest.
Keywords: linker histone, retinoblastoma protein, E2F, transcription, cell cycle, chromatin
Graphical Abstract
Highlights
-
•
The H1.2 linker histone is a major and significant pRb interaction partner
-
•
H1.2 and pRb reside in a chromatin-bound complex on diverse E2F target genes
-
•
H1.2 influences global chromatin association and genome-wide distribution of pRb
-
•
H1.2 enhances transcriptional repression by pRb and facilitates cell-cycle arrest
Munro et al. demonstrate that pRb interacts with linker histone H1.2. The pRb-H1.2 complex is enriched on the chromatin of a subset of E2F target genes associated with cell-cycle control. Moreover, H1.2 influences the genome-wide chromatin-binding properties of pRb and impacts transcriptional repression and cell-cycle control.
Introduction
The replication-dependent linker H1 histones are generally believed to be involved in repressing gene expression through compacting chromatin into higher order structures (Misteli et al., 2000). There are seven somatic H1 subtypes in human cells (H1.1 to H1.5, H1.0, and H1X) that exhibit considerable sequence divergence in the tail regions (Harshman et al., 2013). Because of the differences in intracellular localization and levels between cell types, it has been speculated that H1 subtypes take on different functional roles in addition to the established general effects on chromatin compaction (Biterge and Schneider, 2014). It has been reported that H1.2 associates with a stable protein complex that influences p53 activity (Kim et al., 2012), and murine H1B (equivalent to human H1.2) interacts with the homeobox protein MSX1 to prevent activation of the MYOD gene, thereby delaying myoblast differentiation (Lee et al., 2004), suggestive of gene-specific regulatory effects.
Despite the generally held view that the principal role of H1 histone is to dampen transcription and maintain transcriptional inactivity, some studies have highlighted a role in transcriptional activation (Clausell et al., 2009, Kim et al., 2013). For example, H1-containing chromatin is remodeled by SWI/SNF (switch/sucrose non-fermentable) complexes (Clausell et al., 2009), and H1.2 stably interacts with CUL4A and PAF1 to generate active chromatin (Kim et al., 2013). Thus, it appears that H1 histones are generally dedicated to repressive roles in gene expression, although transcription-factor-specific roles are likely.
The pRb tumor suppressor protein acts as an important gatekeeper in regulating cell-cycle transition through G1 into S phase, and mutation in the Rb gene represents one of the most frequent events in human cancer, contributing to cancer initiation and progression (Munro et al., 2012). Mechanistically, pRb is a transcriptional regulator with its principal target being the E2F family of transcription factors. The E2F family regulates various target genes involved with cell-cycle progression and diverse cell fates, which thereby allows pRb to influence numerous aspects of cell biology.
In this study, we uncover a role for the H1.2 linker histone in directing the genome-wide association of pRb with chromatin. We have found that H1.2 interacts with pRb and thereby facilitates pRb binding near E2F target genes. Our results suggest a selective role for histone H1.2, mediated through modulating the chromatin-binding properties of pRb, which, in turn, allows H1.2 to exert global effects on the E2F gene network and thereby influence cell-cycle control.
Results
Linker H1 Histones in the pRb Interactome
We generated Tet-On stable cell lines that, upon induction, expressed FLAG-pRb 1-379, 379-928, or wild-type (WT) 1-928 (Figure 1A, i and ii). All three pRb derivatives displayed a nuclear localization in induced cells (Figure S1A, i), and caused growth suppression in cell-proliferation assays, with fewer cells evident in the pRb-induced compared to the induced control cell line (Figure 1A, iii). The WT, 379-928, and 1-379 cells each exhibited varying levels of growth inhibition, with WT pRb exhibiting the most significant level (Figure 1A, iii). Notably, pRb 1-379 was also quite active, compared to the control cell line in a colony formation assay (Figure S1B, i and ii).
Figure 1.

pRb Interacts with Linker Histone H1.2
(A) (i) Diagram of pRb with the A and B domains of the pocket shown in blue. The N- and C-terminal regions are also indicated. (ii) U2OS Tet-On-inducible cells expressing pTRE2 control vector, FLAG-pRb 1-379, FLAG-pRb 379-928, and FLAG pRb 1-928 were grown with (+) or without (-) doxycycline (1 μg/mL) for 48 hr. Cell lysates were prepared and immunoblotted with FLAG and actin antibodies. (iii) U2OS Tet-On-inducible cells expressing FLAG-pRb 1-379, FLAG-pRb 379-928, and FLAG pRb 1-928 or pTRE2 empty vector were seeded at a density of 1 × 104. Cell counts were performed at 3 and 5 days (d). Graph indicates average ± SD (n = 3; ∗∗p < 0.01, Student’s t test).
