

ABSTRACT
Haa1 is a transcription factor that adapts Saccharomyces cerevisiae cells to weak organic acid stresses by activating the expression of various genes. Many of these genes encode membrane proteins, such as TPO2 and YRO2. How Haa1 is activated by weak acids is not clear. Here, we show that casein kinase I isoform Hrr25 is an important negative regulator of Haa1. Haa1 is known to be multiply phosphorylated. We found that mutations in HRR25 lead to reduced Haa1 phosphorylation and increased expression of Haa1 target genes and that Hrr25 interacts with Haa1. The other three casein kinase I isoforms, Yck1, Yck2, and Yck3, do not seem to play critical roles in Haa1 regulation. Hrr25 has a 200-residue C-terminal region, including a proline- and glutamine-rich domain. Our data suggest that the C-terminal region of Hrr25 is required for normal inhibition of expression of Haa1 target genes TPO2 and YRO2 and is important for cell growth but is not required for cell morphogenesis. We propose that Hrr25 is an important regulator of cellular adaptation to weak acid stress by inhibiting Haa1 through phosphorylation.
IMPORTANCE Our study has revealed the casein kinase I protein Hrr25 to be a negative regulator of Haa1, a transcription factor mediating the cellular response to stresses caused by weak acids. Many studies have focused on the target genes of Haa1 and their roles in weak acid stress responses, but little has been reported on the regulatory mechanism of Haa1. Weak acids, such as acetic acid, have long been used for food preservation by slowing down the growth of fungal species, including S. cerevisiae. In the biofuel industry, acetic acid in the lignocellulosic hydrolysates limits the production of ethanol, which is undesirable. By understanding how Haa1 is regulated, we can make advances in the field of food sciences to better preserve food and engineer acetic acid-resistant strains that will increase productivity in the biofuel industry.
KEYWORDS: acetic acid stress response, casein kinase I protein, Haa1, Hrr25, Saccharomyces cerevisiae
INTRODUCTION
Cells employ signaling pathways to adapt to rapid changes in their environment. One of these pathways, the weak organic acid stress pathway in Saccharomyces cerevisiae, is mediated by Haa1, a homolog of copper-regulated transcription factor Ace1 (1–5). The protective effect exerted by Haa1 is particularly beneficial against less lipophilic weak acids, such as acetic acid, propionic acid, and lactic acid, while yeast cells receive little to no protection from Haa1 against more lipophilic weak acids, such as octanoic acid (1, 3). Weak acids, such as acetic acid and lactic acid, in growth medium at low pH are able to diffuse through the membrane in their undissociated forms and dissociate once inside the cytoplasm, which has near-neutral pH, leading to an intracellular buildup of protons that lower the intracellular pH. This drop in pH, along with perturbations of the lipid organization and membrane permeability and other changes in cells, is the likely cause of an extended lag phase in yeast growth in response to weak acid stress (reviewed in references 6 and 7). Genome-wide transcriptional analyses have revealed many genes whose expression is affected by weak acid treatment (2–4, 8–11). Different organic acids affect the expression of different sets of genes, involving a number of transcription factors, including Rim101, Msn2, Msn4, Haa1, and War1 (reviewed in reference 7). Induced expression of a number of these genes in response to acetic acid treatment is under the control of transcription factor Haa1. Haa1 target genes such as TPO2 and TPO3, encoding two polyamine transporters (12), and YGP1, encoding a highly glycosylated secreted protein (13), can confer resistance to stresses caused by weak acids (1, 2, 4). It has been reported that over half of the genes that show Haa1-dependent expression in response to acetic acid treatment have the Haa1-responsive element GNN(G/C)(A/C)(A/G)G(A/G/C)G in their promoters (14).
Ethanol production from lignocellulosic hydrolysates by yeast cells is limited by high concentrations of acetic acid and other weak acids (15–17). Yeast strains with increased resistance to acetic acid could be of importance to the biofuel industry. It has been shown that overexpression of Haa1 leads to increased acetic acid resistance, increased expression of Haa1 target genes, and increased yield of bioethanol (18–20). Therefore, it is important to understand how Haa1 activity is regulated in response to acetic acid treatment. Yak1, a serine/threonine protein kinase in the glucose-sensing system, has been reported to regulate acetic acid stress resistance via Haa1 (21), but the underlying mechanism is still unknown. A recent report suggested that Haa1 phosphorylation may play a role in Haa1 regulation (22). Haa1 is found throughout the cell in a multiply phosphorylated state. However, under lactic acid stress, Haa1 translocates into the nucleus and displays a decrease in phosphorylation (22). In the same study, it was also reported that Msn5, a karyopherin important for nuclear export and import of various cargo proteins (23), mediates nuclear export of Haa1. Thus, a working model of the regulation of Haa1 suggests that Haa1 phosphorylation has a negative effect on its activity, possibly by promoting cytoplasmic localization of Haa1. However, the kinase or kinases responsible for Haa1 phosphorylation are unknown. Multiple phosphorylation sites of Haa1 have been revealed through high-throughput studies on protein phosphorylation (24, 25). Some of the phosphorylation sites are the consensus sites of protein kinase A and casein kinase I (PhosphoGRID; https://phosphogrid.org). In this report, we tested the effect of mutations in the genes encoding the four casein kinase I proteins in yeast on the expression of Haa1 target genes and identified the casein kinase I isoform Hrr25 as an important negative regulator of Haa1 function. In hrr25 mutant cells, the activity of Haa1 is increased, which correlates with reduced phosphorylation of Haa1. Our study provides important insights into the regulation of Haa1 and the cellular responses of yeast to acetic acid.
RESULTS
Casein kinase I proteins Yck1, Yck2, and Yck3 do not play significant roles in Haa1 regulation.
Global phosphorylation analysis of yeast proteins has revealed that Haa1 is phosphorylated at 15 sites, including three sites of the casein kinase I motif (24, 25). It has been proposed that Haa1 dephosphorylation correlates with its activation (22). To determine whether casein kinase I regulates the activity of Haa1 through phosphorylation, we set out to examine the expression of Haa1 target genes in wild-type (WT) and casein kinase I mutant cells. To facilitate the analysis of Haa1 target gene expression, we established a reporter gene system by fusing lacZ downstream of the promoter of two well-documented Haa1 targets, TPO2 and YRO2 (1, 5), on a low-copy centromeric plasmid. The resultant plasmids carrying TPO2-lacZ and YRO2-lacZ reporter genes were introduced into a wild-type strain and an haa1Δ mutant, and β-galactosidase activity assays on the transformants showed that they were induced by acetic acid treatment in a largely Haa1-dependent manner (Fig. 1A). Although Haa1 is the major activator of the TPO2-lacZ and YRO2-lacZ reporter genes in response to the acetic acid stress, it is important to note that other transcription factors are also involved in the expression of these two reporter genes since in haa1Δ mutant cells, acetic acid treatment still led to 10.6- and 22.8-fold increases in the expression of the TPO2-lacZ and YRO2-lacZ reporter genes, respectively (Fig. 1A).
FIG 1.

β-Galactosidase activity assays of TPO2-lacZ and YRO2-lacZ reporter genes in wild-type (BY4741) and isogenic haa1Δ mutant (ZLY4043) cells (A), wild-type (LRB341) and isogenic yck1Δ yck2ts double mutant (LRB362) cells (B), and wild-type (BY4741) and yck3Δ mutant (ZLY4260) cells (C). Cells were grown in MM4 without and with 60 mM acetic acid, and β-galactosidase activity assays were carried out as described in Materials and Methods. The error bars indicate the standard deviations of results from independent cultures. Means of the results were compared by t test, and an asterisk indicates significant difference in the means of two groups of data (P < 0.05).
