

Abstract
Energy-dependent proteases, such as ClpXP, are responsible for the regulated destruction of proteins in all cells. AAA+ ATPases in these proteases bind protein substrates and power their mechanical denaturation and subsequent translocation into a secluded degradation chamber where polypeptide cleavage occurs. Here, we show that model unfolded substrates are engaged rapidly by ClpXP and are then spooled into the degradation chamber at a rate proportional to their length. Degradation and competition studies indicate that ClpXP initially binds native and unfolded substrates similarly. However, stable native substrates then partition between frequent release and infrequent denaturation, with only the latter step resulting in committed degradation. During degradation of a fusion protein with three tandem native domains, partially degraded species with one and two intact domains accumulated. These processed proteins were not bound to the enzyme, showing that release can occur even after translocation and degradation of a substrate have commenced. The release of stable substrates and committed engagement of denatured or unstable native molecules ensures that ClpXP degrades less stable substrates in a population preferentially. This mechanism prevents trapping of the enzyme in futile degradation attempts and ensures that the energy of ATP hydrolysis is used efficiently for protein degradation.
Keywords: ClpP, ClpX, energy-dependent proteolysis, protein unfolding, titin I27 domain
Many cellular processes are powered by molecular machines, which convert energy from ATP into mechanical work. The AAA+ superfamily of ATPases represent an important class of these machines, functioning in vesicle fusion, cargo transport, DNA and RNA unwinding, remodeling of the cytoskeleton, DNA replication, transposition, and targeted protein degradation (1). In known energy-dependent proteases, hexameric AAA+ ATPase rings stack against a barrel-shaped peptidase, aligning the central pore of the ATPase with a narrow axial portal of the peptidase. The ATPase ring serves as the control and command center for the proteolytic machine. It binds recognition elements in target proteins, denatures native protein substrates, and translocates the unfolded polypeptide into the proteolytic chamber of the peptidase for degradation (see ref. 2 for review).
We have been interested in understanding the coordination of substrate binding, denaturation, and translocation by the ClpXP protease of Escherichia coli (3-6). Most substrates for this ATP-dependent protease have unstructured recognition sequences or degradation tags at their N or C terminus (7). For example, adding an ssrA tag to the C terminus of a protein during tmRNA-mediated ribosome rescue makes it a substrate for ClpXP and other E. coli proteases (8-10). The ssrA tag appears to bind to the central pore of the ClpX6 ATPase (11), where it serves as a grip or handle that allows the enzyme to apply an unfolding force to the native substrate. Peptide-bond cleavage and product release appear to be fast steps in degradation, whereas denaturation and/or translocation are the slow steps for most native substrates (3-6, 12, 13). After unfolding by ClpXP or the related ClpAP protease, the ssrA tag is the first part of the substrate to enter the protease, suggesting that translocation proceeds in a C- to N-terminal direction (14, 15). The mechanism by which ClpXP or related enzymes denature substrate proteins is not known, but the enzyme may generate an unfolding force by beginning translocation of the degradation tag and attempting to pull the attached native protein into the pore, which is too small for folded proteins to enter (for discussion, see ref. 2).
Studies of ssrA-tagged variants of the I27 domain of the muscle protein titin have been extremely useful in probing individual steps in the processing of protein substrates by ClpXP (5). The ssrA tag of the otherwise wild-type titin substrate is attached to a β-sheet with immense mechanical stability (16), and we have shown that proteolysis rates and ATP utilization during ClpXP degradation can vary dramatically when this β-sheet is destabilized by mutation (5). Importantly, chemical modification of cysteines in the hydrophobic core of titin results in global unfolding, and ClpXP degradation of carboxymethylated (CM) substrates can be used to assess the contribution of steps other than denaturation (5). During degradation of a single 121-residue molecule of ssrA-tagged titin, ClpXP hydrolyzes ≈100 molecules of ATP for translocation and uses an additional 500-600 ATPs for denaturation. The latter result suggests that application of an unfolding force by the enzyme must, on average, be repeated many times to ensure substrate denaturation. Whether the substrate remains bound to the enzyme between successive denaturation attempts or is released and rebound is unknown.