(B) In (i), FLAG-pRb 1-379-inducible cells were grown with (+) or without (-) doxycycline for 48 hr. FLAG immunoprecipitation (IP) was performed followed by elution with FLAG peptide. The eluted protein complexes were subjected to silver staining, and bands of notable difference (indicated by arrows) were excised and subjected to tryptic digestion and nano-liquid chromatography-tandem mass spectrometry (nano-LC-MS/MS). Mol wt, molecular weight. (ii) Chromatin was isolated from FLAG-pRb (1-379) or pTRE2 control cells (both grown in the presence of doxycycline). FLAG immunoprecipitations were performed, followed by elution with FLAG peptide. 20% of each eluted immunocomplexes was analyzed by electrophoresis, and silver staining of the gel was performed. The remaining 80% of the eluates were subjected to in-solution tryptic digestion, and proteins were identified using nano-LC-MS/MS. Comparison between proteins immunoprecipitated from the control vector cell line and pRb 1-379 allowed the identification of proteins that selectively bound to pRb 1-379. (iii) Proteins were identified as pRb 1-379-interacting proteins by mass spectrometry in pRb-associated chromatin, and whole-cell extracts are listed in tabular form, together with Mascot scores. Previously reported interacting proteins are highlighted in red; for example, nucleophosmin, MCM7, and nucleolin (Grinstein et al., 2006, Sterner et al., 1998, Takemura et al., 2002).
(C) In (i), FLAG-pRb 1-379-inducible cells were grown in the presence of doxycycline for 48 hr. Cell lysates were immunoprecipitated with anti-FLAG, followed by immunoblotting with H1.2, H1.4, or FLAG antibodies. (ii) U2OS cells were transfected with HA-pRb 1-379 or control vector. 48 hr post-transfection, cell lysates were prepared, and HA-pRb was then immunoprecipitated, followed by immunoblotting with antibodies against H1.2, H1.4, or HA. (iii) U2OS cells were transfected with FLAG H1.1, FLAG H1.2, or control vector. 48 hr post-transfection, lysates were immunoprecipitated with anti-FLAG, followed by immunoblotting with pRb and FLAG antibodies.
(D) In (i), U2OS cell lysates were immunoprecipitated with non-specific (NS) or pRb antibodies, followed by immunoblotting with H1.2 and pRb antibodies (note that the blot has been spliced between input and IP, as different exposures were required to show input and IP at appropriate levels). (ii) MCF-7 cell lysates were immunoprecipitated with non-specific (NS) or pRb antibodies, followed by immunoblotting with H1.2 and pRb antibodies.
(E) GST-H1.2, GST-H1.4, or GST were incubated in the presence of bacterially expressed His-pRb (1-379) together with Glutathione Sepharose beads. Following extensive washes, immunoblotting was performed with GST and His antibodies.
(F) U2OS cells were transfected with the indicated HA-tagged pRb N-terminal truncations. 48 hr post-transfection, lysates were immunoprecipitated with anti-HA, followed by immunoblotting with H1.2 and HA antibodies.
Given that the ability of pRb to inhibit cell proliferation is principally attributed to its pocket region (379-928), we were intrigued that the N-terminal domain could also suppress cell proliferation, and because the N-terminal domain is poorly characterized, we next used mass spectrometry to identify pRb-interacting proteins, focusing on the pRb 1-379 region. A number of proteins were identified that co-purified with FLAG-pRb 1-379 but not the empty vector control (Figure 1B, iii). We were intrigued by the presence of the linker H1 histone family, including H1.2 and H1.4 (Figure 1B, i and iii), which prompted us to further explore the role of H1. For this, chromatin bound to pRb was purified from FLAG-pRb 1-379-inducible cells, where approximately 5% of the total cellular pRb remained chromatin bound (Figure S1C). Mass spectrometry of the pRb-associated chromatin similarly revealed the presence of H1 histones, including H1.4, H1.2, H1.1, H1.0, and H1x, together with the core histones H2B and H4 (Figure 1B, iii).