We then examined the expression of TPO2-lacZ and YRO2-lacZ reporter genes in casein kinase I mutants. Saccharomyces cerevisiae has four casein kinase I isoforms, Yck1, Yck2, Yck3, and Hrr25, which are involved in multiple cellular pathways, including DNA repair, ribosome biogenesis, cell morphogenesis, glucose sensing, amino acid sensing, vesicular trafficking, and autophagy (26–33). Yck1 and Yck2 perform redundant functions and are essential for cell growth. Because β-galactosidase activity assays require cells to grow for several generations before being collected in the logarithmic phase, we used a temperature-sensitive yck1Δ yck2ts double mutant strain to determine whether Yck1 and Yck2 play a role in the regulation of Haa1. At the nonpermissive temperature (37°C), the yck1Δ yck2ts double mutant fails to grow (27). At the permissive temperature (26°C), the double mutant has been shown to display defects in the phosphorylation of a known Yck1/Yck2 substrate (34). Thus, we chose to grow the double mutant cells carrying a TPO2-lacZ or YRO2-lacZ reporter gene at 30°C, the standard temperature to grow yeast cells. At this temperature in minimal medium at pH 4 (MM4), wild-type cells divided approximately every 200 min, while the isogenic yck1Δ yck2ts mutant doubled approximately every 340 min (data not shown), indicating that the activity of the temperature-sensitive yck2ts allele was indeed compromised. β-Galactosidase activity assays show that TPO2-lacZ expression is slightly increased in yck1Δ yck2ts mutant cells compared to the wild type in the absence of acetic acid treatment (Fig. 1B). In contrast, YRO2-lacZ expression is increased by 4-fold in the double mutant compared to the wild type under the same condition. In the presence of the acetic acid stress, expression of the TPO2-lacZ and YRO2-lacZ reporter genes is reduced by 59% and 56%, respectively, in the yck1Δ yck2ts mutant cells compared to the wild type (Fig. 1B). Because the yck1Δ yck2ts mutant effects on the expression of two Haa1 target genes yielded inconsistent results, we suggest that Yck1 and Yck2 may only play a small role, if any, in the regulation of Haa1. Consistent with this notion, we found that the expression of YGP1, another target gene of Haa1, was actually reduced by ∼2-fold in yck1Δ yck2ts double mutant cells grown in MM4 without acetic acid compared to the wild type, using β-galactosidase activity assays (data not shown). As mentioned above, expression of TPO2-lacZ and YRO2-lacZ reporter genes is mediated not only by Haa1 but also by other transcription factors. The effect of a double yck1 yck2 mutation observed in Fig. 1B could be due to the regulation of these unknown transcriptional regulators by Yck1/Yck2.
We next analyzed the expression of these two reporter genes in a yck3Δ mutant, which unlike the yck1Δ yck2ts mutant does not have a growth defect. Figure 1C shows that yck3Δ has no effect on the expression of TPO2-lacZ and YRO2-lacZ reporter genes in cells grown in MM4 without acetic acid. In cells grown in MM4 with 60 mM acetic acid, yck3Δ has no effect on TPO2-lacZ expression and reduces YRO2-lacZ expression by 18%. These results suggest that Yck3 does not play a critical a role in Haa1 regulation.
Hrr25 negatively regulates the expression of Haa1 target genes.
Hrr25, the fourth isoform of casein kinase I in the budding yeast, has been reported to be an essential protein in the BY4741 background, and the essential function is linked to maturation of the pre-40S ribosomal subunit (33, 35). To examine a potential role of Hrr25 in the regulation of Haa1, we set out to construct a missense mutant allele of HRR25 to determine lacZ reporter gene expression. It has been reported that an hrr25(E52D) mutation (from glutamate 52 to aspartate) leads to viable mutant cells despite the fact that the mutation results in loss of Hrr25 kinase activity toward casein in vitro and reduced phosphorylation of Elp1 in vivo (36). We generated this mutant allele via site-directed mutagenesis by overlap extension using PCR. The recombinant plasmid containing hrr25(E52D) was then introduced into a heterozygous HRR25/hrr25Δ::kanMX4 diploid strain. Transformants were sporulated and tetrad analysis was conducted to generate hrr25Δ mutant cells carrying the plasmid containing the hrr25(E52D) mutant allele. After 3 days of growth on YPD medium (see Materials and Methods), hrr25(E52D) mutant cells exhibited a mild growth defect (Fig. 2A). Surprisingly, after 5 days of growth, hrr25Δ mutant spores without the plasmid containing hrr25(E52D) grew into small colonies (data not shown). We then sporulated HRR25/hrr25Δ::kanMX4 heterozygous mutant cells without the plasmid carrying hrr25(E52D). Tetrad analysis revealed that among the 17 tetrads dissected, 12 showed 2:2 segregations of big and small colonies (Fig. 2B) (data not shown). The remaining five incomplete tetrads yielded no more than two big or two small colonies. All big colonies were Geneticin sensitive (wild type), while the small ones were Geneticin resistant (hrr25Δ::kanMX4). In a different strain background, hrr25Δ leads to an extreme slow-growth phenotype (26). Our data indicate that Hrr25 is important for growth rate but not essential for cell viability in the BY4741 background.
FIG 2.

Mutations in HRR25 increase the expression of the TPO2-lacZ and YRO2-lacZ reporter genes. (A) Generation of hrr25(E52D) missense mutant strains via tetrad analysis. BY4743 hrr25Δ::kanMX4/HRR25 diploid cells containing a centromeric plasmid carrying an hrr25(E52D) mutant allele (pZD359) were sporulated, and tetrads were dissected on a YPD plate. The picture was taken after 3 days. M, hrr25(E52D) mutants; W, wild type. (B) Generation of hrr25Δ mutant strains via tetrad analysis of BY4743 hrr25Δ::kanMX4/HRR25 cells. Small colonies were hrr25Δ mutants. The picture was taken after 6 days. (C and D) The effect of hrr25(E52D) and hrr25Δ mutations on TPO2-lacZ and YRO2-lacZ reporter genes. Wild-type (BY4741) and isogenic hrr25(E52D) (ZLY4467) and hrr25Δ (ZLY4501) mutant cells were grown in MM4 without acetic acid (C) and with 60 mM acetic acid (D). β-Galactosidase activity assays were carried out as described in Materials and Methods.
TPO2-lacZ and YRO2-lacZ reporter genes were then transformed into both the hrr25(E52D) and hrr25Δ mutants, and β-galactosidase activity assays were performed on the transformants. Figure 2C shows that hrr25 mutations lead to increased expression of both reporter genes. Interestingly, hrr25Δ, which causes a more severe growth defect than the hrr25(E52D) mutant allele, also leads to a stronger induction of the two lacZ reporter genes. A higher induction of Haa1 target gene expression by the stronger hrr25Δ mutant allele indicates that Hrr25 negatively regulates Haa1 function. We also examined the effect of hrr25Δ and hrr25(E52D) mutations on TPO2-lacZ and YRO2-lacZ reporter gene expression in cells grown in the presence of 60 mM acetic acid, which activates Haa1. Figure 2D shows that, like cells grown in minimal medium without acetic acid, hrr25 mutations also increase TPO2-lacZ and YRO2-lacZ reporter gene expression in cells stimulated with acetic acid, with the more severe hrr25Δ mutant allele having a stronger effect. Together, these data suggest that Hrr25 negatively regulates Haa1 function under both basal and induced conditions.
Haa1 is required for increased expression of TPO2-lacZ and YRO2-lacZ in hrr25(E52D) mutant cells.
Since Hrr25 is implicated in multiple cellular processes, it is conceivable that hrr25 mutations could activate Haa1 target gene expression in a pathway independent of Haa1. To test this possibility, we introduced an haa1Δ mutation into an hrr25(E52D) mutant to generate an hrr25(E52D) haa1Δ double mutant, which was then transformed with plasmids carrying the TPO2-lacZ and YRO2-lacZ reporter genes. The resulting transformants, along with those of the wild type, an haa1Δ single mutant, and an hrr25(E52D) single mutant, were grown in MM4 with and without supplementation with 60 mM acetic acid, and β-galactosidase activity assays were carried out. Figure 3A and B show that increased expression of TPO2-lacZ and YRO2-lacZ in hrr25(E52D) single mutant cells is largely abolished by haa1Δ, consistent with Hrr25 negatively regulating Haa1 activity.
FIG 3.

Increased expression of TPO2-lacZ and YRO2-lacZ reporter genes due to an hrr25(E52D) mutation requires Haa1. Wild-type (BY4741) and isogenic haa1Δ (ZLY4043), hrr25(E52D) (ZLY4467), and hrr25(E52D) haa1Δ double mutant (ZLY4637) strains carrying centromeric plasmids encoding a TPO2-lacZ or YRO2-lacZ reporter gene were grown in MM4 without (A) and with (B) 60 mM acetic acid to the mid-logarithmic phase. β-Galactosidase activity assays were conducted as described in Materials and Methods. (C and D) An hrr25(E52D) mutation leads to increased nuclear localization of Haa1 in cells without acetic acid treatment. haa1Δ and hrr25(E52D) haa1Δ mutant strains expressing a functional Haa1-GFP fusion from a centromeric plasmid were grown in MM4 without and with 60 mM acetic acid. GFP fluorescence and differential interference contrast (DIC) images were captured as described in Materials and Methods.