Here, we show that the rate of ClpXP degradation of ssrA-tagged proteins containing one to three unfolded titin domains is linearly dependent on substrate length and that any length-independent steps are very fast. These studies indicate that ClpXP engages denatured titin substrates rapidly and effectively irreversibly. Following this committed step, the rate of translocation determines the overall rate of degradation. By contrast, results from degradation and competition experiments indicate that most molecules of native titin undergo many cycles of binding, force application, release, and rebinding until denaturation occurs. As a result, degradation of native titin is much slower than that of unfolded titin. Although ClpXP degradation is generally highly processive, release of partially degraded species with one or two folded domains and a 35- to 40-residue tail was observed for a substrate composed of three native titin domains. Partitioning between denaturation and release of stable native domains is therefore part of the normal enzymatic cycle of ClpXP and plays an important role in substrate selectivity.
Materials and Methods
Genes containing multiple copies of the titin-I27 domain were constructed by purifying an AvaI restriction fragment encoding the I27 domain from pET AvaI (17), ligating this fragment to itself, and then cloning concatenated fragments into a pET AvaI vector modified by PCR to express proteins with MH-GLG and GLG-H6-AANDENYALAA sequences at the N and C terminus, respectively (LG is encoded by the AvaI site; the underlined sequence is the ssrA tag). Transformants were isolated from E. coli XL1-Blue (New England Biolabs), and plasmids encoding different numbers of titin domains were identified by colony PCR. Using PCR mutagenesis, a FLAG-titin3 construct was generated by modifying the N-terminal sequence to encode MHDYKDDDDK-GLG (FLAG epitope in italics).
Proteins were expressed in E. coli JK10 cells (clpP::cat, Δlon, slyD::kan, λDE3) and purified by Ni++ NTA chromatography. Native FLAG-titin3 and truncated fragments were purified by binding to ANTI-FLAG M2-Agarose (Sigma) in 20 mM Hepes (pH 7.5)/500 mM NaCl and eluting with 100 mM glycine (pH 3.5). A buffer of 500 mM Tris·HCl (pH 8.8)/20 mM DTT was immediately added to raise the pH and prevent or reverse cysteine oxidation, and the purified material was dialyzed into 20 mM sodium phosphate (pH 7.5)/150 mM NaCl. Proteins were radiolabeled with 35S as reported in ref. 9 and were purified like unlabeled protein. The molecular weights of all titin variants were verified by MALDI-TOF MS (MIT Biopolymers Laboratory). Carboxymethylation of titin proteins was performed as reported in ref. 5, except for titin10, which required a 1,000-fold molar excess of iodoacetic acid (18). The structures of titin variants were assayed by CD spectroscopy, temperature melts, and tryptophan fluorescence (5). A H6SP-ssrA-tagged variant of GFP (19), ClpX (20), and ClpP (3) were purified as described.
ClpXP degradation reactions were performed at 30°C in 25 mM Hepes-KOH (pH 7.6)/100 mM KCl/10 mM MgCl2/10% glycerol/2 mM DTT with an ATP regeneration mix (5 mM ATP/0.032 mg/ml creatine kinase/16 mM creatine phosphate). For degradation of 35S-labeled substrates other than FLAG-titin3, reactions contained 0.1 μM ClpX6 and 0.3 μM ClpP14 and were assayed by release of TCA-soluble radioactive peptides (9). For degradation of FLAG-titin3, reactions contained 0.3 μM ClpX6, 0.9 μM ClpP14, and 0.3 μM SspB2 (a gift from G. Hersch, Massachusetts Institute of Technology) and were quenched at different times by addition of SDS-loading buffer and run on 15% SDS gels. Following autoradiography, band intensities were determined by using imagequant (Molecular Dynamics) and normalized for the expected specific activities of each fragment. Subtilisin digestion was performed for 30 min at 30°C by using a 1:1,000 (wt/wt) ratio of enzyme to substrate. Inhibition constants (Ki) were calculated for different competitors from changes in 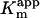 for ClpXP degradation of CM-titin1 by using the equation
for ClpXP degradation of CM-titin1 by using the equation  . Computer simulations were performed by using chemical kinetics simulator 1.01 (IBM).
. Computer simulations were performed by using chemical kinetics simulator 1.01 (IBM).
Results
We constructed and purified fusion proteins containing 1, 2, 3, or 10 tandem titin-I27 domains followed by a His6-tag sequence and a C-terminal ssrA tag. For experiments requiring denatured titin domains, purified proteins were CM by reaction with iodoacetic acid (5). We refer to the native ssrA-tagged fusion molecules as titinX proteins, where X represents the number of repeated titin domains, and to the denatured CM variants as CM-titinX proteins.