We verified that the interaction between pRb and H1 histones occurred in cells, focusing on subtypes H1.2 and H1.4 (we were restricted to investigating these two H1 subtypes due a lack of other suitable subtype-specific antibodies) by immunoprecipitation of pRb from both FLAG-pRb-inducible cells and cells transfected with HA (hemagglutinin)-pRb 1-379, where an interaction was evident (Figure 1C, i and ii). Additionally, endogenous pRb was found to immunoprecipitate with ectopic H1 subtypes (Figure 1C, iii), and an interaction between endogenous pRb and H1.2 was evident in a number of cell types (Figures 1D, i and ii, and S1D). However, because pRb and H1 histones are chromatin associated, it was necessary to rule out that the interaction was mediated indirectly through a separate association of each protein with DNA. We pre-incubated cell lysates with DNase to digest any DNA prior to immunoprecipitation, which actually enhanced the interaction between pRb and H1.2, therefore suggesting that the interaction is not bridged by DNA (Figure S1E). Furthermore, we expressed and purified recombinant GST (glutathione S-transferase)-H1.2, GST-H1.4, and His-pRb 1-379 and performed in vitro GST and Ni-NTA binding assays, where His-pRb bound efficiently to GST-H1.2 or GST-H1.4, but not to GST alone (Figures 1E and S1F), suggesting a specific and direct interaction between pRb and histone H1 subtypes H1.2 and H1.4. We also studied which region of pRb interacts with H1.2. Truncations of HA-tagged pRb were expressed, and the ability to associate with endogenous H1.2 was assessed. While no binding of H1.2 to pRb 1-100 was evident, we observed that H1.2 could associate with other derivatives of pRb (including the low-penetrant point mutant R661W) (Figure S1G). It is consistent with a role for the N-terminal region of pRb for H1.2 binding that deleting the first 126 amino-acid residues of pRb in the context of the full-length protein prevented the interaction with H1.2 (Figure 1F); nuclear accumulation of pRb 126-928 occurred as expected (Figure S1H). Thus, the N-terminal region of pRb is responsible for the interaction with H1.2.
H1 Histones Associate with the Promoters of E2F Target Genes
Because pRb binds to E2F, we next examined whether H1 histones are present in the chromatin environment of E2F target genes. FLAG-tagged H1 subtypes were expressed in U2OS cells, and chromatin immunoprecipitation (ChIP) analysis was performed (Figure S2A, i). All of the H1 subtypes examined were detected in the region of the E2F-binding site on a number of target genes (Figure S2A, ii). We then addressed whether pRb and H1 coexist in a chromatin-bound complex, which we tested by performing sequential ChIP analyses. We detected endogenous pRb and H1.2 as a chromatin-bound complex on E2F target genes in diverse cell types (Figures 2A, i and ii, and S2B). Further, depletion of H1.2 resulted in a reduction in the amount of chromatin-bound pRb on a variety of E2F target genes (Figure 2B). Significantly, the expression of the E2F target genes increased upon H1.2 depletion (Figure 2C). Moreover, H1.2 expression augmented the E2F1 transcriptional repression mediated by pRb in reporter-based assays (Figure S2C). Overall, pRb and H1.2 co-exist in the chromatin environment of E2F target genes, where H1.2 contributes to transcriptional inactivation.
Figure 2.

H1.2 Regulates Chromatin-Bound pRb and E2F Target Gene Expression
(A) In (i), chromatin from T98G cells was immunoprecipitated with control immunoglobulin G (IgG) or pRb antibodies. A sequential re-immunoprecipitation (re-IP) was performed with eluted IgG and pRb material with control IgG or H1.2 antibodies. The presence of the H1/pRb complex on the DHFR promoter was analyzed by qPCR. Graphs indicate average ± SD (n = 3; ∗∗p < 0.01, Student’s t test). (ii) Chromatin from MCF7 cells was immunoprecipitated with control IgG, pRb, or H1.2 antibodies. A sequential re-IP was performed with eluted pRb material with control IgG, H1.2, or E2F-1 antibodies. The binding of the complex to the CDC6 promoter was analyzed by PCR. E2F-1 was included as a positive control for the pRb ChIP-reIP. Quantification of the ChIP signal is shown in graphical format below.
(B) MCF7 cells were transfected with GFP or H1.2 siRNA (20 nM) for 72 hr. ChIP was performed with control IgG and pRb antibodies. ChIP activity on E2F promoters was assessed by qPCR. Corresponding immunoblot is shown on the right. ∗p < 0.05; ∗∗p < 0.01, Student’s t test. Graph indicates mean ± SEM (n = 5).
