

ABSTRACT
Plague is a flea-borne rodent-associated zoonotic disease caused by Yersinia pestis. The disease is characterized by epizootics with high rodent mortalities, punctuated by interepizootic periods when the bacterium persists in an unknown reservoir. This study investigates the interaction between Y. pestis and the ubiquitous soil free-living amoeba (FLA) Acanthamoeba castellanii to assess if the bacterium can survive within soil amoebae and whether intracellular mechanisms are conserved between infection of mammalian macrophages and soil amoebae. The results demonstrate that during coculture with amoebae, representative Y. pestis strains of epidemic biovars Medievalis, Orientalis, and Antiqua are phagocytized and able to survive within amoebae for at least 5 days. Key Y. pestis determinants of the intracellular interaction of Y. pestis and phagocytic macrophages, PhoP and the type three secretion system (T3SS), were then tested for their roles in the Y. pestis-amoeba interaction. Consistent with a requirement for the PhoP transcriptional activator in the intracellular survival of Y. pestis in macrophages, a PhoP mutant is unable to survive when cocultured with amoebae. Additionally, induction of the T3SS blocks phagocytic uptake of Y. pestis by amoebae, similar to that which occurs during macrophage infection. Electron microscopy revealed that in A. castellanii, Y. pestis resides intact within spacious vacuoles which were characterized using lysosomal trackers as being separated from the lysosomal compartment. This evidence for prolonged survival and subversion of intracellular digestion of Y. pestis within FLA suggests that protozoa may serve as a protective soil reservoir for Y. pestis.
IMPORTANCE Yersinia pestis is a reemerging flea-borne zoonotic disease. Sylvatic plague cycles are characterized by an epizootic period during which the disease spreads rapidly, causing high rodent mortality, and an interepizootic period when the bacterium quiescently persists in an unknown reservoir. An understanding of the ecology of Y. pestis in the context of its persistence in the environment and its reactivation to initiate a new epizootic cycle is key to implementing novel surveillance strategies to more effectively predict and prevent new disease outbreaks. Here, we demonstrate prolonged survival and subversion of intracellular digestion of Y. pestis within a soil free-living amoeba. This suggests the potential role for protozoa as a protective soil reservoir for Y. pestis, which may help explain the recrudescence of plague epizootics.
KEYWORDS: free-living amoeba, interepizootic plague, Yersinia pestis
INTRODUCTION
Plague caused by the Gram-negative bacterium Yersinia pestis is a flea-borne rodent-associated zoonotic disease with complex natural sylvatic cycles. These cycles are composed of an interepizootic/quiescent stage that supports the persistence of a Y. pestis reservoir and an epizootic phase characterized by high rodent mortality rates that increases the risk of human disease. The plague is difficult to eradicate because it is widespread in wild rodents and their associated fleas in foci of endemicity even during quiescence. The CDC has classified the plague as a reemerging disease since the early 1990s due to the reoccurrence of numerous plague outbreaks following long absences of the disease. For example, plague has reemerged after 57, 25, and 60 years in Algeria (1), Libya (2), and Madagascar (3), respectively. Understanding the interepizootic cycle of plague may shed light on the environmental reservoir of Y. pestis and its role in plague reemergence.
The plague bacterium, Y. pestis, emerged as a clonal variant of the free-living soilborne gastrointestinal pathogen Yersinia pseudotuberculosis. Comparative genome sequence analysis between these two Yersinia species reveals genomic degeneration of Y. pestis metabolic pathways. Such pathways may normally support a metabolic versatility to survive freely in the environment over prolonged periods (4–7); instead, Y. pestis appears to require a flea or rodent host. This supports the long-standing enzootic host model hypothesis for interepizootic plague persistence, which predicts that plague is maintained naturally in infected fleas feeding on a heterogeneous population of susceptible and relatively resistant hosts (8–10). There are, however, several factors that confound this theory. One is that the bacteremia levels of relatively resistant hosts are usually too low for flea acquisition of Y. pestis from the blood meal, which would be required to perpetuate the flea-rodent transmission cycle (11). In addition, a study that modeled plague reemergence based on a natural gerbil plague ecosystem in Kazakhstan, where epizootics occurred every 4 years, convincingly refutes the idea that a heterogeneous host population can support plague reemergence (9). Furthermore, surveillance of active plague foci during interepizootic periods does not frequently recover infected fleas and seropositive rodents. Finally, reemergence and epizootics are infrequent and can sometimes occur >50 years beyond the life span of fleas and hosts (1, 3).
An alternative but not mutually exclusive hypothesis is that soil serves as an interepizootic Y. pestis reservoir. This has been experimentally tested under conditions that assume that Y. pestis is free-living and metabolically active. These studies have been able to demonstrate the persistence of Y. pestis in soil but only for short periods (8, 10, 12). Ecologists have theorized that ubiquitous free-living soil amoebae could serve as a reservoir host for Y. pestis. Amoebae as a soil reservoir may reconcile the requirement for a host for Y. pestis with a soil niche for persistence (13, 14). This idea has been supported conceptually by two studies: (i) one that demonstrates that Y. pestis is extracellularly associated with the soil free-living amoeba (FLA) Hartmannella rhysodes, and (ii) the other, which reports that bacterivorous FLA are present at active plague foci in Russia (15, 16). More recently, we have demonstrated that Y. pseudotuberculosis, the clonal ancestor of Y. pestis, is also able to survive within A. castellanii trophozoites and cysts (17).
Bacterivorous FLA are unicellular eukaryotic organisms that commonly reside in soil and grow on detritus, from which they absorb organic molecules. Some pathogenic bacteria are, however, resistant to destruction by FLA and instead remain safely harbored within these organisms (18, 19). In such cases, FLA serve as a reservoir for pathogenic bacteria because they provide protection from hostile extracellular environments and can facilitate pathogen transmission to susceptible hosts (18, 20). Several bacterial pathogens, e.g., Legionella pneumophila (19, 21), Mycobacterium marinum (22), and Vibrio cholerae (23), have been demonstrated to be able to survive and replicate in amoebae. These pathogens use conserved mechanisms that favor their adaptation and survival in mammalian host immune cells, including macrophages, to facilitate their survival within FLA. Macrophages and amoebae are similar in that they both engulf bacteria by phagocytosis.
In its interaction with mammalian macrophages, Y. pestis has the ability to survive, replicate, and resist phagocytosis under specific conditions (24–26). The two major determinants that mediate these abilities are the PhoP transcriptional regulator (27–29) and the type III secretion system (T3SS) and Yop effector proteins encoded on the pCD1 plasmid (30, 31). The PhoP regulator is required for intracellular survival and directs the expression of genes involved in resistance to antimicrobial peptides, as well as those involved in acid, oxidative, and hyperosmotic stresses (27, 32, 33). The T3SS is a needle-like injectisome protein complex that serves as a conduit to deliver secreted effector proteins into host cells in order to inhibit phagocytosis (31, 34).
In this study, we address two key questions regarding the Y. pestis-FLA interaction. The first is whether the FLA A. castellanii phagocytizes Y. pestis and whether the engulfed bacterium survives this normally destructive process. The second is to determine whether mechanisms used by Y. pestis to survive within mammalian macrophages are conserved and allow survival within FLA. The outcomes of these experiments are presented and discussed in the context of plague survival in the interepizootic period.