To verify that the role of Haa1 in the increased expression of TPO2-lacZ and YRO2-lacZ in hrr25(E52D) mutant cells is growth phase independent, we generated growth curves for wild-type and haa1Δ, hrr25(E52D), and hrr25(E52D) haa1Δ mutant cells grown in MM4 supplemented with and without 60 mM acetic acid (see Fig. S1 in the supplemental material). β-Galactosidase activity assays were conducted on culture samples at four different growth points. Figure S2 from the supplemental material shows that the expression of TPO2-lacZ and YRO2-lacZ reporter genes is induced in hrr25(E52D) mutant cells, which requires Haa1. Together, these data are consistent with the notion that Hrr25 negatively regulates Haa1.
Lactic acid treatment increases the expression of Haa1 target genes, which correlates with nuclear translocation of Haa1 (22). We asked whether increased expression of TPO2-lacZ and YRO2-lacZ reporter genes in hrr25(E52D) mutant cells is also associated with increased nuclear localization of Haa1. Therefore, we generated an HAA1-green fluorescent protein gene (GFP) fusion on a centromeric plasmid and expressed it in haa1Δ and hrr25(E52D) haa1Δ mutant cells grown in MM4 without and with 60 mM acetic acid. The HAA1-GFP fusion construct was confirmed to be functional based on its ability to rescue the acetic acid sensitivity phenotype of haa1Δ mutant cells and to restore YRO2-lacZ expression in haa1Δ mutant cells to the wild-type level (data not shown). Figure 3C shows that Haa1-GFP is cytoplasmic in wild-type HRR25 cells grown in MM4 without acetic acid treatment. Consistent with the findings of Sugiyama et al. (22), acetic acid treatment leads to nuclear localization of Haa1-GFP (Fig. 3C). In hrr25(E52D) mutant cells, nuclear localization of Haa1-GFP was easily observed, even in the absence of acetic acid treatment (Fig. 3D). These data suggest that increased nuclear translocation of Haa1 is responsible for higher levels of expression of Haa1 target genes in hrr25(E52D) mutant cells.
Reduced Haa1 phosphorylation in hrr25E52D mutant cells.
Haa1 has been reported to be a phosphoprotein, and its reduced phosphorylation correlates with its activation (22). Our data on a negative regulatory role of Hrr25 on Haa1 prompted us to determine whether a mutation in HRR25 affects the phosphorylation state of Haa1. Therefore, we generated a plasmid encoding a three-hemagglutinin (3×HA) tag at the C terminus of Haa1 to facilitate immunodetection of Haa1-HA using anti-HA antibodies. We confirmed that the Haa1-HA fusion under the control of the endogenous promoter was functional based on its ability to rescue the acetic acid sensitivity phenotype of haa1Δ mutant cells (data not shown). We then introduced the plasmid containing HAA1-HA into haa1Δ single and hrr25(E52D) haa1Δ double mutant cells, and transformants were grown in minimal medium with and without 60 mM acetic acid. Total cellular proteins were isolated, separated by SDS-PAGE, and transferred to a nitrocellulose membrane for Western blotting. Figure 4A shows that Haa1 migrates as diffuse mobility forms in wild-type cells, consistent with published results (22). The diffuse migration pattern of Haa1 on Western blots has been attributed to phosphorylation. In hrr25(E52D) mutant cells in comparison to the wild type, the level of slower-mobility forms of Haa1-HA is reduced, suggesting that Haa1 is less phosphorylated in hrr25(E52D) mutant cells. Lactic acid treatment has been reported to lead to reduced Haa1 phosphorylation (22). However, we failed to detect Haa1 dephosphorylation in cells treated with acetic acid in both wild-type and hrr25(E52D) mutant cells (Fig. 4A). Rather, we detected a slight increase in the overall Haa1-HA protein level after acetic acid treatment. To confirm that a mobility shift of Haa1-HA in hrr25(E52D) mutant cells is due to changes in Haa1-HA phosphorylation, total cellular proteins were prepared and treated with λ protein phosphatase (PPase). Figure 4B shows that phosphatase treatment leads to the appearance of the same-size bands of Haa1-HA from wild-type and hrr25(E52D) mutant cell extracts. Phosphatase inhibitors reduce the effect of λ PPase, indicating that mobility shifts of Haa1-HA on SDS-PAGE are due to changes in phosphorylation.
FIG 4.

A mutation in HRR25, but not in YCK1/YCK2 or in YCK3, leads to reduced phosphorylation of Haa1. (A) An hrr25(E52D) mutation increases Haa1-HA mobility on SDS-PAGE. haa1Δ single (ZLY4043) and hrr25(E52D) haa1Δ double mutant (ZLY4637) strains with a centromeric plasmid carrying HA epitope-tagged HAA1 were grown in MM4 without and with 60 mM acetic acid. Total cellular proteins were prepared and separated by SDS-PAGE, and Haa1-HA was detected by Western blotting. Ilv5 (acetohydroxyacid reductoisomerase) was included as a loading control. (B) Phosphatase treatment of Haa1-HA from wild-type HRR25 and hrr25(E52D) mutant cells results in bands of the same size on Western blots. The strains used in panel A were grown in MM4 without and with 60 mM acetic acid, and TCA-precipitated total cellular proteins were treated with λ protein phosphatase (PPase) as indicated. When required, phosphatase inhibitors were added to the reaction mixtures to inhibit λ PPase. After treatment, proteins were separated by SDS-PAGE and Haa1-HA was detected by Western blotting. The arrowhead indicates a nonspecific band that is detected by anti-HA antibody. (C and D) Haa1-HA mobility is not affected by mutations in YCK1/YCK2 (C) and in YCK3 (D). Haa1-HA mobility in the indicated strains was analyzed as described for panel A. 3-Phosphoglycerate kinase (Pgk1) was included as a loading control.
We next examined whether mutations in YCK1/YCK2 and YCK3 affect phosphorylation of Haa1-HA despite the apparent lack of critical roles of these three casein kinase I isoforms in Haa1 regulation (Fig. 1). We generated a yck1Δ yck2ts haa1Δ triple mutant and a yck3Δ haa1Δ double mutant and transformed them with a plasmid carrying HAA1-HA. Transformants were grown in MM4 with and without 60 mM acetic acid, and Haa1-HA was analyzed by immunoblotting. Figure 4C and D show that mutations in YCK1/YCK2 and YCK3 have no obvious effect on Haa1-HA phosphorylation. As in Fig. 4A, acetic acid treatment results in a slight increase in the Haa1-HA protein level (Fig. 4C and D). Together, these data suggest that Hrr25 is the primary casein kinase I isoform that mediates Haa1 phosphorylation.
The C-terminal region of Hrr25 is differentially required for different cellular processes.
Hrr25 has an N-terminal kinase domain (residues 1 to 295) and a C-terminal 200-residue region, including a proline/glutamine-rich domain (residues 395 to 494) (Fig. 5A). Protein kinases often use regions outside the kinase domain for interaction with their substrates and/or cellular localization. Previously, it has been shown that the fission yeast Schizosaccharomyces pombe has two Hrr25 homologs: one has a C-terminal proline- and glutamine-rich domain, and the other does not (37). The former can restore normal cell growth to S. cerevisiae hrr25Δ mutant cells, which are extremely slow growing, while the latter does not (37). To determine whether the C-terminal region of Hrr25 is required for its function in the Haa1 pathway, we generated a plasmid encoding myc-tagged Hrr25 kinase domain [Hrr25(KD), comprising residues 1 to 302] to determine whether expression of HRR25(KD) under the control of the endogenous promoter in hrr25Δ mutant cells can lower TPO2-lacZ and YRO2-lacZ expression to wild-type levels. Accordingly, plasmids encoding myc-tagged full-length Hrr25 or Hrr25(KD) were transformed into hrr25Δ mutant cells carrying a TPO2-lacZ or YRO2-lacZ reporter gene. We confirmed that myc-tagged Hrr25 and Hrr25(KD) were expressed by immunoblotting, using antibody against the myc tag (Fig. 5B). The kinase domain of Hrr25 was expressed at a reduced level compared with full-length Hrr25. Figure 5C shows that, as expected, expression of full-length Hrr25 from a centromeric plasmid in hrr25Δ mutant cells reduced the expression of the TPO2-lacZ and YRO2-lacZ reporter genes to wild-type levels. Interestingly, expression of the Hrr25 kinase domain in hrr25Δ mutant cells significantly reduced the expression of both reporter genes, indicating that the kinase domain of Hrr25 alone is partially functional in inhibiting Haa1. Consistently, we found that the kinase domain alone of Hrr25 largely restores Haa1 phosphorylation to hrr25Δ mutant cells (Fig. 5D). Similar to an hrr25(E52D) mutation, hrr25Δ also leads to reduced Haa1 phosphorylation. However, unlike in hrr25(E52D) cells, the steady-state level of Haa1-HA seems to be significantly reduced in hrr25Δ mutant cells (compare Fig. 4A and 5D). While growing cultures for β-galactosidase activity assays, we noticed that the kinase domain of Hrr25 only marginally improved cell growth for the hrr25Δ mutant cells. A plate assay on the growth of hrr25Δ mutant cells with a plasmid carrying HRR25 or HRR25(KD) or an empty vector yielded similar results (Fig. 5E). It has been reported that cell morphology was abnormal in hrr25Δ cells (26). Therefore, we examined whether Hrr25's C-terminal region is required for the role of Hrr25 in cell morphogenesis. As reported, we easily detected elongated and multibudded cells among hrr25Δ mutant cells (Fig. 5F). Expression of the kinase domain of Hrr25 in hrr25Δ mutant cells fully restored normal cell morphology, suggesting that the C-terminal region of Hrr25 is dispensable for this process. Together, these results suggest that the C-terminal region of Hrr25 is differentially required for three different cellular processes: it is dispensable for cell morphogenesis, important for cell growth, and partially required for Haa1 regulation.