Length Dependence of Degradation of Unfolded Titin Substrates. If the rate-limiting step in ClpXP degradation of unfolded titin is translocation (5), then a linear relationship should exist between the degradation rate and the number of titin repeats in a substrate. To test this model, initial rates for ClpXP-degradation of 35S-substrates with one, two, or three unfolded titin domains were measured by release of acid-soluble radioactive peptides. For each substrate, rates determined at different substrate concentrations fit well to a simple Michaelis-Menten kinetic model (Fig. 1A). Km values for these substrates were similar (0.5 ± 0.2 μM) but were roughly 3-fold lower than reported in ref. 5. The His6-tag sequence adjacent to the ssrA tag in the substrates used here, which was absent in the original single-domain substrate, probably accounts for this difference. Maximal turnover rates (kcat = Vmax/Etotal) for the one-, two-, and three-domain substrates were 3.4 ± 0.3 min-1, 1.8 ± 0.2 min-1, and 1.3 ± 0.1 min-1, respectively (Fig. 1 A and Table 1). We also assayed ClpXP degradation of the CM-titin10 substrate. Although aggregation at concentrations above 1 μM precluded determination of reliable kinetic parameters, rates at low substrate concentrations were consistent with a Km of 0.38 μM and a kcat of 0.58 ± 0.3 min-1.
Fig. 1.

Degradation of native and unfolded titinx proteins. (A) Michaelis-Menten plots for ClpXP degradation of unfolded titin substrates. (B) Length dependence of the average time for ClpXP degradation of unfolded titin substrates. (C) Competitive ClpXP degradation of native and unfolded single-domain titin substrates (10 μM each). (D) Lineweaver-Burke analysis of the degradation rate of different concentrations of 35S-labeled CM-titin1 in the presence of unlabeled titin proteins (1 μM each). Degradation reactions contained 0.1 μM ClpX6 and 0.3 μM ClpP14.
Table 1. Steady-state ClpXP degradation and inhibition parameters.
| ssrA-tagged titin variant | Length residues | kcat, min-1·[ClpXP]6-1 | Km, μM | kcat/Km, μM-1·min-1 | Ki, μM |
|---|---|---|---|---|---|
| Titin1 | 113 | 0.17 ± 0.02 | 0.50 ± 0.2 | 0.34 | 1.0 ± 0.14 |
| CM-titin1 | 113 | 3.4 ± 0.3 | 0.60 ± 0.2 | 5.7 | 0.5 ± 0.11 |
| CM-titin2 | 205 | 1.8 ± 0.2 | 0.48 ± 0.2 | 3.8 | 0.38 ± 0.10 |
| CM-titin3 | 297 | 1.3 ± 0.1 | 0.45 ± 0.2 | 2.9 | |
| CM-titin10 | 941 | 0.58 ± 0.3 | 0.38 ± 0.2 | 1.5 | 0.13 ± 0.05 |
The average time required for ClpXP degradation of titin constructs with one, two, or three unfolded domains is plotted as a function of substrate length in Fig. 1B. Two points are notable. First, the plot is linear, indicating that each unfolded titin repeat adds a constant value to the time required for ClpXP degradation. Second, the y-intercept, which represents the time required for any length-independent steps in the degradation of unfolded titin, is 0.01 min (error limits 0-0.04 min). Hence, substrate-processing steps other than translocation appear to be too fast to contribute significantly to the degradation rate of these unfolded substrates. The inverse slope of the plot is 387 ± 25 residues per min. Assuming that translocation is the length-dependent step (see Discussion), this value represents the average rate of spooling of unfolded titin into the degradation chamber of ClpP. This rate predicts a kcat of 0.4 min-1 for CM-titin10, which is within error of the experimental value (Table 1).
Preferential Degradation of Unfolded Substrates. Km for ClpXP degradation of native titin1 (0.5 μM) was similar to that for unfolded CM-titin1 (0.6 μM), but kcat for the native substrate (0.17 min-1) was much slower than for the unfolded substrate (3.4 min-1; Table 1). These steady-state kinetic parameters predict that ClpXP should degrade the unfolded substrate much faster than the native substrate. To test this proposal, 10 μM concentrations of titin1 and CM-titin1 were mixed and subjected to ClpXP degradation (Fig. 1C). The native substrate was 35S-labeled in one experiment and the denatured substrate was 35S-labeled in an otherwise identical experiment to allow degradation of both substrates to be assayed under competitive conditions. ClpXP degraded the unfolded substrate rapidly, whereas degradation of native titin1 in the mix was at or near the detection limit. Because these substrates have similar Km values, the enzyme must engage the native substrate far more slowly than the denatured substrate.