RESULTS
Y. pestis is phagocytized and exhibits prolonged survival within A. castellanii trophozoites.
The ability of Y. pestis to be phagocytized by or invade amoebae is referred to here as invasion frequency and was determined 3 h post-coculture with initial gentamicin treatment (Fig. 1). The interaction of amoebae with Y. pestis was unknown; hence, multiplicity of infection (MOI) values of 1 (low) and 100 (high) were assessed in coculture assays. At an MOI of 100, all strains were phagocytized by amoebae at a similar frequency of ∼1% or greater, except for the ΔphoP mutant strain, which exhibited a significantly lower invasion frequency. This lower invasion frequency was restored in its isogenic phoP-complemented strain ΔphoP pLG::phoP (Fig. 1). The invasion frequency at an MOI of 1 relative to that at an MOI of 100 was significantly lower for all strains, with the exception of the KIM6+ and Kuma D7 mutants, which showed no change in invasion frequency (Fig. 1).
FIG 1.

Invasion frequency of Y. pestis strains cocultured with A. castellanii ATCC 30010. White bars indicate an MOI of 1 and gray bars an MOI of 100. Error bars represent mean ± standard deviation (SD) of the results from 3 to 6 independent experiments. Statistically significant differences are indicated by ** for P < 0.01 and by *** for P < 0.001, and ns means not statistically significant.
Survival assays were conducted to determine if phagocytized bacteria were able to survive and/or replicate in amoebae. For these assays, cocultures were maintained in gentamicin. At MOIs of 1 and 100, all the strains, except for the ΔphoP mutant, were able to survive over the maximal testing period of 120 h (Fig. 2A and B; see also Fig. S1 in the supplemental material). In coculture assays conducted at 25°C using an MOI of 100, a significant, approximately 1-log10 decrease in CFU occurs in the first 24 h in all strains, except for CO92 and the phoP-complemented strain (Fig. 2A). A further small and gradual decrease in the number of intracellular bacteria was observed by 5 days of coculture; nevertheless, intracellular CFU were maintained between 103 and 104 CFU/ml for all strains by 5 days of coculture. In contrast, at an MOI of 1, there was no significant decrease in bacterial CFU in the first 24 h, and no difference in the bacterial CFU was noted between 72 h and 120 h (Fig. S1).
FIG 2.
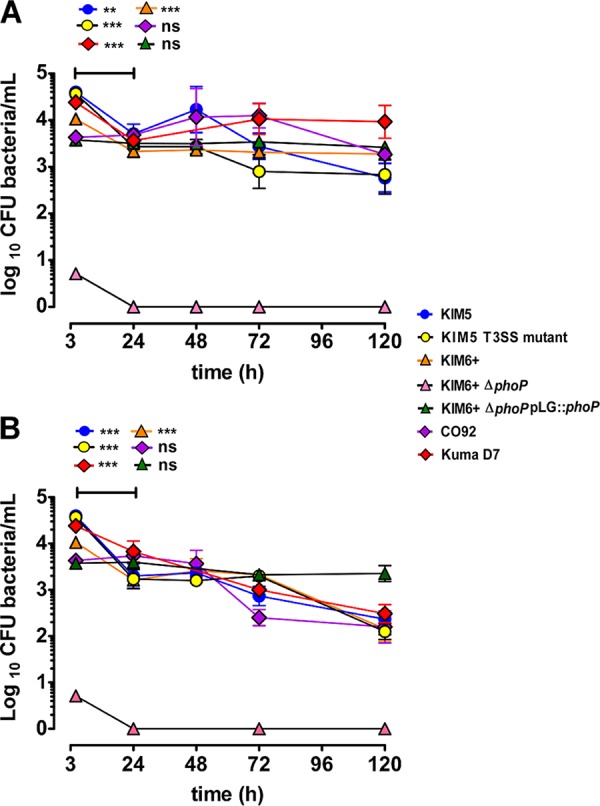
Prolonged intra-amoebal survival of Y. pestis strains. Intracellular survival of Y. pestis in cocultures maintained in gentamicin at 25°C (A) and 4°C (B). Error bars represent the mean ± SD of the results from 3 to 6 independent experiments. Horizontal bars indicate comparisons between two time points. Statistically significant differences are indicated by ** for P < 0.01 and by *** for P < 0.001, and ns means not statistically significant.
Coculture assays conducted at 4°C (Fig. 2B) were aimed at assessing prolonged intra-amoebal survival of Y. pestis under temperature conditions that mimicked burrows during interepizootic winter months. For these assays (Fig. 2B), a trend similar to that described above for 25°C incubation assays was observed, except that here all strains survived within a range of 103 to 102 CFU per ml at 5 days post-coculture. We do observe a tendency for infected amoebae to slough off more easily during washing steps after prolonged coculture incubations at 4°C than at 25°C. This results in some loss of infected amoebae during washing steps.
The rapid decrease in bacterial CFU in the first 24 h at an MOI of 100 (Fig. 2A and B) versus no decrease at an MOI of 1 (Fig. S1) may suggest that phagocytic saturation of amoebae occurs at high MOIs, limiting bacterial uptake and resulting in bacterial exocytosis and subsequent extracellular killing by gentamicin. The Y. pestis strains used in this study are able to efficiently replicate at similar rates in protease peptone-yeast extract-glucose (PYG) medium (data not shown). As such, we reasoned that if phagocytic saturation occurs, in the absence of gentamicin maintenance, exocytosed bacteria would be able replicate extracellularly and reinvade amoebae. At the higher MOI of 100, we would then expect constant intracellular bacterial CFU, but using an MOI of 1, bacterial CFU should increase until saturation is achieved. This was tested in coculture assays without gentamicin maintenance dosing in representative KIM5, KIM6+, and KIM5 T3SS mutant strains. Consistent with our predictions, no significant difference in intracellular bacterial CFU was noted between 2 h and 120 h using an MOI of 100. In contrast, using an MOI of 1, an increase in CFU was observed for both KIM5 and the KIM5 T3SS mutant, with no subsequent significant difference in CFU for either of these strains between MOI 1 and 100 cocultures by 24 h and 120 h post-coculture (Fig. 3). Light microscopy was used to monitor coculture experiments to ensure the amoebae remained in trophozoite form during coculture.
FIG 3.
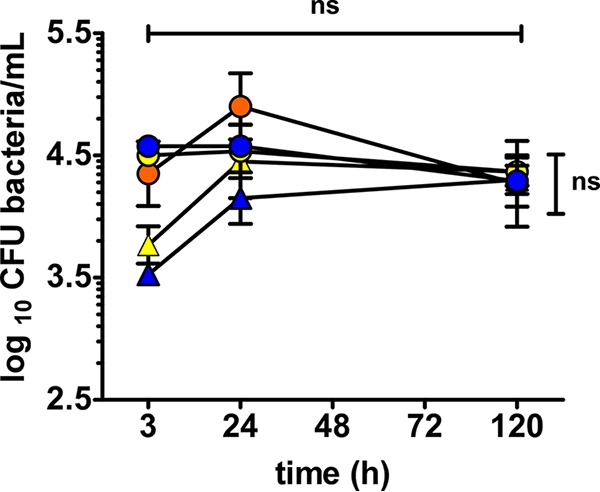
Prolonged intra-amoebal survival of Y. pestis KIM strains in the absence of gentamicin. Intracellular survival of Y. pestis excluding gentamicin maintenance of cocultures at 25°C. Error bars represent the mean ± SD of the results from 3 to 6 independent experiments. For assays with KIM5 (blue), KIM6+ (orange), and the KIM5 T3SS mutant (yellow), horizontal bar indicates comparison between two time points at an MOI of 100, while vertical bar indicates comparisons between MOI of 1 (triangles) and 100 (circles). ns means not statistically significant.