(C) U2OS cells were transfected with GFP or H1.2 siRNA (20 nM) for 72 hr. E2F target gene RNA levels were assessed by qPCR. Transcript levels were normalized to housekeeping gene GAPDH. Corresponding immunoblot is shown on the right. Graph indicates average ± SD (n = 3; ∗p < 0.05; ∗∗p < 0.01, Student’s t test).
(D) Venn diagram showing the overlap of pRb peaks between treatment conditions; namely, siGFP control conditions (+ H1.2) and H1.2 knockdown (−H1.2). The siGFP treatment (red) yielded 1,650 pRb peaks, and the siH1.2 treatment (blue) yielded 708 peaks. There were 670 shared peaks (purple) between the two conditions, and 980 and 38 peaks unique to each condition, respectively.
(E) Motif analysis of the most enriched de novo peak identified in the siGFP and siH1.2 condition and among peaks unique to the siGFP condition.
In order to clarify whether H1 histones have a global effect on pRb, we performed a genome-wide chromatin-binding analysis by ChIP sequencing (ChIP-seq). We used MCF7 cells, which express WT pRb, and studied the genome-wide distribution of the pRb ChIP complex, as well as the impact that H1.2 has on the genomic distribution of pRb. Initially, we confirmed that the ChIP enrichment was specific to pRb by generating MCF7 CRISPR cells that lack pRb; there was no detectable pRb signal apparent upon the E2F1 promoter in pRb−/− cells compared to their WT counterparts (Figure S2D). Endogenous pRb was immunoprecipitated from MCF7 cells that had been treated with either a control or an H1.2 small interfering (si)RNA (siRNA). Chromatin was isolated, libraries prepared, and the DNA was subjected to deep sequencing. We identified 1,650 and 708 specific peaks in the control siRNA and the H1.2 siRNA, respectively, with an overlap of 670 peaks (Figure 2D). As anticipated, a very strong enrichment of the E2F binding-site motif was identified in all conditions (Figure 2E). Minor binding-site differences were apparent when the “siGFP-only” condition was analyzed, the significance of which has yet to be explored.
Aligning the peaks across the human genome revealed that the majority of the pRb peaks mapped to promoter and intergenic regions (Figures S3A and S3B). The number of pRb peaks observed at promoter regions decreased upon H1.2 depletion, which was accompanied by an increased association of pRb at intergenic regions (Figures S3A and S3B). Moreover, this was consistent with H1.2 depletion, which caused reduced pRb enrichment at transcription start sites (Figures 3A and 3B, i and iii). Importantly, there was a significant reduction in the association of pRb with E2F target genes in the absence of H1.2 (from 1,056 to 283 peaks, which represents a 73% decrease; Figures 3B, ii, and 3C), contrasting with the effect on non-E2F regions, where the absence of H1.2 caused a proportional increase in the binding of pRb (Figure 3C); in the absence of H1.2, pRb binding to non-E2F genomic regions increased from 36% to 60%. Moreover, for certain E2F target genes, the level of pRb binding was reduced in the absence of H1.2—for example, E2F1, E2F2, CCNA2, CCNE2, EIF2S1, and BRCA2 (Figure 3D, i)—whereas for others such as PRIM1, CDT1, and MCM4, binding was minimally affected (Figure 3D, ii). Overall, these results indicate that H1.2 influences the ability of pRb to associate with the promoter regions of E2F target genes.
Figure 3.

H1.2 Regulates the Genome-wide Association of pRb with Chromatin
(A) Heatmap analysis of pRb binding around transcription start sites (TSSs) under siH1.2 or siGFP treatment. Each panel represents 2,000 bp upstream and 2,000 bp downstream of the transcription start site.
(B) Normalized coverage plots of either siGFP (red) or siH1.2 (blue) treatment around the transcription start site (i), around all E2F-binding motifs identified computationally (ii), or around all peaks from the experiment (iii).
(C) Total number of pRb peaks for siH1.2 (708) and siGFP (1,650) treatment, and peaks unique to siGFP (970) condition. Peaks corresponding to the E2F-binding motif (blue) or not (green) are indicated.
(D) Coverage tracks around E2F target gene promoters that show a decrease in peak size with siH1.2 treatment (E2F1, E2F2, EIF2S1, CCNA2, CCNE2, and BRCA1) (i) or no significant change (PRIM1, CDT1, and MCM4) (ii).