FIG 5.

Differential requirement of the C-terminal region of Hrr25 for Haa1 target gene expression, cell growth, and cell morphology. (A) Schematic representation of Hrr25. (B) Western blot analysis of myc-tagged Hrr25 and Hrr25(KD). hrr25Δ mutant cells (ZLY4479) carrying control vector (pRS415) or centromeric plasmids carrying HRR25-myc (pZL3338) and HRR25(KD)-myc (pZL3361) were grown in MM4, and total cellular proteins were prepared. Hrr25-myc and Hrr25(KD)-myc were detected by Western blotting. 3-Phosphoglycerate kinase (Pgk1) was included as a loading control. (C) The Hrr25 kinase domain alone is partially functional in mediating Haa1 target gene expression. Strains described in panel B were transformed with centromeric plasmids carrying TPO2-lacZ or YRO2-lacZ. Transformants were grown in MM4 without and with 60 mM acetic acid as indicated, and β-galactosidase activity assays were carried out as described in Materials and Methods. (D) Expression of the Hrr25 kinase domain in hrr25Δ mutant cells largely restores Haa1-HA phosphorylation. haa1Δ hrr25Δ double mutant cells (ZLY5046) expressing Haa1-HA and myc-tagged full-length Hrr25 (F) or the kinase domain of Hrr25(KD) were grown in MM4 without and with 60 mM acetic acid. Haa1-HA was detected by Western blotting. (E) The Hrr25 kinase domain alone is not sufficient to support cell growth. Strains described for panel B were streaked on the SD plate, and the picture was taken after 3 days of growth at 30°C. (F) The Hrr25 kinase domain is fully functional in supporting normal cell morphology in hrr25Δ mutant cells. The strains described for panel B were grown in MM4 liquid medium to the mid-logarithmic phase. Cells were imaged using differential interference contrast microscopy.
Hrr25 interacts with Haa1.
Our data suggest that Hrr25 mediates Haa1 phosphorylation. This phosphorylation could be conducted either directly by Hrr25 or indirectly through another kinase. We first attempted immunoprecipitation assays to examine whether Hrr25 and Haa1 directly interact. However, we failed to detect an interaction between Hrr25-myc and Haa1-HA. As an alternative approach, we then used the yeast two-hybrid assay to examine their interaction. We generated plasmids encoding fusion proteins of the Gal4 DNA binding domain (GBD) and Hrr25 and of the Gal4 transcriptional activation domain (GAD) and Haa1 and introduced them into the yeast two-hybrid strain AH109, which carries ADE2 and HIS3 reporter genes under the control of Gal4-dependent promoters. Figure 6 shows that AH109 cells coexpressing GBD-HRR25 and GAD-HAA1 on synthetic dextrose (SD) medium supplemented with histidine and adenine are white, while cells expressing GBD and GAD, GBD and GAD-HAA1, or GBD-HRR25 and GAD are red, indicating that there is an interaction between Hrr25 and Haa1. On complete supplement mixture (CSM) medium without histidine, AH109 cells expressing GBD-HRR25 and GAD-HAA1 are able to grow, while cells coexpressing GBD and GAD or GAD-HAA1 are not (Fig. 6). AH109 cells coexpressing GBD-HRR25 and GAD are also able to grow, albeit worse than cells coexpressing GBD-HRR25 and GAD-HAA1, suggesting that GBD-Hrr25 can weakly activate reporter expression on its own. Addition of 3 mM 3-amino-1,2,4-triazole (3-AT) to CSM medium to inhibit His3 effectively eliminates the growth of AH109 cells coexpressing GBD-HRR25 and GAD but not GBD-HRR25 and GAD-HAA1. The kinase domain of Hrr25 is partially functional in inhibiting the expression of Haa1 target genes (Fig. 5C), raising the possibility that Hrr25(KD) is sufficient for an interaction with Haa1. Accordingly, we constructed a plasmid encoding a fusion protein of GBD and Hrr25(KD) and transformed it into AH109 cells expressing GAD or GAD-HAA1. On CSM medium without histidine, cells coexpressing GBD-Hrr25(KD) with GAD-HAA1, but not with GAD, are able to grow, indicating that Hrr25(KD) interacts with Haa1. Together, these data provide support for an interaction between Hrr25 and Haa1.
FIG 6.
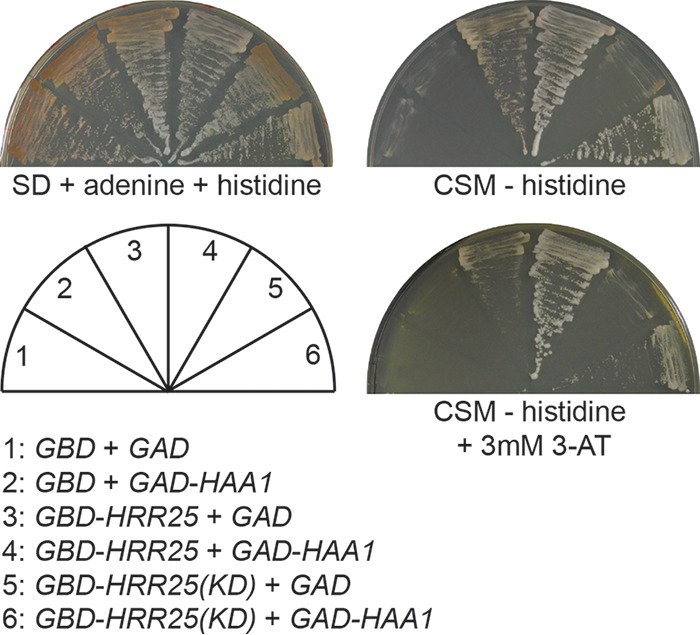
Yeast two-hybrid analysis of an interaction between Hrr25 and Haa1. AH109 cells expressing GBD, GAD, GBD-HRR25, GBD-HRR25(KD), or GAD-HAA1 as indicated were streaked on SD medium with histidine and adenine and CSM dropout medium without histidine and with or without 3 mM 3-AT. Pictures were taken after 4 days at 30°C.
DISCUSSION
Haa1 is an important transcriptional regulator that mediates adaptation to weak organic acids in yeast. How Haa1 is regulated is largely unknown. In this report, we identified the casein kinase I isoform Hrr25 as a negative regulator of Haa1. Our conclusion was based on the observation that the expression of Haa1 target genes is increased in hrr25 mutant cells, which requires Haa1. There are four isoforms of casein kinase I proteins in yeast, and our data suggest that Hrr25 may be the primary casein kinase I protein that regulates the activity of Haa1.