Y. pestis is localized in spacious vacuoles within A. castellanii trophozoites.
Electron microscopy was used to identify the intracellular niche of phagocytized Y. pestis. Vacuoles and pseudopodia (used to phagocytize food particles) are visible in both the uninfected (Fig. 4A) and infected (Fig. 4B and C) trophozoites, indicating that amoebae were not compromised upon infection with the bacteria and that they remain metabolically active. Lactate dehydrogenase release, an indicator of cytotoxicity, was not different between monocultures of bacteria or amoebae or in cocultures (Fig. S2). The KIM5 strain cultured with amoebae at an MOI of 100 at 24 h (Fig. 4B) and 7 days (Fig. 4C) showed that these bacteria are located within large spacious vacuoles surrounded by an intact membrane.
FIG 4.

Transmission electron micrographs of A. castellanii ATCC 30010 infected with Y. pestis KIM5. (A) An uninfected amoeba trophozoite. (B and C) Amoeba trophozoite infected with Y. pestis localized within spacious vacuoles at 24 h (B) and 7 days (C) post-coculture using MOI of 100. White arrows point to intracellular Y. pestis. V, vacuole; M, mitochondria; N, nucleus; P, pseudopodia.
Induction of the T3SS prevents phagocytic uptake of Y. pestis by A. castellanii.
The T3SS is known to be a major antiphagocytic determinant that prevents phagocytosis of Y. pestis by mammalian macrophages (24, 31). To determine if this function is conserved in the Y. pestis-amoeba interaction, the invasion frequency and early survival of the KIM5 (T3SS positive) and a KIM5 T3SS mutant were compared after growing these strains under conditions known to induce T3SS expression (low Ca2+ and 37°C). Under T3SS-inducing conditions, significantly fewer to no KIM5 bacteria were recovered from amoebae (Fig. 5A) relative to the bacterial recovery observed for the KIM5 T3SS mutant.
FIG 5.
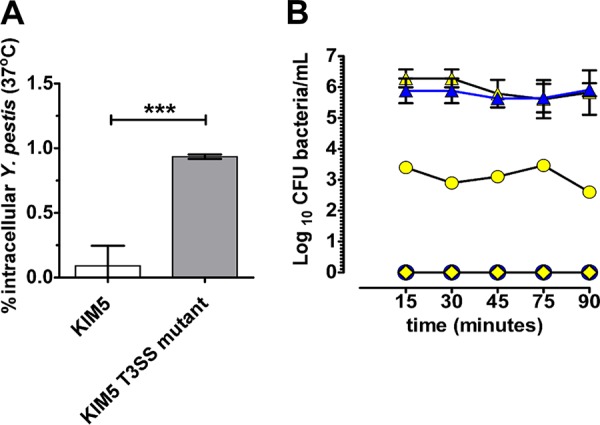
A. castellanii ATCC 30010 phagocytosis of Y. pestis KIM5 and KIM5 T3SS mutant grown under T3SS-inducing conditions. (A) Invasion frequency and early survival (3 h postinfection) of Y. pestis KIM5 and KIM5 T3SS mutant cocultured with A. castellanii ATCC 30010 under T3SS-inducing conditions. (B) Early intracellular survival of KIM5 (blue) and KIM5 T3SS mutant (yellow). ●, intracellular CFU of bacteria per milliliter ; ▲, CFU of bacteria per milliliter before wash; ◆, CFU of bacteria per milliliter after wash. Error bars represent means ± SD of the results from at least 3 independent experiments. Statistically significant differences are indicated by *** for P < 0.001.
One explanation for the inability to recover intracellular KIM5 bacteria during T3SS induction could be that these bacteria are being killed. To further explore this idea, we assessed intracellular survival of KIM5 and the KIM5 T3SS mutant at earlier time points (Fig. 5B), with the expectation that greater CFU of KIM5 bacteria would be recovered at much earlier time points if they were being killed by 3 h postinfection. Additionally, this assay was conducted in the absence of gentamicin to allow continuous bacterial phagocytosis to ensue without extracellular killing, to essentially enumerate newly phagocytized bacteria. At 15-min intervals for the first 1.5 h of coculture, the KIM5 T3SS mutant was consistently recovered but not the KIM5 strain (Fig. 5B).
In an alternate experiment that aimed to visually determine if KIM5 bacteria were subject to intracellular killing during T3SS induction, the percentage of bacteria that were located intracellularly versus extracellularly when associated with amoebae was determined. An immunofluorescence microscopy staining technique (35) which was based on colocalization of indirect Alexa Fluor 594 fluorescence from bound Y. pestis-specific antibodies (αrF1) with green fluorescent protein (GFP)-expressing Y. pestis was used. Antibody accessibility to Y. pestis cells depended on their localization and the permeability of infected amoebae. Almost 100% of the KIM5 bacteria identified to be associated with amoebae appeared to reside extracellularly (Fig. 6A and B), while no KIM5 bacteria were detected intracellularly (Fig. 6A). In contrast, only a few induced KIM5 T3SS mutant bacteria appeared to reside extracellularly, while between 80 and 100% localized intracellularly (Fig. 6A and B). Overall, it was more difficult to find bacteria associated with the amoebae in cocultures with KIM5 than with the KIM5 T3SS mutant.
FIG 6.
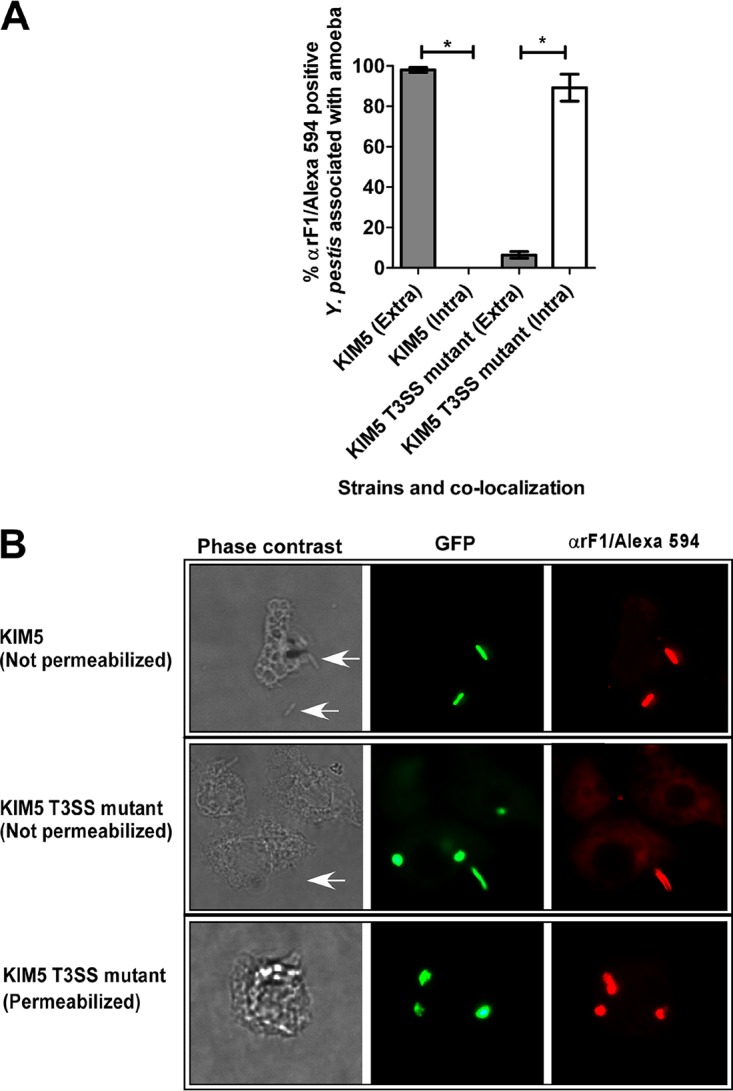
Intra-amoebal localization of GFP-expressing Y. pestis KIM5 and KIM5 T3SS mutant strains grown under T3SS-inducing conditions. Percent extracellular (Extra; nonpermeabilized infected amoebae, gray bars) or intracellular (Intra; permeabilized infected amoebae, white bars) Y. pestis. (A) Localization of bacteria was determined in 50 permeabilized and nonpermeabilized amoebae associated with GFP-expressing Y. pestis in each of two independent experiments per strain. Values are the means, and error bars represent the SD. Statistically significant differences are indicated by * for P < 0.0001. (B) Infected amoebae visualized using phase-contrast light microscopy and fluorescence microscopy showing colocalization or not of GFP-expressing bacteria and Alexa Fluor 594 red fluorescence (αrF1 antibody binding) to indicate extracellular (nonpermeabilized) or intracellular (permeabilized) localization of bacteria in association with amoebae. White arrows indicate Y. pestis cells.