(E) GO analysis of pRb association with promoters in the presence and absence of H1.2. Comparison of pRb binding to promoter regions common to both conditions (siGFP and siH1.2) and promoter regions unique to siGFP treatment alone. Analysis was performed using HOMER (v.4.8).
Cyclin/Cdk Activity Regulates the H1-pRb Interaction
Gene Ontology (GO) analysis revealed that the most significant group of pRb target genes affected by the loss of H1.2 was connected with the cell cycle (Figure 3E). Therefore, we surmised that the chromatin-bound H1-pRb interaction could be influenced by the cell cycle, which we examined in cells that had been growth arrested by either serum starvation or treatment with CDK inhibitors. We performed a sequential ChIP analysis (anti-pRb followed by anti-H1.2 or anti-pan H1 antibody) where, in serum-starved MCF7 cells, an increased level of the chromatin-associated H1-pRb complex on the CDC6 promoter was evident, compared to asynchronous cultures of growing cells (Figures 4A and S4A, i) (as a negative control, ChIP-binding activity to the actin promoter was assessed; Figure S4A, ii and iii). Further, in MCF7 cells growth arrested by treatment with the CDK inhibitor roscovitine (Meijer et al., 1997), the H1-pRb complex was more evident in CDK-inhibitor-treated cells (Figures 4B and S4B). These results indicate that the chromatin-bound H1.2-pRb complex is influenced by cell-cycle progression, with its appearance enhanced in growth-arrested cells. This is compatible with the biological role of pRb, which is principally exerted at the G1-to-S phase transition (Munro et al., 2012).
Figure 4.

H1.2 Facilitates the Growth Regulatory Properties of pRb
(A) MCF7 cells were grown in normal growth conditions or under conditions of serum starvation for 72 hr. Chromatin extracts were immunoprecipitated with control IgG (IgG) or pRb antibodies. ChIP re-IP was performed with the eluted IgG and pRb material with control IgG, H1.2, or H1 antibodies. Binding of the complex to the CDC6 promoter was analyzed by qPCR.
(B) MCF7 cells were treated with roscovitine (20 μM) for 16 hr or were untreated. Chromatin extracts were immunoprecipitated with control IgG or pRb antibodies. ChIP re-IP was performed with the eluted pRb material with control IgG, H1.2, or H1 antibodies. Binding of the complex to the CDC6 promoter was analyzed by qPCR.
(C) In (i), pRb ChIPs in U2OS WT (WT) and U2OS H1.2−/− CRISPR cell lines. ChIP analysis was performed with either control IgG or pRb antibodies. Binding to the indicated E2F promoters was assessed by qPCR. (ii) U2OS WT and U2OS H1.2−/− CRISPR cell lines were transfected with expression vectors encoding E2F-1 and CDC6-luciferase for 48 hr, and pCMV-βGal was included to monitor transfection efficiency. Relative luciferase activity (luciferase units per unit of βGal) is shown.
(D) U2OS WT and U2OS H1.2−/− cells were seeded in triplicate. Cell counts were performed at 3, 5, and 7 days post-seeding. Corresponding immunoblots are shown on the right.
(E) U2OS WT, U2OS pRb−/−, U2OS H1.2−/−, and double-knockout U2OS pRb−/−/H1.2−/− cells were seeded at a density of 2 × 104 cells in triplicate. Cell counts were performed 3, 5, and 7 days post-seeding. Corresponding immunoblots for each cell line are shown below.
(F) U2OS WT and U2OS H1.2−/− cells were transfected with E2F1 siRNA (20 nM) or control siRNA (20 nM). 24 hr later, cells were trypsinized, counted, and seeded at a density of 1 × 104 cells in triplicate. Cell counts were performed 3, 5, and 7 days post-seeding. Corresponding immunoblots are shown below.
(G) In (i), SAOS2 WT and SAOS2 H1.2−/− CRISPR cell lines were transfected with either control plasmid or HA-pRb. 48 hr later, cells were harvested for flow-cytometry analysis. Graph shows the proportion of cells in G1 phase of the cell cycle. (ii) Corresponding immunoblots for (i).
(H) SAOS2 WT and SAOS2 H1.2−/− CRISPR cell lines were transfected with either control plasmid, HA-pRb, or HA-pRb (126-928). 48 hr later, cells were harvested for flow cytometry analysis. Graph shows the fold change in cells in G1 phase of the cell cycle.