Mutations in HRR25 lead to activation of Haa1 target gene expression, reduced phosphorylation of Haa1, and increased Haa1 localization to the nucleus, providing a potential mechanism for the negative regulation of Haa1. It has been reported that reduced phosphorylation of Haa1 due to lactic acid treatment correlates with increased activity of Haa1 (22). Our result is consistent with this notion. However, there is an important difference between our study and the study by Sugiyama et al. (22). In cells stimulated with acetic acid, which activates Haa1, we failed to detect reduced phosphorylation of Haa1. Instead, we detected a slight increase in the Haa1 protein level in cells treated with acetic acid. This discrepancy could be due to the difference in the stress conditions used in these two studies: acetic acid in this study and lactic acid in the study by Sugiyama et al. (22). Overexpression of Haa1 leads to increased expression of its target genes (18). When Haa1 is mildly overexpressed from a centromeric plasmid under the control of endogenous promoter in wild-type cells, the expression of Haa1 target genes is only partially induced (our unpublished result). We propose that both the Haa1 protein level and changes in the phosphorylation state of Haa1 under certain conditions might contribute to the activation of the pathway.
A yeast two-hybrid analysis revealed an interaction between Hrr25 and Haa1. We hypothesize that Hrr25 directly binds to and phosphorylates Haa1 to regulate Haa1 activity. We generated mutations at the three casein kinase I consensus sites uncovered in global protein phosphorylation studies (24, 25), but we failed to see any change in the activity of Haa1 (our unpublished results). It is possible that other sites may be targeted by Hrr25 or that combinations of phosphorylation sites are required for the regulation of Haa1. One possible role of phosphorylation in Haa1 regulation could be linked to its export out of the nucleus. Msn5 has been reported to be required for Haa1 nuclear export, and for many nuclear cargo proteins, including Pho4, Aft1, HO endonuclease, Mig1, and Crz1 (23, 38–43), their phosphorylation is required for both an interaction with Msn5 and nuclear export. Therefore, Haa1 phosphorylation may play a similar role in its nuclear export. Indeed, reduced phosphorylation of Haa1 in hrr25 mutant cells correlates with its increased nuclear localization (Fig. 3D and 4A). In wild-type cells treated with acetic acid, Haa1 is localized in the nucleus, but its phosphorylation state does not seem to change based on its mobility on SDS-PAGE (Fig. 4). This discrepancy could be explained by the possibility that dephosphorylation at one or a small number of residues that mediate Haa1 nuclear translocation may be masked by the bulk phosphorylation state of Haa1. It is also possible that different mechanisms are implicated in the nuclear translocation of Haa1 in hrr25 mutant cells without acetic acid stress and in wild-type cells treated with acetic acid.
Hrr25 has a 200-residue C-terminal region, including a proline- and glutamine rich domain. Our data suggest that the Hrr25 sequence outside the kinase domain is differentially required for three different cellular processes. Despite high similarity in their kinase domains, the four casein kinase I proteins have no significant sequence homology in their C-terminal regions. Both Yck1 and Yck2 have a glutamine-rich domain at their C-terminal regions, while the C-terminal region of Yck3 is enriched in serine and asparagine residues. Yck1, Yck2, and Yck3 all have a Cys-Cys dipeptide at their C termini, which is required for palmitoylation and their subsequent membrane targeting (44–48). The involvement of Hrr25 in multiple cellular processes is also reflected in its cellular localization. It has been reported that it localizes in the nucleus, spindle pole bodies, the bud neck, the septin ring, the Golgi complex, and the P-bodies (49–51). Different sequence elements of the C-terminal region of Hrr25 are likely to be required for interaction with different partners in various cellular processes or different cellular localization. We hypothesize that the C-terminal region of Hrr25 may mediate an interaction with a target protein or proteins important for growth based on data presented in Fig. 5E. It remains possible that the poor growth is associated with the observation that the tailless Hrr25 is not expressed as well as full-length Hrr25 (Fig. 5B). However, the C-terminal region is completely dispensable for cell morphogenesis (Fig. 5F), which could be related to its function and localization to the bud neck and/or septin rings. The role of the C-terminal region in the regulation of Haa1 is complicated by the fact that the kinase domain alone is expressed at a lower level than the full-length protein (Fig. 5B). The kinase domain itself, even at a reduced level, can partially complement hrr25Δ in mediating the expression of Haa1 target genes, suggesting that the C-terminal region of Hrr25 is partially required, if not dispensable, for Haa1 regulation (Fig. 5C). Consistent with this result, the kinase domain alone maintains an interaction with Haa1 (Fig. 6). The mechanism behind the reduced expression level of the kinase domain itself is still unclear. Construction and characterization of fine, nested truncation alleles in the C-terminal region of Hrr25 will provide valuable insights into the role of its C-terminal domain in multiple cellular processes.
MATERIALS AND METHODS
Strains, plasmids, growth media, and growth conditions.
The yeast strains and plasmids used in this study are listed in Tables 1 and 2, respectively. To generate the TPO2-lacZ reporter gene, two primers (5′-gtcaGGATCCACCAGTTCCTGAAACCT-3′ and 5′-gtcaAAGCTTCAACAGATTCTTGATCACTCAT-3′) (lowercase letters indicate nucleotides that are added to facilitate the digestion of PCR products by restriction endonucleases, and underlining indicates the introduced restriction sites) were used to amplify the DNA sequence from positions −1926 to +22 in the promoter region of TPO2 using BY4741 genomic DNA as the template. The resulting PCR product was cleaved with BamHI and HindIII and fused in frame to the Escherichia coli lacZ gene in the centromeric plasmid pWEJ (52) to form plasmid pZL3158. To generate the YRO2-lacZ reporter gene, two primers (5′-gtcaGAATTCCCGATATAACTACCAC-3′ and 5′-gtcaGTCGACCAACATAATCAGACATTTTGATGC-3′) were used to amplify the DNA sequence from positions −1929 to +16 in the promoter region of YRO2. The PCR product was cleaved with EcoRI and SalI and cloned into pWEJ to form plasmid pZL3164. Yeast strains were grown at 30°C in YPD medium (1% Bacto yeast extract, 2% Bacto peptone, 2% glucose), YNBcasD medium (0.67% yeast nitrogen base, 1% Casamino Acids, 2% dextrose), synthetic dextrose minimal medium (SD) (0.67% yeast nitrogen base without amino acids, 2% glucose), complete supplement mixture (CSM) medium (0.67% yeast nitrogen base without amino acids, 2% glucose, 0.6 g/liter CSM minus histidine, leucine, and tryptophan), or MM4 (0.17% yeast nitrogen base without amino acids and ammonium sulfate, 0.265% ammonium sulfate, 2% glucose, adjusted to pH 4) with or without 60 mM acetic acid. SD medium was supplemented with leucine, histidine, methionine, lysine, and uracil at standard concentrations to cover auxotrophic requirements if required (53). For yeast two-hybrid analysis involving the HIS3 reporter gene under the control of Gal4, 3-amino-1,2,4-triazole was added to the CSM medium. Agar was added at a final concentration of 2% for plate medium.
TABLE 1.
S. cerevisiae strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| LRB341 | MATa ura3-52 leu2 his3 | 58 |
| LRB362 | MATa ura3-52 leu2 his3 yck1-1::ura3 yck2-2ts | 58 |
| BY4741 | MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 | Saccharomyces Genome Deletion Project |
| ZLY4260 | BY4741 yck3Δ::kanMX4 | Saccharomyces Genome Deletion Project |
| ZLY962 | BY4743 hrr25Δ::kanMX4/HRR25 | Saccharomyces Genome Deletion Project |
| ZLY4467 | BY4741 hrr25Δ::kanMX4 [pRS415-HRR25(E52D)] | This study |
| ZLY4501 | BY4741 hrr25Δ::kanMX4 | This study |
| ZLY4043 | BY4741 haa1Δ::kanMX4 | Saccharomyces Genome Deletion Project |
| ZLY4637 | BY4741 hrr25Δ::kanMX4 haa1::HIS3 [pRS415-HRR25(E52D)] | This study |
| ZLY5057 | LRB341 haa1::HIS3 | This study |
| ZLY5058 | LRB362 haa1::HIS3 | This study |
| ZLY5173 | BY4741 yck3Δ::kanMX4 haa1::HIS3 | This study |
| ZLY5046 | MATα ura3 leu2 his3 met15 lys2 hrr25Δ::kanMX4 haa1::HIS3 | This study |
| ZLY4479 | MATα ura3 leu2 his3 met15 lys2 hrr25Δ::kanMX4 | This study |
| AH109 | MATa ura3-52 his3-200 trp1-901 leu2-3,112 gal4Δ gal80Δ URA3::MEL1UAS-MEL1TATA-lacZ GAL2UAS-GAL2TATA-ADE2 LYS2::GAL1UAS-GAL1TATA-HIS3 | Clontech Laboratories, Inc. |
TABLE 2.