The Y. pestis containing vacuole does not fuse with the lysosome.
To understand if Y. pestis survives by subverting intracellular destruction, trafficking of the Yersinia-containing vacuole (YCV) through the endocytic pathway was characterized. To mark the lysosome-endosome trafficking processes of A. castellanii, bovine serum albumin conjugated with colloidal gold nanoparticles (BSA-CGN) was used (27, 36). The BSA-CGN are first pinocytosed by the cell and traffic to localize within lysosomal compartments (27). Following the preloading of lysosomal compartments with BSA-CGN, amoebae were cocultured with either 1.1-μm latex beads, which serve as a positive control for trafficking via the canonical endocytic pathway, or with the KIM5 strain. At 2 h postinfection, >90% of latex beads (Fig. 7A and B) were observed to be colocalized with BSA-CGN in a single compartment; in contrast, <10% of Y. pestis bacteria were similarly colocalized with BSA-CGN (Fig. 7A and D). A similar trend of colocalization prevailed at 24 h of coculture (Fig. 7A, C, and E). Consistent with the above-mentioned observations, a qualitative analysis of a coculture of amoebae with latex beads and Y. pestis simultaneously revealed the presence and absence of BSA-CGN colocalization with latex beads and bacteria, respectively (Fig. S3).
FIG 7.

Phagosome trafficking of Y. pestis KIM5 using gold nanoparticles to track lysosomes. (A) Percent colocalization of Y. pestis or 1.1-μm latex beads with 10 nm of BSA conjugated with colloidal gold nanoparticles (BSA-CGN) in A. castellanii ATCC 30010 vacuoles was determined from 50 infected phagosomes at 2 h and 24 h postinfection for each of 2 independent experiments. All data are given as means ± SD. Statistically significant differences are indicated by **** for P < 0.0001. (B and C) Colocalization of BSA-CGN with latex beads at 3 h (B) and 24 h (C). (D and E) Lack of colocalization of BSA-CGN with Y. pestis KIM5 at 3 h (D) and 24 h (E). Black asterisks indicate latex beads in panels B and C and Y. pestis in panels D and E. Arrows indicate lysosomal compartments loaded with BSA-CGN.
DISCUSSION
This study demonstrates that Y. pestis strains belonging to different epidemic biovars (37, 38) are phagocytized and can survive within trophozoites of the ubiquitous soil amoeba A. castellanii by subverting intracellular digestion in phagosomal compartments. Collectively, our coculture data in the absence and presence of gentamicin use indicate that Y. pestis survives for a prolonged period of time in amoebae without replicating and can likely exit amoebae and replicate extracellularly to reinvade.
The molecular mechanisms employed by some bacterial pathogens to infect mammalian macrophages are conserved during interactions with their amoeba hosts, most notably demonstrated with L. pneumophila (18, 19, 39, 40). This was similarly observed in the interaction of Y. pestis and amoebae. The inability of the ΔphoP mutant to survive in FLA trophozoites is consistent with its phenotype in murine macrophages (27–29). PhoP is therefore a conserved factor required for intracellular survival in A. castellanii. PhoP is a transcriptional regulator, which together with PhoQ, a sensory histidine kinase, comprises a two-component phosphorelay signal transduction system that couples the sensing of an environmental stimulus with coordinately responding to it (27, 32, 41). In Y. pestis, PhoP directs the regulation of a global stress response that facilitates adaptation of the bacterium to physiologically challenging conditions (e.g., low pH, osmotic stress, oxidative stress, low Mg2+) and resistance to antimicrobial peptides in macrophages (27, 28). Whether or not comparable physiological stresses occur in the amoeba phagosome is unknown. Similarly, while the amoeboid protozoa Entamoeba histolytica and Naegleria fowleri produce antimicrobial peptides called amoebapores (42), the A. castellanii antimicrobial response represents a knowledge gap.
Our data show that the T3SS exhibits a conserved ability to confer resistance to Y. pestis phagocytic uptake whether in amoebae or in mammalian macrophages (31). Intracellular killing was ruled out as a contributing factor to the absence of recovery of Y. pestis from KIM5 cells that were cultured under T3SS-inducing conditions, because at earlier time points post-coculture, bacteria were neither recovered nor observed to be localized within the amoebae. As with mammalian macrophages, when these factors are maximally expressed, Y. pestis KIM5 avoids being taken up by phagocytic amoebae, a phenotype that is lost in the T3SS mutant.
Additionally, an incidental observation regarding another antiphagocytic factor has been revealed in our experiments in which we used the anti-F1 antibody to determine the localization of Y. pestis during T3SS induction. The anti-F1 antibody is raised against the F1 capsule which surrounds the bacteria during growth at 37°C. The F1 capsule contributes to blocking phagocytic uptake of Y. pestis in macrophages at a significantly lower rate than the T3SS (43). Our study suggests that although the F1 capsule is expressed by Y. pestis during coculture with amoebae, it may not effectively block phagocytosis, as Y. pestis cells expressing this factor are found intracellularly in the absence of T3SS production. The mechanism of inhibition of phagocytosis by the F1 capsule is not defined, but it is suggested that it shields a receptor that is required for active phagocytosis by macrophages (43); perhaps a similar mechanism of phagocytic uptake of Y. pestis is not present in amoebae.
Nonetheless, the T3SS-inducing temperature of 37°C is not expected to be reached within the subterranean rodent burrow, which is negligibly affected by climatic fluctuations (44, 45); as such, there is minimal likelihood for this factor to be activated in rodent burrows. In the natural environmental interaction of Y. pestis with FLA, the absence of T3SS induction would serve to enhance phagocytosis of Y. pestis.