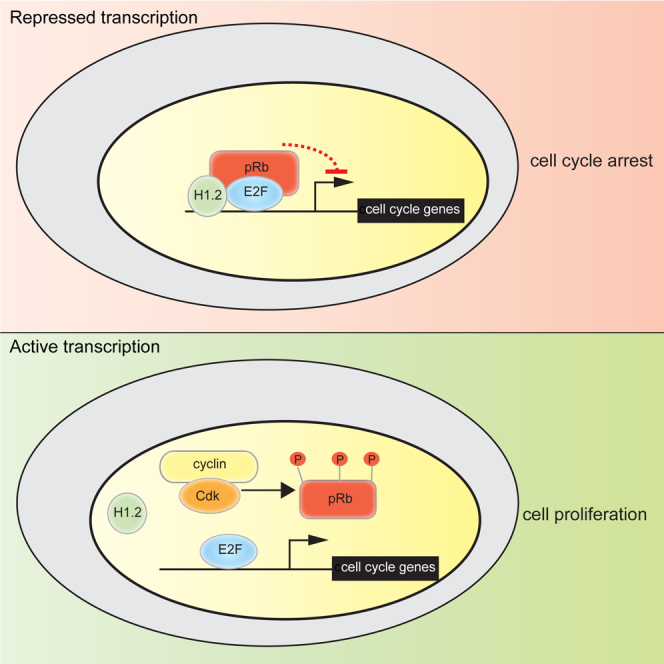
(I) Model depicting the relationship between pRb and histone H1.2. H1.2 associates with pRb at E2F-regulated promoters and augments the ability of pRb to silence transcription, potentially resulting in scenarios such as cell-cycle arrest, differentiation, or senescence. Under conditions favorable to cell growth, pRb is phosphorylated by cyclin-CDK, resulting in the dissociation of the pRb-H1.2 complex from chromatin and active transcription of cell-cycle-associated genes by E2F and cell-cycle progression.
Data in (A)–(H) indicate average ± SD (n = 3). ∗p < 0.05; ∗∗p < 0.01, Student’s t test.
H1 Histones Influence Cell Growth
Since the ability of pRb to control cell proliferation requires pRb-dependent regulation of E2F activity (Frolov and Dyson, 2004), and because the transcription properties of pRb are influenced by the interaction with H1.2, we reasoned that H1.2 may impact on cell growth. Therefore, we developed CRISPR cell lines, derived from U2OS (expressing WT pRb), MCF7 (expressing WT pRb), and SAOS2 (expressing MT pRb) cells, in which we disrupted the endogenous H1.2 gene; immunoblotting and immunostaining confirmed that H1.2 protein was undetectable (Figures S4C–S4E). An analysis of chromatin-associated pRb by ChIP analysis confirmed the earlier results (Figure 2C) that H1.2 augments pRb binding, as pRb ChIP activity was diminished in the H1.2−/− cells, compared to their WT counterparts (Figure 4C, i). Concomitant with decreased pRb binding, the transcriptional activity of the E2F target gene, CDC6, was enhanced in the H1.2−/− cells (Figure 4C, ii).
It was necessary to rule out the possibility that H1.2 depletion affected the association of any transcription factor or chromatin-associated protein with DNA and to confirm that the observed effects were specific to pRb binding at localized regions. To this end, ChIP analysis of SP1 and RNA polymerase II (PolII; POLR2A) binding was performed in H1.2−/− cells; significantly, there was no effect upon H1.2 depletion on the association of either SP1 or PolII with chromatin (Figure S4F, i and ii). We additionally performed ChIP analysis of p53 binding to p53 target genes (where significant levels of H1 were present; Figure S4G, i) and found that H1.2 depletion did not affect p53 association with chromatin (Figure S4G, ii), thus confirming the specificity of the effect of H1.2 on pRb.
An analysis of the growth properties of the H1.2−/− U2OS and H1.2−/− MCF7 cells indicated that the H1.2−/− cells grow faster than their WT counterparts (at 5 and 7 days), reflecting a shorter doubling time of the H1.2−/− cells (U2OS cells: 23.8 hr compared to 27.6 hr; Figures 4D and S4H). As anticipated, Rb−/− cells also grew faster than WT cells (Figure 4E). Moreover, a similar level of increased proliferation was observed in cells that lack H1.2, or both H1.2 and pRb (Figure 4E), which is consistent with pRb and H1.2 acting through a shared mechanism and implying that H1.2 is functionally involved in pRb-dependent growth control. Further, the increased growth rate of H1.2−/− cells was dependent on E2F1 activity, because the increase in growth rate was no longer evident in H1.2−/− cells with depleted E2F1 levels (Figure 4F).