Competition by Native and Unfolded Substrates. To determine the ability of native or unfolded titinX molecules to bind ClpXP, regardless of their eventual fate, we assayed degradation rates for different concentrations of 35S-CM-titin1 in the presence or absence of unlabeled competitor proteins at concentrations of 1 μM. The resulting data fit a simple competitive model in which the unlabeled substrates increase the apparent Km for the radiolabeled substrate but do not change kcat (Fig. 1D). Inhibition constants (Ki) calculated from these data were 1.0 ± 0.14 μM for native titin1, 0.5 ± 0.11 μM for CM-titin1, 0.38 ± 0.10 μM for CM-titin2, and 0.13 ± 0.05 μM for CM-titin10 (Table 1).
The initial interaction of denatured and native titin proteins with ClpXP is mediated by the ssrA tag and thus should be similar for all substrates. Hence, to the extent that kcat is reciprocally related to the time that a substrate is bound to the enzyme, one would expect titin substrates with smaller kcat values to be better competitors and vice versa. Indeed, longer unfolded titin substrates have smaller kcat values and are better inhibitors than shorter unfolded substrates. This result makes sense if these molecules are bound and irreversibly engaged by ClpXP with similar kinetics, because length-dependent translocation would ensure that a longer molecular occupied the enzyme for a prolonged period of time. However, native titin1 was a poorer inhibitor than unfolded CM-titin1, despite having a similar KM and a 20-fold lower value of kcat. As demonstrated by the modeling discussed below, this result can be explained if native substrates are bound and released many times, on average, before finally being denatured and irreversibly engaged by the enzyme.
Partial Degradation of a Multidomain Native Titin Substrate. Previous studies have shown that ClpXP and related proteases can produce partially degraded protein products when it stalls while attempting to denature a second folded domain within a multidomain substrate (14, 21-24). It has not been clear, however, whether the enzyme irreversibly stalls or sometimes succeeds in denaturing and degrading the second domain. Similarly, whether these partially degraded proteins are released from the enzyme has not been established. To investigate these questions, we assayed ClpXP degradation of a titin3 substrate with an N-terminal FLAG epitope in both native and unfolded states. 35S-labeled substrates were incubated with ClpXP for varying times, and degradation was assayed by SDS/PAGE and autoradiography (Fig. 2 A and B). For technical reasons, these experiments were performed in the presence of SspB, an adaptor that strengthens binding of ssrA-tagged substrates to ClpX (25).
Fig. 2.

Degradation and properties of FLAG-titin3. (A) Kinetics of degradation of native 35S-FLAG-titin3 (0.3 μM) assayed by SDS/PAGE. (B) Degradation of denatured 35S-CM-FLAG-titin3 (2 μM). (C) Native domains in the titin1 and titin3 proteins have similar melting temperatures. (D) Western blots show that partially degraded titin proteins have the FLAG epitope but lack the ssrA tag. Degradation reactions contained 0.3 μM ClpX6, 0.9 μM ClpP14, and 0.3 μM SspB2.
Degradation of native FLAG-titin3 resulted in the appearance of two partially degraded species, with sizes consistent with the loss of one and two titin domains from the substrate (Fig. 2 A). Similar results were obtained when ClpXP degradation was performed in the absence of SspB (data not shown). Most one- and two-domain species remained refractory to further degradation, even at the latest time points. This result was not caused by inactivation of ClpXP or depletion of ATP, because addition of fresh enzyme and/or additional ATP did not stimulate further degradation (data not shown). Partial degradation was not observed during ClpXP degradation of the corresponding unfolded substrate (Fig. 2B), demonstrating that the native titin fold plays an important role in the genesis of these protein fragments. Each domain in native titin3 exhibited essentially the same melting temperature as in titin1 (Fig. 2C), suggesting that incomplete ClpXP degradation of this protein did not result from any special stability properties introduced by domain-domain interactions.
Western blots using antibodies against the N-terminal FLAG epitope or C-terminal ssrA tag (Fig. 2D) showed that the partially degraded species were C-terminal truncations of the intact protein, consistent with C → N degradation. Mass spectroscopy revealed major fragments with molecular weights of 15,294 and 25,269, consistent with a final ClpXP cleavage between Trp-34 and Lys-35 of either the central or C-terminal titin domain in FLAG-titin3. Cleavage at these positions produced tails of ≈37 residues appended to the C terminus of the remaining native titin domain(s). This length is sufficient to span the distance from the entry of the ClpX pore to one of the active sites within ClpP (14).