Another conserved strategy that intracellular pathogens employ to avoid being digested within macrophages and amoebae is to disrupt the canonical endocytic trafficking pathways. For example, phagosome-lysosome fusion is inhibited by pathogens L. pneumophila (46, 47), Francisella tularensis (36), and Campylobacter jejuni (48), which reside in phagosomes within macrophages and amoebae. By virtue of the lack of colocalization of Y. pestis with BSA-CGN, our data implicate a similar strategy of inhibition of phagosome-lysosome fusion for Y. pestis within A. castellanii. This may differ from the Y. pestis interaction within mammalian macrophages. Within murine macrophages, Y. pestis strains reside mainly in immature phagolysosomes (28), which exhibit partial acidification. Acidification is suggested to optimize the phagosome-lysosome fusion event (28, 49). In the case that bacteria are phagocytized by macrophages, they usually replicate and are able to evade intracellular digestion and exit quickly to continue an extracellular infectious cycle in the mammalian host within 24 h postinfection (26, 50, 51). Residence of Y. pestis within macrophages is therefore short-lived, whereas amoebae may serve as a refuge for Y. pestis in the environment, and prolonged exposure to an acidified phagosomal compartment may not favor prolonged intracellular survival of Y. pestis.
Collectively, our data envisage a model (Fig. 8) for how Y. pestis persists in the environment within protozoa and how this interaction may serve to activate an epizootic event. At plague foci, protozoa may be found within warmer, humid, and richly organic soil in rodent burrows. During epizootics, Y. pestis initially enters the burrow soil from a decomposing carcass of a rodent or flea that has succumbed to plague or from infected flea feces and is subsequently phagocytized by ubiquitous soil protozoa. Once phagocytized, bacteria can survive in the same trophozoite host or be taken up by the same or other trophozoites after exocytosis. During winter months or following rodent die-off from plague, protozoa serve as a Y. pestis reservoir, with a limited possibility of encountering rodents. Once favorable environmental conditions (e.g., warmer temperatures and increased humidity) resume during spring through early autumn, there is an increase in rodents and fleas and a possibility for renewal of the epizootic disease phase. The large number of active and foraging rodents may acquire Y. pestis-infected trophozoites via intranasal transmission or through contact with an open wound. Transmission to mice and subsequent development of enhanced pulmonary infection have been experimentally shown in a study that introduced L. pneumophila-infected amoebae to mice intratracheally (20). Once an infected rodent acquires Y. pestis by flea bite, the flea could in turn transmit the pathogen to another rodent host. At this point, reemergence of plague from its quiescent phase can initiate the epizootic cycle.
FIG 8.

Hypothetical interepizootic plague cycle depicting the putative role of amoebae during these periods. A black question mark is used to indicate hypothetical infection routes. Refer to the Discussion for details.
In an alternate hypothetical scenario, soil protozoa may phagocytize Y. pestis while seeking nutrition from infected flea feces. In turn, protozoa infected with Y. pestis may be consumed by flea larvae feeding on adult feces. Amoeba trophozoites have been found in the hindgut of fleas (52), supporting this postulation. Although there is no evidence for transstadial transmission of Y. pestis, these infected larvae, once they have emerged into adult fleas, may support propagation of Y. pestis and development of a transmissible infection.
The duration of prolonged intra-amoebal survival of Y. pestis that may support our hypothetical model for how FLA may serve as a reservoir host for Y. pestis is unknown. It is likely heavily impacted by complex seasonal dynamics between the biota and abiotic components within the burrow environment. Further testing in the context of a burrow ecosystem over months or even years may be required to determine Y. pestis prolonged viability in FLA naturally. It is also tempting to speculate that Y. pestis can survive within amoeba cysts over prolonged periods and that plague reemergence may then occur coincidentally with excystation of amoebae upon return of favorable environmental conditions. Accordingly, we have demonstrated that Y. pseudotuberculosis can survive in cysts of A. castellanii over a short period of time (17). Nevertheless, our data serve as a proof or principle that is consistent with the non-mutually exclusive hypothesis that FLA may serve as a reservoir host for Y. pestis during the interepizootic period.
Species of Acanthamoeba and other FLA are ubiquitous in plague foci in the Caspian region in Russia and Colorado, USA (16, 53). The lack of detection of soilborne Y. pestis in areas endemic for the organism during interepizootic periods may be related to the bacteria surviving within a protozoan host rather than freely in the soil. A confounding factor in detection may be an overall low prevalence of intra-amoebal Y. pestis and inaccessibility due to its occurrence only deep within the rodent burrows. Indeed, a <1% rate of persistence of bacteria was observed in our coculture assays. As such, it is foreseeable that specialized sample acquisition, preparation, and determination of the lower limits of detection of Y. pestis may be required to detect intraprotozoal Y. pestis at plague foci. Our findings could serve to inform the development of new methods for plague surveillance and detection. Importantly, this study may open a new avenue of investigation into the mystery of how plague persists in between epizootics.
MATERIALS AND METHODS
Bacterial cultures.
For this study, attenuated Y. pestis variants of strains KIM, CO92, and Kuma belonging to epidemic biovars Medievalis, Orientalis, and Antiqua (37, 38), respectively, were used. Y. pestis was first cultured overnight in brain heart infusion (BHI) broth from −80°C glycerol stocks at 25°C with shaking aeration and then subcultured at a 1:10 dilution until mid-log phase (∼4 h) at 25°C or 37°C in the same medium for subsequent experiments. To understand if known antiphagocytic and intracellular survival factors essential for Y. pestis interaction with macrophages were conserved in its interaction with A. castellanii, a KIM5 ΔyopE-sycE::kan ΔyscV mutant, which essentially renders the bacterium T3SS deficient (KIM5 T3SS mutant) (54), and a PhoP mutant (KIM6+ ΔphoP) (41) were used in coculture studies. In addition, strains were also transformed with the plasmid pMMB207gfp3.1 (28), which encodes green fluorescent protein (GFP), to monitor Y. pestis-amoeba interactions using fluorescence microscopy. All strains are described in Table 1.
TABLE 1.
Yersinia pestis strains used in this studyc
| Y. pestis strain | Biovar | Intracellular factor profile | Reference(s) |
|---|---|---|---|
| KIM5 Δpgm pCD1+ pMT1+ pPCP1+a | Medievalis | phoP+, T3SS+ | 63 |
| KIM5 Δpgm pCD1+ pMT1+ pPCP1+ (ΔyopE-sycE::kan ΔyscV)b | Medievalis | phoP+, T3SS− | 54 |
| KIM6+ pgm+ pCD1− pMT1+ pPCP1+ | Medievalis | phoP+, T3SS− | 64, 65 |
| KIM6+ ΔphoP pgm+ pCD1− pMT1+ pPCP1+ | Medievalis | ΔphoP, T3SS− | 41 |
| KIM6+ ΔphoP pLG::phoP pgm+ pCD1− pMT1+ pPCP1+ | Medievalis | phoP+, T3SS− | 41 |
| Colorado 92 (CO92) pgm+ pCD1− pMT1+ pPCP1+ | Orientalis | phoP+, T3SS− | 66 |
| Kuma D7 pgm+ pMT1+ pPCP1+ pCD1− | Antiqua | phoP+, T3SS− | 67 |
Designated KIM5 for this study.
Designated KIM5 T3SS mutant for this study.
“−” indicates the absence of a plasmid or the T3SS; “+” indicates the presence of a plasmid, gene, or T3SS.