Plasmids used in this study
| Plasmid | Descriptiona | Source |
|---|---|---|
| pZL3158 | pRS416-TPO2-lacZ, expressing TPO2-lacZ reporter gene with 1,926-bp TPO2 promoter sequence fused to lacZ coding sequence | This study |
| pZL3164 | pRS416-YRO2-lacZ, expressing YRO2-lacZ reporter gene with 1,929-bp YRO2 promoter sequence fused to lacZ coding sequence | This study |
| pZD359 | pRS415-HRR25(E52D), expressing missense HRR25 mutant allele substituting for glutamate residue 52 with aspartate | This study |
| pST428 | pRS416-HAA1-GFP, expressing HAA1-GFP from endogenous promoter, with GFP fused at C-terminal end of Haa1 | This study |
| pZL3210 | pRS416-HAA1-HA, expressing HAA1 from endogenous promoter with 3×HA epitope tag at C terminus | This study |
| pZL3338 | pRS415-HRR25-myc, expressing HRR25 from endogenous promoter with 3×myc epitope tag at C terminus | This study |
| pZL3361 | pRS415-HRR25(KD)-myc, expressing N-terminal kinase domain of Hrr25 (residues 1–302) from endogenous promoter with 3×myc epitope tag at C terminus | This study |
| pACTII | Yeast 2-hybrid vector encoding GAD | Clontech Laboratories, Inc. |
| pZL3539 | pRS415-TEF-GAD, with coding sequence of GAD from pACT-II cloned into XbaI site of pRS415-TEF | This study |
| pZL3554 | pRS415-TEF-GAD-HAA1, with HAA1 open reading frame cloned into BamHI and XhoI sites of pZL3539 | This study |
| pGBKT7 | Yeast 2-hybrid vector encoding GBD | Clontech Laboratories, Inc. |
| pZL3542 | pRS424-ADH1-GBD, with coding sequence of GBD from pGBKT7 cloned into XbaI site of pRS424-ADH1 | This study |
| pZL3557 | pRS424-ADH1-GBD-HRR25, with HRR25 open reading frame cloned into BamHI and SalI sites of pZL3542 | This study |
| pZL3736 | pRS424-ADH1-GBD-HRR25(KD), with DNA sequence encoding N-terminal kinase domain of Hrr25 (residues 1–302) cloned into BamHI and SalI sites of pZL3542 |
GAD, Gal4 transcriptional activation domain; GBD, Gal4 DNA-binding domain.
Yeast transformation and β-galactosidase assays.
For transformation, yeast cells were grown in YPD medium and transformed using a high-efficiency method (54). YNBcasD plates or SD plates with appropriate amino acids were used to select for yeast transformants. For β-galactosidase assays, yeast strains were grown in MM4 with or without 60 mM acetic acid at 30°C for at least 6 generations to reach an optical density at 600 nm (OD600) of 0.5 to 0.8 before collection. Cells were collected by centrifugation, and β-galactosidase activity assays were conducted as described previously (53). Two to four independent cultures were grown, and assays were carried out in duplicate for each sample. Means of β-galactosidase assay results from independent cultures of strain-plasmid combinations were compared by t test using the GraphPad Prism 5 software. An asterisk in a figure indicates a significant difference in the means of two groups of data (P < 0.05).
Cell extract preparation, phosphatase treatment, and Western blotting.
Total cellular proteins were prepared from yeast cells treated with 7.5% β-mercaptoethanol and 1.85 N NaOH solution and precipitated with trichloroacetic acid (TCA) as described previously (55). For phosphatase treatment, neutralized TCA-precipitated total cellular proteins were incubated with 400 U of λ protein phosphatase (PPase) (New England BioLabs, Inc.) in a final volume of 20 μl at 30°C for 90 min. When indicated, phosphatase inhibitors (1 μM sodium orthovanadate, 10 mM β-glycerol phosphate, 10 mM sodium pyrophosphate, 10 mM NaF) were added to inhibit λ PPase activity. Pellets from TCA precipitation were resuspended in 1× SDS-PAGE loading buffer with 100 mM dithiothreitol (DTT). Protein samples were boiled for 3 min before being separated by SDS-PAGE. Proteins were then transferred to nitrocellulose membrane for immunoblotting. Haa1 with a C-terminal 3×HA epitope tag was probed with rat monoclonal high-affinity anti-HA antibody (3F10 [Roche]) followed by goat anti-rat horseradish peroxidase-conjugated polyclonal secondary antibody (Jackson ImmunoResearch Laboratories, Inc.). Ilv5 was probed with rabbit anti-Ilv5 polyclonal antibodies. Chemiluminescence images of Western blots were captured using Bio-Rad Chemi-Doc photo documentation system and processed using the Bio-Rad Quantity One software.
Generation of an hrr25(E52D) mutant allele.
A glutamate-to-aspartate mutation at residue 52 was introduced via site-directed mutagenesis by overlap-extension PCR as described previously (56). Briefly, a primer pair (5′-gtcaTCTAGACCAGTGTCTGAGTCATG-3′ and 5′-TTAAGTATCTGTAGACGCGGGAGTCATAGTCCAATTGAGGATGTCT-3′) was used to amplify an 841-bp HRR25 promoter and the coding sequence to Leu 59, including the E52D mutation, using high-fidelity Pfu DNA polymerase. Another primer pair (5′-AGACATCCTCAATTGGACTATGACTCCCGCGTCTACAGATACTTAA-3′ and 5′-gtcaGTCGACGTCTTCTCAGAGGCCCTCCT-3′) was used to amplify the coding sequence from Arg 45 and a 332-bp sequence downstream of the open reading frame. These two PCR products were purified and subjected to overlap extension in the presence of the primers (5′-gtcaTCTAGACCAGTGTCTGAGTCATG-3′ and 5′-gtcaGTCGACGTCTTCTCAGAGGCCCTCCT-3′). The final PCR product carrying the E52D mutation was digested with XbaI and SalI restriction endonucleases and cloned into XbaI and XhoI sites of the vector pRS415.
Yeast two-hybrid analysis.
The HAA1 open reading frame was amplified by PCR and cloned into the BamHI and XhoI sites of pZL3539 (pRS415-TEF-GAD), a Gal4 transcriptional activation domain (GAD) vector to generate pRS415-TEF-GAD-HAA1. pZL3539 was constructed by cloning the coding sequence of Gal4 transcriptional activation domain from pACT2 (Clontech Laboratories, Inc.) into the XbaI site of pRS415-TEF (57). The HRR25 open reading frame was cloned into the BamHI and SalI sites of pZL3542 (pRS424-ADH1-GBD), a Gal4 DNA binding domain (GBD) vector, to generate pRS424-ADH1-GBD-HRR25. pZL3542 was constructed by cloning the coding sequence of the GBD from pGBKT7 (Clontech Laboratories, Inc.) into the XbaI site of pRS424-ADH1 (57). The yeast two-hybrid strain AH109 (Clontech Laboratories, Inc.) was transformed with two plasmids expressing GBD and GAD, GBD and GAD-Haa1, GBD-Hrr25 and GAD, or GBD-Hrr25 and GAD-Haa1, and transformants were selected on SD medium without leucine and tryptophan. Transformants were streaked on SD plate supplemented with adenine and histidine for the analysis of an ADE2 reporter gene under the control of the GAL2 promoter. CSM medium without leucine, tryptophan, and histidine was used for the analysis of a HIS3 reporter gene under the control of the GAL1 promoter. 3-Amino-1,2,4-triazole (3-AT), an inhibitor of His3, was added to CSM medium when required.
Fluorescence microscopy analysis of GFP-tagged proteins.
Cells expressing Haa1-GFP were grown in MM4 without and with 60 mM acetic acid overnight to an OD600 of 0.6 to 0.8. Cells were “concentrated” by applying 3-μl cell cultures onto the slide and letting cells settle down for 3 min before coverslips were applied. GFP fluorescence and differential interference contrast (DIC) images of live cells were immediately captured using a Nikon Eclipse E800 microscope equipped with an HBO 100 W/2 mercury arc lamp, a Nikon Plan Fluor 100× objective lens, and a Nikon B-2E/C medium band excitation bandpass filter set (excitation light, 465 to 495 nm; emission light, 515 to 555 nm). Images were acquired with a Photometrics Coolsnap fx charge-coupled device (CCD) camera and Metamorph imaging software (Molecular Devices, Sunnyvale, CA) and processed using ImageJ (National Institutes of Health) and Adobe Photoshop (Mountain View, CA) software.