ClpXP Releases the Partially Processed Titin Proteins. Partially degraded proteins might be released from ClpXP during the normal enzymatic cycle but not rebind because they lack a degradation tag. Alternatively, they might only be released during preparation of samples for SDS/PAGE. Multiple results support the first model. For example, partially degraded products purified from degradation reactions under native conditions by anti-FLAG affinity were not contaminated by ClpX or ClpP (data not shown). Moreover, the concentration of substrate fragments produced after 3 h of ClpXP degradation increased in a linear fashion with the initial FLAG-titin3 concentration (Fig. 3A). If these products were not released from ClpXP, dead-end proteolytic complexes should have accumulated and resulted in nonlinear generation of products.
Fig. 3.

Partially degraded titin species are released by ClpXP. (A) Production of partially degraded species by ClpXP degradation of different initial concentrations of FLAG-titin3.(B) SDS/PAGE autoradiogram of 35S-labeled titin proteins and fragments. Lanes 1-3, purified titin1, titin2, and titin3 standards; lanes 4 and 5, purified FLAG-titin3 degradation products ± subtilisin; lanes 6 and 7, FLAG-titin3 degradation products after 60 min of ClpXP degradation ± subtilisin. (C) After 4 or 60 min of incubation of 0.3 μM ClpX6/0.9 μM ClpP14/0.3 μM SspB2 with (+) or without (-) FLAG-titin3 (0.3 μM), GFP-ssrA (0.3 μM) was added and its degradation was monitored by loss of fluorescence.
Digestion with another protease was also used to assess the accessibility of the tails of the partially degraded FLAG-titin3 substrates after purification or in the original degradation mix with ClpXP. In both cases, addition of a small amount of subtilisin removed ≈20 residues based on changes in SDS/PAGE mobility (Fig. 3B, lanes 4-7). If the tails of the partially degraded fragments were threaded through the ClpX pore and into ClpP, some protection from subtilisin cleavage would have been expected because no significant degradation of ClpXP was observed (data not shown). Thus, this experiment supports the normal release model.
As a final test for the release of partially degraded titin molecules from ClpXP, we assayed the concentration of free enzyme early and late in the degradation reaction. For these experiments, FLAG-titin3 and ClpXP were mixed, and degradation was allowed to proceed for 4 or 60 min. ssrA-tagged GFP was then added, and its degradation by ClpXP was assayed by loss of fluorescence. After 4 min of FLAG-titin3 degradation, ClpXP degraded GFP at ≈65% of the rate of a control reaction lacking the titin substrate (Fig. 3C). Thus, about 35% of ClpXP was engaged in degrading FLAG-titin3. After 60 min, by contrast, ClpXP degraded GFP at the same rate as the control reaction lacking the titin substrate (Fig. 3C), indicating that no partially degraded fragments remained bound to the enzyme.
Modeling of ClpXP Proteolysis of Titin Substrates. To assess the validity of our qualitative conclusions, we developed models for ClpXP degradation of titinX substrates and tested whether they could reproduce our degradation and competition results. A basic model depicts degradation of native titin1 and unfolded titinX substrates (Fig. 4A). Based on results presented above, the translocation rate was set at 400/n min-1 (n = residue number). The rate constants for other steps were varied by trial-and-error until computer-simulated and experimental results were similar. The kinetic scheme and constants shown in Fig. 4A predicted Ki and kcat values that matched experimental parameters very well and Km values that matched moderately well for four native and denatured titin substrates (Fig. 4C). Notably, the rate constants for partitioning of ES* and for length-dependent translocation in this model determine the distinct properties of these different titin molecules as ClpXP substrates and competitors.
Fig. 4.

Kinetic modeling of ClpXP degradation. (A) Model for ClpXP degradation of native titin1 and denatured CM-titinX variants. Units for rate constants are μM-1·min-1 for the E + S → ES step and min-1 for all other steps. For steps where the rate constant for the native and unfolded substrates differs, the native value is in bold and the denatured value is in italics. Denaturation of the native substrate occurs in the ES* → ET step. The ET → E + P step is unrealistic because product formation and release probably begin as soon as 35 residues of the polypeptide have been translocated and well before ClpX is free to engage another substrate; practically, however, this simplification does not grossly affect the results. (B) Model for degradation of the FLAG-titin3 substrate. Units are as defined in A. Because denaturation is much slower than translocation, these steps are combined into a single step for the downstream domains. (C) Comparison of experimental values (light gray bars) with simulated values (black bars) calculated by using the model in A.(D) Changes in substrate and partially degraded protein concentrations during ClpXP degradation of FLAG-titin3. The symbols represent experimental values (see Fig. 2 A). The solid lines were simulated by using the model in B.