We then used SAOS2 cells, which undergo G1 arrest upon the expression of ectopic WT pRb (Li et al., 1995). The level of G1 cells apparent upon WT pRb expression was compromised in H1.2−/− SAOS2 cells, compared to H1.2-expressing cells (Figures 4G, 4H, and S4I), thus establishing a role for H1.2 in pRb-dependent growth control. Furthermore, expression of pRb (126-928), which is unable to bind to H1.2 (Figure 1F), demonstrated a diminished ability to elicit G1 arrest, compared to WT pRb in H1.2-expressing cells (Figure 4H), thus supporting the hypothesis that the H1.2 interaction is important for pRb-dependent growth control. In sum, these results suggest that the linker histone, H1.2, is functionally important in mediating the growth-regulating effects of pRb.
Discussion
H1 histones have traditionally been regarded as widespread, if not general, repressors of global transcription, mediated through their ability to compact chromatin (Misteli et al., 2000). There are, however, an increasing number of reports that suggest that H1 histones exert gene-specific effects, which can be either positive or negative in how they influence gene expression (Biterge and Schneider, 2014).
We identified linker histones as interaction partners for pRb. The interaction between H1 histone and pRb occurs on chromatin and augments the binding of pRb to E2F target gene promoters. We focused on the role of H1.2, which, by genome-wide analysis of the chromatin-binding properties of pRb by ChIP-seq, highlighted a requirement for H1.2 for the efficient recruitment of pRb to the global network of E2F target genes, with genes involved in cell-cycle progression being particularly sensitive to the influence of H1.2. We propose, therefore, that the H1.2-pRb interaction facilitates the regulation, at a global level, of the E2F target gene network. The enhanced chromatin association, which occurs in arrested cells, is consistent with a model in which H1.2 augments transcriptional repression by pRb and thereby assists cell-cycle arrest (Figure 4I).
In conclusion, our results advance our understanding of the biological role of H1 histones by describing a new interaction with pRb, which enables H1.2 to influence the E2F pathway and the expression of downstream target genes and, consequently, impact cellular proliferation. Our study supports the idea that H1 histones, while able to mediate general repressive effects on transcription by facilitating chromatin compaction, are, in addition, endowed with selective interaction partners, such as pRb, which enables them to preferentially target and regulate key gene networks, like the extensive network controlled by the pRb-E2F pathway.
Experimental Procedures
Expanded details of methods are listed in the Supplemental Experimental Procedures.
Cell Culture and Transfection
U2OS, HeLa, MCF7, T98G, and SAOS2 cells were cultured in DMEM (GIBCO) supplemented with 5% fetal bovine serum (FBS) and penicillin-streptomycin (GIBCO) at 37°C in 5% CO2. Cell lines were transfected with GeneJuice (Novagen). Transfections included pCMV-βGal (β-galactosidase) as an internal control to normalize transfection efficiency. For siRNA-knockdown experiments, cells were transfected with 20 nM siRNA using Oligofectamine Transfection Reagent (Invitrogen). siRNA sequences are available upon request.
Antibodies
The following antibodies were used: anti-FLAG peptide monoclonal antibody M2 (Sigma), anti-FLAG peptide monoclonal antibody M2-coupled agarose beads (Sigma), anti-HA11 monoclonal antibody (Covance), E2F-1 (C20 and KH95, Santa Cruz Biotechnology), pRb (4H1, Cell Signaling Technology), G3-245 (Becton Dickinson) and IF8 (Santa Cruz), GAPDH (V18, Santa Cruz) and β-Actin (Sigma), anti-H1.2 (ab17677 and ab4086, Abcam), anti H1.4 (from Millipore), anti-H1 (sc-8030, Santa Cruz), SP1 (sc59, Santa Cruz), and RNA PolII (sc55492, Santa Cruz).
Author Contributions
S.M. designed and performed the experiments, analyzed the data, and wrote the paper. M.F. and S.M.C. performed additional experiments. E.S.H. and U.O. performed and analyzed the ChIP-seq experiment. R.K. and B.M.K. performed the mass spectrometry sample analysis. N.B.L.T. directed the research and wrote the paper.