In studies requiring induction of the T3SS, bacteria were subcultured twice in succession at 37°C in BHI medium supplemented with MgOx (20 mM sodium oxalate and 20 mM MgCl2) (37, 55). The presence of the pCD1 plasmid in the KIM5 strains was confirmed by PCR analysis using a pCD1-specific primer set obtained from BEI Resources (catalog no. NR-9562). To confirm that the KIM5 strain maintained the T3SS phenotype, bacteria were grown on BHI supplemented with MgOx and incubated at 26°C and 37°C, as previously described (55). To confirm that the pgm-positive strains retained this phenotype, these strains were grown on Congo red agar, as previously described (56).
Cultivation of A. castellanii.
A. castellanii (Douglas) Page amoebae (ATCC 30010) were maintained axenically in protease peptone-yeast extract-glucose (PYG) medium (ATCC recipe [http://www.atcc.org/products/all/30010.aspx#culturemethod]) at ambient room temperature (25°C) in culture flasks. The adherent amoebae were harvested by scraping the flasks to dislodge amoebae and were centrifuged (300 × g, 5 min). Amoebae were then washed and resuspended in nutrient-rich PYG medium. Amoebae were enumerated using a Petroff-Hausser counting chamber prior to seeding of these cells in a 24-well tissue culture plate. The viability of A. castellanii trophozoites was confirmed using trypan blue exclusion staining (57).
For coculture studies of bacteria with amoebae, approximately 2.5 × 105 viable trophozoites/ml were seeded per well of the tissue culture plate and allowed to adhere to the well overnight at 25°C. All coculture studies were performed at 25°C, except for when indicated that coculture was conducted at 4°C.
Bacterial entry and intracellular survival assays in PYG medium.
The lower (4°C) and upper (25°C) limits of the Acanthamoeba species growth (58) range are expected in subterranean rodent burrows during seasonal interepizootic and epizootic periods, respectively (59). The phagocytosis of Y. pestis by FLA is expected to initially occur in burrows during epizootic plague events when this pathogen is abundant. As such, to determine if Y. pestis can invade or be phagocytized by amoebae, bacteria were cocultured with amoebae at a multiplicity of infection (MOI) of 100 (2.5 × 107 bacterial/ml) or 1 (2.5 × 105 bacteria/ml) at 25°C. This temperature coincides with optimal in vitro laboratory growth conditions for Y. pestis. Following coculture of Y. pestis and amoebae for 2 h, 100 μg/ml gentamicin was added to the medium, and incubation was continued for a further 1 h. We determined that this gentamicin dose was minimally sufficient to kill extracellular bacteria in the PYG medium used for coculture (data not shown). The infected amoebae were then washed thrice to remove dead bacterial cells and debris, after which they were lysed with 0.2% Triton X-100 for 10 min to release intracellular bacteria. The lysates were immediately serially diluted and plated on BHI agar. The same was done for the washes to ensure that total extracellular bacterial killing occurred. Following incubation for 48 h at 28°C, the CFU per milliliter were enumerated. The CFU of Y. pestis per milliliter determined in this part of the assay was used to determine the invasion frequency and early survival of Y. pestis in amoebae at 3 h postinfection.
To assess if Y. pestis strains that have been phagocytized could survive and replicate intracellularly in FLA trophozoites over time, intracellular bacteria were processed as described above after incubation of the coculture for 24 h, 48 h, 72 h, and 120 h after the initial 1-h treatment with gentamicin. Five days (120 h) was defined as a “prolonged period,” given that the typical time frame for the Y. pestis intracellular life stage in macrophages is suggested to be <1 h to 24 h (50, 51, 60). Assays to determine prolonged survival of intra-amoebal Y. pestis were conducted at temperatures of 25°C and 4°C to reflect the epizootic and seasonal interepizootic cycles of Y. pestis.
These assays were performed with a gentamicin maintenance regimen comprising maintenance of the coculture in gentamicin that was refreshed every 24 h. The appropriate effective gentamicin concentration was determined for each strain (data not shown): KIM5, 6.5 μg/ml; KIM6+, 20 μg/ml; CO92, 5 μg/ml; and Kuma, 25 μg/ml. To assess intracellular survival in the absence of extracellular bacterial killing, assays using an MOI of 100 and an MOI of 1 were also conducted for the KIM5, the KIM6+, and the KIM5 T3SS mutant strains. Here, the initial 100 μg/ml gentamicin treatment for 1 h that followed the initial coculture at 2 h was carried out but with no subsequent gentamicin maintenance dosing.
Survival at time points earlier than that used to determine invasion frequency was used to assess the effect of T3SS induction on the ability of Y. pestis to be phagocytized in the KIM5 and KIM5 T3SS mutant strains. This assay was performed in a manner similar to that described above, except that gentamicin was not added, and lysis was conducted every 15 min for the initial 1.5 h of coculture. Extracellular medium was plated before washing and lysis to enumerate extracellular bacteria.
All experiments were performed using 3 to 6 independent biological replicates, each composed of three technical replicates.
Phagosome trafficking assays using BSA-CGN.
To monitor phagosome trafficking by Y. pestis in amoebae, 10-nm BSA-CGN (optical density of 2 at A520; Electron Microscopy Sciences) was used (27). BSA-CGN was first dialyzed (SpectraPor; molecular weight cutoff [MWCO], 6 to 8 kDa) in 1× phosphate-buffered saline (PBS) overnight to remove sodium azide. FLA trophozoites at a density of 2.5 × 105 cells/cm2 were incubated in T25 culture flasks overnight at 25°C. The cells were washed twice and incubated with 1.5 ml of dialyzed BSA-CGN solution supplemented with 10% PYG medium for 4 h (pulse), allowing the pinocytic internalization of these particles by endosomes. Extracellular BSA-CGN was removed by washing amoebae twice with 1× PBS, and incubation was allowed to continue for a further 2 h (chase) in PYG medium to allow accumulation of the BSA-CGN in lysosomes. Following this step, cells were washed twice again and inoculated for 2 h and 24 h with 1.1-μm polystyrene latex beads (LB11; Sigma), or Y. pestis KIM5 at an MOI of 100. All samples were treated with gentamicin in a manner similar to the description above for the invasion frequency and survival assays.
Electron microscopy.
To prepare samples for transmission electron microscopy (TEM), the amoebae infected at an MOI of 100 were first detached from culture flasks by shaking and were concentrated by centrifugation (800 × g, 5 min). The supernatant was discarded, and samples were fixed in 4% paraformaldehyde (PFA) for 1 h and postfixed overnight in 2% paraformaldehyde and 1.25% glutaraldehyde (Acros, Morris Plains, NJ) in 0.1 M sodium cacodylate. Samples were stained using the lipid stain 2% osmium tetroxide for 24 h and dehydrated after two rinses with buffer in an increasing ethanol series. Dehydrated samples were embedded in a resin and cut into ultrathin sections (∼70 to 100 nm), which were stained with uranyl acetate and Reynold's lead citrate. Samples were examined with a Philips CM100 transmission electron microscope (TEM T20; FEI, at the Franceschi Microscopy and Imaging Center at WSU) operating at 80 kV (17).
Immunofluorescence microscopy to distinguish extracellular from intracellular bacteria associated with amoebae.
For immunofluorescence microscopy, glass coverslips were laid down in each well prior to seeding of amoebae and coculture, as described above. Bacteria harboring the GFP-encoding plasmid were used. Following a 1-h coculture under conditions of T3SS induction, amoeba monolayers infected with KIM5 or the KIM5 T3SS mutant at an MOI of 1 were washed thrice with 1× PBS and then fixed with 4% PFA at 37°C for 20 min. To determine if bacteria were extracellular, infected amoeba monolayers were incubated with a 1:500 dilution of a polyclonal Y. pestis-specific anti-F1-antigen (αrF1, NR-31024; BEI Resources) without permeabilization for 1 h, followed by three washes with 1× PBS. Next, the infected amoeba monolayers were incubated with a rabbit anti-goat IgG Alexa Fluor 594 secondary antibody (Thermo Fisher).