For the titin3 substrate, the basic model was extended to include two additional intermediates (ES2* and ES3*) at which native domains could dissociate or be denatured, and the denaturation and translocation steps for each domain were represented as a single step (Fig. 4B). The constants for the initial association/dissociation and substrate release steps are slightly different from those in Fig. 4A because of the presence of SspB. Simulations based on this extended model predicted the kinetics with which full-length FLAG-titin3 disappeared and partially degraded species accumulated with good accuracy (Fig. 4D). Hence, denaturation/release reactions for each successive titin domain at the ES1*, ES2*, and ES3* intermediates accounts nicely for the experimental results.
Discussion
By studying ClpXP degradation of ssrA-tagged proteins containing unfolded titin domains, we found that kcat was a simple linear function of the number of residues in each substrate. This length dependence should reflect the average substrate translocation rate through ClpX and into the degradation chamber of ClpP (≈400 residues per min), as the subsequent peptide-bond cleavage and product-release steps are much faster reactions (12-13). Our results place a lower limit on the combined rates (≥25 min-1) of any steps in ClpXP degradation that are not length dependent. At saturating substrate concentrations, translocation of a single unfolded titin domain takes about 15-20 s and is the rate-limiting step in degradation, whereas other steps take only a small fraction of this time.
ClpXP degradation of native titin1 takes 5-6 min when substrate is saturating. Once ClpXP has denatured this molecule, translocation should require the same 15-20 s as the unfolded substrate. How is the remaining time spent? Because the ssrA tag mediates binding to ClpXP, the native substrate should bind ClpX at a rate similar to unfolded substrates. Two alternative models are then possible: (i) the native substrate remains bound to the enzyme until denaturation and translocation occur; or (ii) the native substrate dissociates and rebinds multiple times on average before these committed enzymatic steps. Our results support the second model and are inconsistent with the first model, which incorrectly predicts that native titin should compete for ClpXP binding much better than the unfolded substrate. In fact, native titin is a slightly poorer inhibitor.
A basic kinetic model (Fig. 4A) explains the properties of the native and unfolded titin molecules, both as substrates and inhibitors of ClpXP. Although other kinetic schemes might work equally well, examination of the model is instructive. Other than the length-dependence of translocation, the differences in the properties of the different native and unfolded titin substrates arise solely from differences in the rate at which the ES* species partitions. For unfolded substrates, no dissociation of ES* is required to account for the experimental data, and the translocating species (ET) forms rapidly. By contrast, native substrates in the ES* complex dissociate much faster than they are denatured to form the committed ET species. As a result, many dissociation and rebinding events are required before the chance of denaturation becomes significant. Dissociation of native substrates from ES* probably occurs because the unfolding force strains the interaction between the enzyme and substrate as well as the native fold of the substrate (2, 5).
ClpXP has a remarkable ability to unfold and degrade titin and other very stable proteins (3-7, 14, 19, 21), which our results suggest occurs largely through repeated cycles of substrate binding, force application, and substrate release. In the end, this dogged enzymatic persistence leads to substrate denaturation, but in a process that is expensive in time and ATP consumption. Although the Fig. 4A model does not explicitly include ATP, we assume that all steps after the initial binding of substrate to enzyme involve ATP hydrolysis. With this assumption, an ATP hydrolysis rate of ≈600 min-1 for translocation and a rate ≈4-fold slower for all other steps nicely accounts for the observed ATP consumption during ClpXP degradation of native and denatured titin substrates (for discussion, see ref. 5). Each round of titin binding, force application, and release would then be accompanied by the hydrolysis of 15-20 ATPs by the ClpX hexamer. Only four ATPs are consumed during the degradation of a short ssrA-tagged peptide (unpublished results), and thus the binding and ES → ES* steps of the model are unlikely to consume most of the ATP during a single round of titin binding, attempted denaturation, and release. Hence, roughly 15 ATPs appear to be hydrolyzed before ES* dissociates, indicating that multiple denaturation attempts probably precede each release event.