Acknowledgments
This work was supported by Cancer Research UK Programme Award 300/A13058 and a Medical Research Council (MRC) grant (to N.B.L.T.). U.O. is supported by Arthritis Research UK program grant 20522 and Rosetrees Trust.
Published: June 13, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.05.053.
Accession Numbers
The accession number for the sequencing data reported in this paper is GEO: GSE98728.
Supplemental Information
References
- Biterge B., Schneider R. Histone variants: key players of chromatin. Cell Tissue Res. 2014;356:457–466. doi: 10.1007/s00441-014-1862-4. [DOI] [PubMed] [Google Scholar]
- Clausell J., Happel N., Hale T.K., Doenecke D., Beato M. Histone H1 subtypes differentially modulate chromatin condensation without preventing ATP-dependent remodeling by SWI/SNF or NURF. PLoS ONE. 2009;4:e0007243. doi: 10.1371/journal.pone.0007243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolov M.V., Dyson N.J. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J. Cell Sci. 2004;117:2173–2181. doi: 10.1242/jcs.01227. [DOI] [PubMed] [Google Scholar]
- Grinstein E., Shan Y., Karawajew L., Snijders P.J., Meijer C.J., Royer H.D., Wernet P. Cell cycle-controlled interaction of nucleolin with the retinoblastoma protein and cancerous cell transformation. J. Biol. Chem. 2006;281:22223–22235. doi: 10.1074/jbc.M513335200. [DOI] [PubMed] [Google Scholar]
- Harshman S.W., Young N.L., Parthun M.R., Freitas M.A. H1 histones: current perspectives and challenges. Nucleic Acids Res. 2013;41:9593–9609. doi: 10.1093/nar/gkt700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K., Jeong K.W., Kim H., Choi J., Lu W., Stallcup M.R., An W. Functional interplay between p53 acetylation and H1.2 phosphorylation in p53-regulated transcription. Oncogene. 2012;31:4290–4301. doi: 10.1038/onc.2011.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K., Lee B., Kim J., Choi J., Kim J.M., Xiong Y., Roeder R.G., An W. Linker Histone H1.2 cooperates with Cul4A and PAF1 to drive H4K31 ubiquitylation-mediated transactivation. Cell Rep. 2013;5:1690–1703. doi: 10.1016/j.celrep.2013.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H., Habas R., Abate-Shen C. MSX1 cooperates with histone H1b for inhibition of transcription and myogenesis. Science. 2004;304:1675–1678. doi: 10.1126/science.1098096. [DOI] [PubMed] [Google Scholar]
- Li W., Fan J., Hochhauser D., Banerjee D., Zielinski Z., Almasan A., Yin Y., Kelly R., Wahl G.M., Bertino J.R. Lack of functional retinoblastoma protein mediates increased resistance to antimetabolites in human sarcoma cell lines. Proc. Natl. Acad. Sci. USA. 1995;92:10436–10440. doi: 10.1073/pnas.92.22.10436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L., Borgne A., Mulner O., Chong J.P., Blow J.J., Inagaki N., Inagaki M., Delcros J.G., Moulinoux J.P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- Misteli T., Gunjan A., Hock R., Bustin M., Brown D.T. Dynamic binding of histone H1 to chromatin in living cells. Nature. 2000;408:877–881. doi: 10.1038/35048610. [DOI] [PubMed] [Google Scholar]
- Munro S., Carr S.M., La Thangue N.B. Diversity within the pRb pathway: is there a code of conduct? Oncogene. 2012;31:4343–4352. doi: 10.1038/onc.2011.603. [DOI] [PubMed] [Google Scholar]
- Sterner J.M., Dew-Knight S., Musahl C., Kornbluth S., Horowitz J.M. Negative regulation of DNA replication by the retinoblastoma protein is mediated by its association with MCM7. Mol. Cell. Biol. 1998;18:2748–2757. doi: 10.1128/mcb.18.5.2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura M., Ohoka F., Perpelescu M., Ogawa M., Matsushita H., Takaba T., Akiyama T., Umekawa H., Furuichi Y., Cook P.R., Yoshida S. Phosphorylation-dependent migration of retinoblastoma protein into the nucleolus triggered by binding to nucleophosmin/B23. Exp. Cell Res. 2002;276:233–241. doi: 10.1006/excr.2002.5523. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.