To determine if bacteria resided intracellularly, infected amoeba monolayers were first permeabilized with 0.2% Triton X-100 for 3 min, incubated in ice-cold 100% methanol for 2 min, and then fixed and stained as described above. The coverslips were mounted on slides and examined by epifluorescence microscopy using a Leica DMi8 and a 40× objective. A camera was used to sequentially capture Alexa Fluor 594 (red, tetramethyl rhodamine isocyanate [TRITC] cube, excitation [Ex] 590/50, emission [Em] 617/50), GFP (green, Ex 470/40, Em 525/50), and phase-contrast images. Similar assays have been published (35, 61, 62).
The percentage of extracellular bacteria was determined from nonpermeabilized infected amoeba monolayers by counting the number of bacteria that fluoresced red (bound antibody) out of 100 (50 each from two independent experiments) amoebae that associated with GFP-expressing bacteria. The number of intracellular bacteria was determined similarly in permeabilized infected amoeba monolayers.
Statistical analysis.
The GraphPad Prism 5.0 software was used for statistical analysis of all data. For invasion assays, a one-way analysis of variance (ANOVA) and a post hoc Bonferroni's multiple-comparison test were conducted. For survival assays, a one-way ANOVA and a Bonferroni multiple-comparison posttest were performed to determine differences between relevant time points. Student's t tests were used to determine differences between MOIs at different time points and for differences in extracellular and intracellular bacteria in immunofluorescence assays. For phagosome trafficking assays, statistical analyses were performed using a two-way ANOVA with a Bonferroni multiple-comparison posttest to compare replicate means.
Supplementary Material
ACKNOWLEDGMENTS
This investigation was supported by WSU CVM intramural grants and a Morris Animal Foundation grant (D16ZO-014) to V.V.
We are indebted to Leigh Knodler for the valuable guidance on design, data analysis, and interpretation of trafficking assays. We are grateful to Jennifer Santos and Angie Hinz for sharing their experience with working with amoeba and Y. pestis cocultures. We thank Greg Plano and Robert Perry for providing us with strains and Jim Bliska, Guy Palmer, Anders Omsland, and Alex Bobrov for critical comments.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00593-17.
REFERENCES
- 1.Bertherat E, Bekhoucha S, Chougrani S, Razik F, Duchemin JB, Houti L, Deharib L, Fayolle C, Makrerougrass B, Dali-Yahia R, Bellal R, Belhabri L, Chaieb A, Tikhomirov E, Carniel E. 2007. Plague reappearance in Algeria after 50 years, 2003. Emerg Infect Dis 13:1459–1462. doi: 10.3201/eid1310.070284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cabanel N, Leclercq A, Chenal-Francisque V, Annajar B, Rajerison M, Bekkhoucha S, Bertherat E, Carniel E. 2013. Plague outbreak in Libya, 2009, unrelated to plague in Algeria. Emerg Infect Dis 19:230–236. doi: 10.3201/eid1902.121031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrianaivoarimanana V, Kreppel K, Elissa N, Duplantier JM, Carniel E, Rajerison M, Jambou R. 2013. Understanding the persistence of plague foci in Madagascar. PLoS Negl Trop Dis 7:e2382. doi: 10.1371/journal.pntd.0002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Navid A, Almaas E. 2009. Genome-scale reconstruction of the metabolic network in Yersinia pestis, strain 91001. Mol Biosyst 5:368–375. doi: 10.1039/b818710j. [DOI] [PubMed] [Google Scholar]
- 5.Chain PSG, Carniel E, Larimer FW, Lamerdin J, Stoutland PO, Regala WM, Georgescu AM, Vergez LM, Land ML, Motin VL, Brubaker RR, Fowler J, Hinnebusch J, Marceau M, Medigue C, Simonet M, Chenal-Francisque V, Souza B, Dacheux D, Elliott JM, Derbise A, Hauser LJ, Garcia E. 2004. Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Proc Natl Acad Sci U S A 101:13826–13831. doi: 10.1073/pnas.0404012101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hinnebusch BJ, Chouikha I, Sun YC. 2016. Ecological opportunity, evolution, and the emergence of flea-borne plague. Infect Immun 84:1932–1940. doi: 10.1128/IAI.00188-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, Carniel E. 1999. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc Natl Acad Sci U S A 96:14043–14048. doi: 10.1073/pnas.96.24.14043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eisen RJ, Gage KL. 2009. Adaptive strategies of Yersinia pestis to persist during inter-epizootic and epizootic periods. Vet Res 40:1. doi: 10.1051/vetres:2008039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmid BV, Jesse M, Wilschut LI, Viljugrein H, Heesterbeek JAP. 2012. Local persistence and extinction of plague in a metapopulation of great gerbil burrows, Kazakhstan. Epidemics 4:211–218. doi: 10.1016/j.epidem.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Boegler KA, Graham CB, Montenieri JA, MacMillan K, Holmes JL, Petersen JM, Gage KL, Eisen RJ. 2012. Evaluation of the infectiousness to mice of soil contaminated with Yersinia pestis-infected blood. Vector Borne Zoonotic Dis 12:948–952. doi: 10.1089/vbz.2012.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lorange EA, Race BL, Sebbane F, Hinnebusch BJ. 2005. Poor vector competence of fleas and the evolution of hypervirulence in Yersinia pestis. J Infect Dis 191:1907–1912. doi: 10.1086/429931. [DOI] [PubMed] [Google Scholar]
- 12.Ayyadurai S, Houhamdi L, Lepidi H, Nappez C, Raoult D, Drancourt M. 2008. Long-term persistence of virulent Yersinia pestis in soil. Microbiology 154:2865–2871. doi: 10.1099/mic.0.2007/016154-0. [DOI] [PubMed] [Google Scholar]
- 13.Wimsatt J, Biggins DE. 2009. A review of plague persistence with special emphasis on fleas. J Vector Borne Dis 46:85–99. [PubMed] [Google Scholar]
- 14.Gage KL, Kosoy MY. 2005. Natural history of plague: perspectives from more than a century of research. Annu Rev Entomol 50:505–528. doi: 10.1146/annurev.ento.50.071803.130337. [DOI] [PubMed] [Google Scholar]
- 15.Nikul'shin SV, Onatskaia TG, Lukanina LM, Bondarenko AI. 1992. Associations of the soil amoeba Hartmannella rhysodes with the bacterial causative agents of plague and pseudotuberculosis in an experiment. Zh Mikrobiol Epidemiol Immunobiol 9-10:2–5. (In Russian.) [PubMed] [Google Scholar]
- 16.Koshel EI, Anisimova LV, Novichkova LA, Vidyaeva NA, Guseva NP, Eroshenko GA, Kutyrev VV. 2015. A study on the taxonomy of soil amoebas from Caspian plague foci based on an analysis of ribosomal operon sequences. Russ J Genet 51:33–38. doi: 10.1134/S1022795415010056. [DOI] [PubMed] [Google Scholar]