What happens when ClpXP begins degradation of a substrate but then encounters a stable structural element that impedes further polypeptide translocation? In our studies of a three-domain native titin substrate, partially degraded species accumulate. These species consist of one or two N-terminal titin domains followed by a 37-residue tail from the partially degraded domain, indicating that ClpXP stalls at some frequency when it encounters the second or third titin domain during translocation. Partial degradation products, often with tails of similar length, also arise from incomplete proteolysis of other multidomain substrates by ClpXP, ClpAP, and the eukaryotic proteasome (14, 21-24). For instance, the proteasome incompletely degrades NF-κB in a reaction that appears to depend on the stability of a refractory domain (23).
Two additional facts emerge from our studies of partial degradation of the titin3 substrate. First, the partially processed protein fragments are released from ClpXP. Because these experiments were performed with purified enzyme and substrate, the ability to release partially degraded species with intact native domains must be an intrinsic activity of the enzyme. Second, ClpXP degrades the second and third native domains of FLAG-titin3 to some extent. Indeed, modeling suggests that denaturation and degradation of these domains is twice as likely as release (Fig. 4B). Hence, upon encountering these downstream folded domains during polypeptide translocation, the enzyme partitions between a denaturation step and a release step. This event is obviously reminiscent of the partitioning between denaturation and release that occurs when ClpXP encounters the first native domain in a multidomain substrate or in a single-domain substrate. Several differences between these two classes of release/denaturation events are worth noting. First, release before denaturation of the first domain permits rebinding to the enzyme via the ssrA tag of the undegraded substrate, whereas tagless protein fragments released after degradation of the first domain would not rebind. Second, differences in the nature of the substrate sequence serving as the grip for ClpX probably affect the applied denaturation force and/or the release rate. The ssrA tag occupies the ClpX pore when the first domain is encountered, whereas a different amino acid sequence plays this role when the second and third native domains are encountered.
How do the partially degraded titin proteins, which contain a tail that must be threaded through the ClpX pore and into ClpP, escape from the enzyme? One possibility is that the attempt to denature a stable substrate also distorts ClpX. This structural strain could disrupt the ClpX hexamer, allowing release of substrate both from the ATPase and from ClpP. This model is plausible, because any denaturing force applied to distort the substrate would also distort the enzyme, and ClpX hexamers are only moderately stable to denaturation. A second possibility is that the ClpX ATPase motor runs in reverse for a short period and actively translocates the tail out of the pore. Reverse translocation may be possible, because unfolded polypeptides trapped in the chamber of inactive ClpP can be released in a reaction that requires ClpX and ATP hydrolysis (3). By either model, the trigger for substrate release must be a stochastic event linked to the process of substrate denaturation, because partially degraded species were not observed during degradation of a three-domain unfolded substrate. Whether ClpXP is used to process substrates in the cell through partial degradation is not currently known.
Substrate selection by energy-dependent proteases is generally discussed in terms of the specificity and strength of substrate binding. For example, sequence-specific recognition of degradation tags and/or the tethering sequences of adaptor proteins are clearly important in proteolytic targeting (7, 9, 10, 25). However, the stability of a native protein can be equally important in substrate selectivity. For instance, ClpXP degrades unstructured titin ≈20-fold more rapidly than native titin, despite the fact that both molecules bear the same, highly efficient ssrA degradation tag.
From a biological perspective, the ability of ClpXP to release difficult-to-denature substrates may be important for two reasons. First, it is possible that ClpXP is unable to denature and degrade some proteins with degradation tags. The ability to release such proteins would prevent the enzyme from becoming trapped in a futile endeavor. Second, this release mechanism would allow the enzyme to continually sample a population of substrates for those that could be readily engaged and degraded at the lowest energetic cost. In the cell, for example, some ClpXP substrates are probably incomplete ssrA-tagged proteins without structure, whereas other substrates are stable native proteins (7, 8, 26). In such a mixture, ClpXP would preferentially degrade the unfolded and metastable substrates and postpone degradation of the difficult-to-denature substrates. Because degradation of the former substrates would consume less ATP, this strategy would be cost-effective when cellular energy stores were low but the demand for protein degradation was high.
Acknowledgments
We are grateful to Jennifer Hou for performing initial experiments and thank Steve Bell, Randy Burton, Greg Hersch, and Frank Solomon for helpful discussions and materials. This work was supported, in part, by National Institutes of Health Grant AI-15706. T.A.B. is an employee of the Howard Hughes Medical Institute.
Abbreviation: CM, carboxymethylated.