Supplementary Material
ACKNOWLEDGMENTS
We thank Lucy Robinson for yeast strains, Zhejun Dong, Manika Bhondeley, and Natasha M. Bourgeois for technical support, the W. M. Keck Foundation for the Keck Facility, and Robin Rowe for sequencing.
This research was supported by grants from the NIH (1R15GM121998-01) and the Office of Research and Sponsored Programs of University of New Orleans to Z.L. and the Louisiana Governor's Biotechnology Initiative to M.E.C.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00672-17.
REFERENCES
- 1.Fernandes AR, Mira NP, Vargas RC, Canelhas I, Sa-Correia I. 2005. Saccharomyces cerevisiae adaptation to weak acids involves the transcription factor Haa1p and Haa1p-regulated genes. Biochem Biophys Res Commun 337:95–103. doi: 10.1016/j.bbrc.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 2.Simoes T, Mira NP, Fernandes AR, Sa-Correia I. 2006. The SPI1 gene, encoding a glycosylphosphatidylinositol-anchored cell wall protein, plays a prominent role in the development of yeast resistance to lipophilic weak-acid food preservatives. Appl Environ Microbiol 72:7168–7175. doi: 10.1128/AEM.01476-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abbott DA, Suir E, van Maris AJ, Pronk JT. 2008. Physiological and transcriptional responses to high concentrations of lactic acid in anaerobic chemostat cultures of Saccharomyces cerevisiae. Appl Environ Microbiol 74:5759–5768. doi: 10.1128/AEM.01030-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mira NP, Becker JD, Sa-Correia I. 2010. Genomic expression program involving the Haa1p-regulon in Saccharomyces cerevisiae response to acetic acid. OMICS 14:587–601. doi: 10.1089/omi.2010.0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keller G, Ray E, Brown PO, Winge DR. 2001. Haa1, a protein homologous to the copper-regulated transcription factor Ace1, is a novel transcriptional activator. J Biol Chem 276:38697–38702. doi: 10.1074/jbc.M107131200. [DOI] [PubMed] [Google Scholar]
- 6.Piper P, Calderon CO, Hatzixanthis K, Mollapour M. 2001. Weak acid adaptation: the stress response that confers yeasts with resistance to organic acid food preservatives. Microbiology 147:2635–2642. doi: 10.1099/00221287-147-10-2635. [DOI] [PubMed] [Google Scholar]
- 7.Mira NP, Teixeira MC, Sa-Correia I. 2010. Adaptive response and tolerance to weak acids in Saccharomyces cerevisiae: a genome-wide view. OMICS 14:525–540. doi: 10.1089/omi.2010.0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teixeira MC, Fernandes AR, Mira NP, Becker JD, Sa-Correia I. 2006. Early transcriptional response of Saccharomyces cerevisiae to stress imposed by the herbicide 2,4-dichlorophenoxyacetic acid. FEMS Yeast Res 6:230–248. doi: 10.1111/j.1567-1364.2006.00041.x. [DOI] [PubMed] [Google Scholar]
- 9.Mira NP, Lourenco AB, Fernandes AR, Becker JD, Sa-Correia I. 2009. The RIM101 pathway has a role in Saccharomyces cerevisiae adaptive response and resistance to propionic acid and other weak acids. FEMS Yeast Res 9:202–216. doi: 10.1111/j.1567-1364.2008.00473.x. [DOI] [PubMed] [Google Scholar]
- 10.Abbott DA, Knijnenburg TA, de Poorter LM, Reinders MJ, Pronk JT, van Maris AJ. 2007. Generic and specific transcriptional responses to different weak organic acids in anaerobic chemostat cultures of Saccharomyces cerevisiae. FEMS Yeast Res 7:819–833. doi: 10.1111/j.1567-1364.2007.00242.x. [DOI] [PubMed] [Google Scholar]
- 11.Kawahata M, Masaki K, Fujii T, Iefuji H. 2006. Yeast genes involved in response to lactic acid and acetic acid: acidic conditions caused by the organic acids in Saccharomyces cerevisiae cultures induce expression of intracellular metal metabolism genes regulated by Aft1p. FEMS Yeast Res 6:924–936. doi: 10.1111/j.1567-1364.2006.00089.x. [DOI] [PubMed] [Google Scholar]
- 12.Tomitori H, Kashiwagi K, Asakawa T, Kakinuma Y, Michael AJ, Igarashi K. 2001. Multiple polyamine transport systems on the vacuolar membrane in yeast. Biochem J 353:681–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Destruelle M, Holzer H, Klionsky DJ. 1994. Identification and characterization of a novel yeast gene: the YGP1 gene product is a highly glycosylated secreted protein that is synthesized in response to nutrient limitation. Mol Cell Biol 14:2740–2754. doi: 10.1128/MCB.14.4.2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mira NP, Henriques SF, Keller G, Teixeira MC, Matos RG, Arraiano CM, Winge DR, Sa-Correia I. 2011. Identification of a DNA-binding site for the transcription factor Haa1, required for Saccharomyces cerevisiae response to acetic acid stress. Nucleic Acids Res 39:6896–6907. doi: 10.1093/nar/gkr228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maiorella B, Blanch HW, Wilke CR. 1983. By-product inhibition effects on ethanolic fermentation by Saccharomyces cerevisiae. Biotechnol Bioeng 25:103–121. doi: 10.1002/bit.260250109. [DOI] [PubMed] [Google Scholar]
- 16.Sanchez i Nogue V, Narayanan V, Gorwa-Grauslund MF. 2013. Short-term adaptation improves the fermentation performance of Saccharomyces cerevisiae in the presence of acetic acid at low pH. Appl Microbiol Biotechnol 97:7517–7525. doi: 10.1007/s00253-013-5093-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmqvist E, Grage H, Meinander NQ, Hahn-Hagerdal B. 1999. Main and interaction effects of acetic acid, furfural, and p-hydroxybenzoic acid on growth and ethanol productivity of yeasts. Biotechnol Bioeng 63:46–55. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka K, Ishii Y, Ogawa J, Shima J. 2012. Enhancement of acetic acid tolerance in Saccharomyces cerevisiae by overexpression of the HAA1 gene, encoding a transcriptional activator. Appl Environ Microbiol 78:8161–8163. doi: 10.1128/AEM.02356-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inaba T, Watanabe D, Yoshiyama Y, Tanaka K, Ogawa J, Takagi H, Shimoi H, Shima J. 2013. An organic acid-tolerant HAA1-overexpression mutant of an industrial bioethanol strain of Saccharomyces cerevisiae and its application to the production of bioethanol from sugarcane molasses. AMB Express 3:74. doi: 10.1186/2191-0855-3-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakihama Y, Hasunuma T, Kondo A. 2015. Improved ethanol production from xylose in the presence of acetic acid by the overexpression of the HAA1 gene in Saccharomyces cerevisiae. J Biosci Bioeng 119:297–302. doi: 10.1016/j.jbiosc.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 21.Malcher M, Schladebeck S, Mosch HU. 2011. The Yak1 protein kinase lies at the center of a regulatory cascade affecting adhesive growth and stress resistance in Saccharomyces cerevisiae. Genetics 187:717–730. doi: 10.1534/genetics.110.125708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugiyama M, Akase SP, Nakanishi R, Horie H, Kaneko Y, Harashima S. 2014. Nuclear localization of Haa1, which is linked to its phosphorylation status, mediates lactic acid tolerance in Saccharomyces cerevisiae. Appl Environ Microbiol 80:3488–3495. doi: 10.1128/AEM.04241-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshida K, Blobel G. 2001. The karyopherin Kap142p/Msn5p mediates nuclear import and nuclear export of different cargo proteins. J Cell Biol 152:729–740. doi: 10.1083/jcb.152.4.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swaney DL, Beltrao P, Starita L, Guo A, Rush J, Fields S, Krogan NJ, Villen J. 2013. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat Methods 10:676–682. doi: 10.1038/nmeth.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Albuquerque CP, Smolka MB, Payne SH, Bafna V, Eng J, Zhou H. 2008. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol Cell Proteomics 7:1389–1396. doi: 10.1074/mcp.M700468-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoekstra MF, Liskay RM, Ou AC, DeMaggio AJ, Burbee DG, Heffron F. 1991. HRR25, a putative protein kinase from budding yeast: association with repair of damaged DNA. Science 253:1031–1034. doi: 10.1126/science.1887218. [DOI] [PubMed] [Google Scholar]