References
- 1.Neuwald, A. F., Aravind, L., Spouge, J. L. & Koonin, E. V. (1999) Genome Res. 9, 27-43. [PubMed] [Google Scholar]
- 2.Sauer, R. T., Bolon, D. N., Burton, B. M., Burton, R. E., Flynn, J. M., Grant, R. A., Hersch, G. L., Joshi, S. A., Kenniston, J. A., Levchenko, I., et al. (2004) Cell 119, 9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim, Y. I., Burton, R. E., Burton, B. M., Sauer, R. T. & Baker, T. A. (2000) Mol. Cell 5, 639-648. [DOI] [PubMed] [Google Scholar]
- 4.Burton, R. E., Siddiqui, S. M., Kim, Y. I., Baker, T. A. & Sauer, R. T. (2001) EMBO J. 20, 3092-3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kenniston, J. A., Baker, T. A., Fernandez, J. M. & Sauer, R. T. (2003) Cell 114, 511-520. [DOI] [PubMed] [Google Scholar]
- 6.Kenniston, J. A., Burton, R. E., Siddiqui, S. M., Baker, T. A. & Sauer R. T. (2004) J. Struct. Biol. 146, 130-140. [DOI] [PubMed] [Google Scholar]
- 7.Flynn, J. M., Neher, S. B., Kim, Y. I., Sauer, R. T. & Baker, T. A. (2003) Mol. Cell 11, 671-683. [DOI] [PubMed] [Google Scholar]
- 8.Keiler, K. C., Waller, P. R. H. & Sauer, R. T. (1996) Science 271, 990-993. [DOI] [PubMed] [Google Scholar]
- 9.Gottesman, S., Roche, E., Zhou, Y. & Sauer, R. T. (1998) Genes Dev. 12, 1338-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herman, C., Thevenet, D., Bouloc, P., Walker, G. C. & D'Ari, R. (1998) Genes Dev. 12, 1348-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siddiqui, S. M., Sauer, R. T. & Baker, T. A. (2004) Genes Dev. 18, 369-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson, M. W. & Maurizi, M. R. (1994) J. Biol. Chem. 269, 18201-18208. [PubMed] [Google Scholar]
- 13.Thompson, M. W., Singh, S. K. & Maurizi, M. R. (1994) J. Biol. Chem. 269, 18209-18215. [PubMed] [Google Scholar]
- 14.Lee, C., Schwartz, M. P., Prakash, S., Iwakura, M. & Matouschek, A. (2001) Mol. Cell 7, 627-637. [DOI] [PubMed] [Google Scholar]
- 15.Reid, B. G., Fenton, W. A., Horwich, A. L. & Weber-Ban, E. U. (2001) Proc. Natl. Acad. Sci. USA 98, 3768-3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li, H., Carrion-Vasquez, M., Oberhauser, A. F., Marszalek, P. E. & Fernandez, J. M. (2000) Nat. Struct. Biol. 7, 1117-1120. [DOI] [PubMed] [Google Scholar]
- 17.Carrion-Vasquez, M., Oberhauser, A. F., Fowler, S. B., Marszalek, P. E., Broedel, S. E., Clarke, J. & Fernandez, J. M. (1999) Proc. Natl. Acad. Sci. USA 96, 3694-3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Creighton, T. E. (1974) J. Mol. Biol. 87, 579-602. [DOI] [PubMed] [Google Scholar]
- 19.Bolon, D. N., Grant, R. A., Baker, T. A. & Sauer, R. T. (2004) Mol. Cell 16, 343-350. [DOI] [PubMed] [Google Scholar]
- 20.Burton, R. E., Baker, T. A. & Sauer, R. T. (2003) Protein Sci. 12, 893-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoskins, J. R., Yanagihara, K., Mizuuchi, K. & Wickner, S. (2002) Proc. Natl. Acad. Sci. USA 99, 11037-11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ortega, J., Lee, H. S., Maurizi, M. R. & Steven, A. C. (2002) EMBO J. 21, 4938-4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin, L. & Kobayashi, M. (2003) J. Biol. Chem. 278, 31479-31485. [DOI] [PubMed] [Google Scholar]
- 24.Prakash, S., Tian, L., Ratliff, K. S., Lehotzky, R. E. & Matouschek, A. (2004) Nat. Struct. Mol. Biol. 11, 830-837. [DOI] [PubMed] [Google Scholar]
- 25.Wah, D. A., Levchenko, I., Rieckhof, G. E., Bolon, D. N., Baker, T. A. & Sauer, R. T. (2003) Mol. Cell 12, 355-363. [DOI] [PubMed] [Google Scholar]
- 26.Roche, E. D. & Sauer, R. T. (1999) EMBO J. 18, 4579-4589. [DOI] [PMC free article] [PubMed] [Google Scholar]