- 27.Robinson LC, Hubbard EJ, Graves PR, DePaoli-Roach AA, Roach PJ, Kung C, Haas DW, Hagedorn CH, Goebl M, Culbertson MR. 1992. Yeast casein kinase I homologues: an essential gene pair. Proc Natl Acad Sci U S A 89:28–32. doi: 10.1073/pnas.89.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abdel-Sater F, El Bakkoury M, Urrestarazu A, Vissers S, Andre B. 2004. Amino acid signaling in yeast: casein kinase I and the Ssy5 endoprotease are key determinants of endoproteolytic activation of the membrane-bound Stp1 transcription factor. Mol Cell Biol 24:9771–9785. doi: 10.1128/MCB.24.22.9771-9785.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moriya H, Johnston M. 2004. Glucose sensing and signaling in Saccharomyces cerevisiae through the Rgt2 glucose sensor and casein kinase I. Proc Natl Acad Sci U S A 101:1572–1577. doi: 10.1073/pnas.0305901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ray P, Basu U, Ray A, Majumdar R, Deng H, Maitra U. 2008. The Saccharomyces cerevisiae 60 S ribosome biogenesis factor Tif6p is regulated by Hrr25p-mediated phosphorylation. J Biol Chem 283:9681–9691. doi: 10.1074/jbc.M710294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mochida K, Ohsumi Y, Nakatogawa H. 2014. Hrr25 phosphorylates the autophagic receptor Atg34 to promote vacuolar transport of alpha-mannosidase under nitrogen starvation conditions. FEBS Lett 588:3862–3869. doi: 10.1016/j.febslet.2014.09.032. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka C, Tan LJ, Mochida K, Kirisako H, Koizumi M, Asai E, Sakoh-Nakatogawa M, Ohsumi Y, Nakatogawa H. 2014. Hrr25 triggers selective autophagy-related pathways by phosphorylating receptor proteins. J Cell Biol 207:91–105. doi: 10.1083/jcb.201402128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghalei H, Schaub FX, Doherty JR, Noguchi Y, Roush WR, Cleveland JL, Stroupe ME, Karbstein K. 2015. Hrr25/CK1delta-directed release of Ltv1 from pre-40S ribosomes is necessary for ribosome assembly and cell growth. J Cell Biol 208:745–759. doi: 10.1083/jcb.201409056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Z, Thornton J, Spirek M, Butow RA. 2008. Activation of the SPS amino acid-sensing pathway in Saccharomyces cerevisiae correlates with the phosphorylation state of a sensor component, Ptr3. Mol Cell Biol 28:551–563. doi: 10.1128/MCB.00929-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Güldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kötter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 36.Mehlgarten C, Jablonowski D, Breunig KD, Stark MJ, Schaffrath R. 2009. Elongator function depends on antagonistic regulation by casein kinase Hrr25 and protein phosphatase Sit4. Mol Microbiol 73:869–881. doi: 10.1111/j.1365-2958.2009.06811.x. [DOI] [PubMed] [Google Scholar]
- 37.Dhillon N, Hoekstra MF. 1994. Characterization of two protein kinases from Schizosaccharomyces pombe involved in the regulation of DNA repair. EMBO J 13:2777–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ueta R, Fujiwara N, Iwai K, Yamaguchi-Iwai Y. 2007. Mechanism underlying the iron-dependent nuclear export of the iron-responsive transcription factor Aft1p in Saccharomyces cerevisiae. Mol Biol Cell 18:2980–2990. doi: 10.1091/mbc.E06-11-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taberner FJ, Quilis I, Igual JC. 2009. Spatial regulation of the start repressor Whi5. Cell Cycle 8:3010–3018. doi: 10.4161/cc.8.18.9621. [DOI] [PubMed] [Google Scholar]
- 40.Kaffman A, Rank NM, O'Neill EM, Huang LS, O'Shea EK. 1998. The receptor Msn5 exports the phosphorylated transcription factor Pho4 out of the nucleus. Nature 396:482–486. doi: 10.1038/24898. [DOI] [PubMed] [Google Scholar]
- 41.DeVit MJ, Johnston M. 1999. The nuclear exportin Msn5 is required for nuclear export of the Mig1 glucose repressor of Saccharomyces cerevisiae. Curr Biol 9:1231–1241. doi: 10.1016/S0960-9822(99)80503-X. [DOI] [PubMed] [Google Scholar]
- 42.Boustany LM, Cyert MS. 2002. Calcineurin-dependent regulation of Crz1p nuclear export requires Msn5p and a conserved calcineurin docking site. Genes Dev 16:608–619. doi: 10.1101/gad.967602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bakhrat A, Baranes-Bachar K, Reshef D, Voloshin O, Krichevsky O, Raveh D. 2008. Nuclear export of Ho endonuclease of yeast via Msn5. Curr Genet 54:271–281. doi: 10.1007/s00294-008-0216-8. [DOI] [PubMed] [Google Scholar]
- 44.Roth AF, Feng Y, Chen L, Davis NG. 2002. The yeast DHHC cysteine-rich domain protein Akr1p is a palmitoyl transferase. J Cell Biol 159:23–28. doi: 10.1083/jcb.200206120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng Y, Davis NG. 2000. Akr1p and the type I casein kinases act prior to the ubiquitination step of yeast endocytosis: Akr1p is required for kinase localization to the plasma membrane. Mol Cell Biol 20:5350–5359. doi: 10.1128/MCB.20.14.5350-5359.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Babu P, Deschenes RJ, Robinson LC. 2004. Akr1p-dependent palmitoylation of Yck2p yeast casein kinase 1 is necessary and sufficient for plasma membrane targeting. J Biol Chem 279:27138–27147. doi: 10.1074/jbc.M403071200. [DOI] [PubMed] [Google Scholar]
- 47.Roth AF, Wan J, Bailey AO, Sun B, Kuchar JA, Green WN, Phinney BS, Yates JR III, Davis NG. 2006. Global analysis of protein palmitoylation in yeast. Cell 125:1003–1013. doi: 10.1016/j.cell.2006.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun B, Chen L, Cao W, Roth AF, Davis NG. 2004. The yeast casein kinase Yck3p is palmitoylated, then sorted to the vacuolar membrane with AP-3-dependent recognition of a YXXPhi adaptin sorting signal. Mol Biol Cell 15:1397–1406. doi: 10.1091/mbc.E03-09-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lord C, Bhandari D, Menon S, Ghassemian M, Nycz D, Hay J, Ghosh P, Ferro-Novick S. 2011. Sequential interactions with Sec23 control the direction of vesicle traffic. Nature 473:181–186. doi: 10.1038/nature09969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lusk CP, Waller DD, Makhnevych T, Dienemann A, Whiteway M, Thomas DY, Wozniak RW. 2007. Nup53p is a target of two mitotic kinases, Cdk1p and Hrr25p. Traffic 8:647–660. doi: 10.1111/j.1600-0854.2007.00559.x. [DOI] [PubMed] [Google Scholar]
- 51.Zhang B, Shi Q, Varia SN, Xing S, Klett BM, Cook LA, Herman PK. 2016. The activity-dependent regulation of protein kinase stability by the localization to P-bodies. Genetics 203:1191–1202. doi: 10.1534/genetics.116.187419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chelstowska A, Liu Z, Jia Y, Amberg D, Butow RA. 1999. Signalling between mitochondria and the nucleus regulates the expression of a new d-lactate dehydrogenase activity in yeast. Yeast 15:1377–1391. [DOI] [PubMed] [Google Scholar]
- 53.Amberg DC, Burke DJ, Strathern JN. 2005. Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 54.Gietz D, St Jean A, Woods RA, Schiestl RH. 1992. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res 20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yaffe MP, Schatz G. 1984. Two nuclear mutations that block mitochondrial protein import in yeast. Proc Natl Acad Sci U S A 81:4819–4823. doi: 10.1073/pnas.81.15.4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 57.Mumberg D, Muller R, Funk M. 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- 58.Panek HR, Stepp JD, Engle HM, Marks KM, Tan PK, Lemmon SK, Robinson LC. 1997. Suppressors of YCK-encoded yeast casein kinase 1 deficiency define the four subunits of a novel clathrin AP-like complex. EMBO J 16:4194–4204. doi: 10.1093/emboj/16.14.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.