- 17.Santos-Montañez J, Benavides-Montaño JA, Hinz AK, Vadyvaloo V. 2015. Yersinia pseudotuberculosis IP32953 survives and replicates in trophozoites and persists in cysts of Acanthamoeba castellanii. FEMS Microbiol Lett 362:fnv091. doi: 10.1093/femsle/fnv091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greub G, Raoult D. 2004. Microorganisms resistant to free-living amoebae. Clin Microbiol Rev 17:413–433. doi: 10.1128/CMR.17.2.413-433.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Quadan T, Price CT, Abu Kwaik Y. 2012. Exploitation of evolutionarily conserved amoeba and mammalian processes by Legionella. Trends Microbiol 20:299–306. doi: 10.1016/j.tim.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brieland JK, Fantone JC, Remick DG, LeGendre M, McClain M, Engleberg NC. 1997. The role of Legionella pneumophila-infected Hartmannella vermiformis as an infectious particle in a murine model of Legionnaires' disease. Infect Immun 65:5330–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cirillo JD, Falkow S, Tompkins LS. 1994. Growth of Legionella pneumophila in Acanthamoeba castellanii enhances invasion. Infect Immun 62:3254–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kennedy GM, Morisaki JH, DiGiuseppe Champion PA. 2012. Conserved mechanisms of Mycobacterium marinum pathogenesis within the environmental amoeba Acanthamoeba castellanii. Appl Environ Microbiol 78:2049–2052. doi: 10.1128/AEM.06965-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abd H, Saeed A, Weintraub A, Nair GB, Sandstrom G. 2007. Vibrio cholerae O1 strains are facultative intracellular bacteria, able to survive and multiply symbiotically inside the aquatic free-living amoeba Acanthamoeba castellanii. FEMS Microbiol Ecol 60:33–39. doi: 10.1111/j.1574-6941.2006.00254.x. [DOI] [PubMed] [Google Scholar]
- 24.Ke Y, Chen Z, Yang R. 2013. Yersinia pestis: mechanisms of entry into and resistance to the host cell. Front Cell Infect Microbiol 3:106. doi: 10.3389/fcimb.2013.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang XZ, Lindler LE. 2004. The pH 6 antigen is an antiphagocytic factor produced by Yersinia pestis independent of Yersinia outer proteins and capsule antigen. Infect Immun 72:7212–7219. doi: 10.1128/IAI.72.12.7212-7219.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pujol C, Bliska JB. 2003. The ability to replicate in macrophages is conserved between Yersinia pestis and Yersinia pseudotuberculosis. Infect Immun 71:5892–5899. doi: 10.1128/IAI.71.10.5892-5899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grabenstein JP, Fukuto HS, Palmer LE, Bliska JB. 2006. Characterization of phagosome trafficking and identification of PhoP-regulated genes important for survival of Yersinia pestis in macrophages. Infect Immun 74:3727–3741. doi: 10.1128/IAI.00255-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pujol C, Klein KA, Romanov GA, Palmer LE, Cirota C, Zhao Z, Bliska JB. 2009. Yersinia pestis can reside in autophagosomes and avoid xenophagy in murine macrophages by preventing vacuole acidification. Infect Immun 77:2251–2261. doi: 10.1128/IAI.00068-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oyston PCF, Dorrell N, Williams K, Li SR, Green M, Titball RW, Wren BW. 2000. The response regulator PhoP is important for survival under conditions of macrophage-induced stress and virulence in Yersinia pestis. Infect Immun 68:3419–3425. doi: 10.1128/IAI.68.6.3419-3425.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cornelis GR. 2002. The Yersinia Ysc-Yop ‘type III’ weaponry. Nat Rev Mol Cell Biol 3:742–752. doi: 10.1038/nrm932. [DOI] [PubMed] [Google Scholar]
- 31.Viboud GI, Bliska JB. 2005. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol 59:69–89. doi: 10.1146/annurev.micro.59.030804.121320. [DOI] [PubMed] [Google Scholar]
- 32.Vadyvaloo V, Viall AK, Jarrett CO, Hinz AK, Sturdevant DE, Joseph Hinnebusch B. 2015. Role of the PhoP-PhoQ gene regulatory system in adaptation of Yersinia pestis to environmental stress in the flea digestive tract. Microbiology 161:1198–1210. doi: 10.1099/mic.0.000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han Y, Zhou D, Pang X, Zhang L, Song Y, Tong Z, Bao J, Dai E, Wang J, Guo Z, Zhai J, Du Z, Wang X, Wang J, Huang P, Yang R. 2005. Comparative transcriptome analysis of Yersinia pestis in response to hyperosmotic and high-salinity stress. Res Microbiol 156:403–415. doi: 10.1016/j.resmic.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 34.Straley SC, Harmon PA. 1984. Growth in mouse peritoneal macrophages of Yersinia pestis lacking established virulence determinants. Infect Immun 45:649–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Celli J, Gorvel JP. 2004. Organelle robbery: Brucella interactions with the endoplasmic reticulum. Curr Opin Microbiol 7:93–97. doi: 10.1016/j.mib.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 36.El-Etr SH, Margolis JJ, Monack D, Robison RA, Cohen M, Moore E, Rasley A. 2009. Francisella tularensis type A strains cause the rapid encystment of Acanthamoeba castellanii and survive in amoebal cysts for three weeks postinfection. Appl Environ Microbiol 75:7488–7500. doi: 10.1128/AEM.01829-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perry RD, Fetherston JD. 1997. Yersinia pestis–etiologic agent of plague. Clin Microbiol Rev 10:35–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anisimov AP, Lindler LE, Pier GB. 2004. Intraspecific diversity of Yersinia pestis. Clin Microbiol Rev 17:434–464. doi: 10.1128/CMR.17.2.434-464.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Portier E, Zheng H, Sahr T, Burnside DM, Mallama C, Buchrieser C, Cianciotto NP, Hechard Y. 2015. lroT/mavN, a new iron-regulated gene involved in Legionella pneumophila virulence against amoebae and macrophages. Environ Microbiol 17:1338–1350. doi: 10.1111/1462-2920.12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Al-Quadan T, Abu Kwaik Y. 2011. Molecular characterization of exploitation of the polyubiquitination and farnesylation machineries of Dictyostelium discoideum by the AnkB F-box effector of Legionella pneumophila. Front Microbiol 2:23. doi: 10.3389/fmicb.2011.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rebeil R, Jarrett CO, Driver JD, Ernst RK, Oyston PC, Hinnebusch BJ. 2013. Induction of the Yersinia pestis PhoP-PhoQ regulatory system in the flea and its role in producing a transmissible infection. J Bacteriol 195:1920–1930. doi: 10.1128/JB.02000-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herbst R, Marciano-Cabral F, Leippe M. 2004. Antimicrobial and pore-forming peptides of free-living and potentially highly pathogenic Naegleria fowleri are released from the same precursor molecule. J Biol Chem 279:25955–25958. doi: 10.1074/jbc.M401965200. [DOI] [PubMed] [Google Scholar]
- 43.Du Y, Rosqvist R, Forsberg A. 2002. Role of fraction 1 antigen of Yersinia pestis in inhibition of phagocytosis. Infect Immun 70:1453–1460. doi: 10.1128/IAI.70.3.1453-1460.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ben-Ari T, Neerinckx S, Gage KL, Kreppel K, Laudisoit A, Leirs H, Stenseth NC. 2011. Plague and climate: scales matter. PLoS Pathog 7:e1002160. doi: 10.1371/journal.ppat.1002160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Šumbera R, Chitaukali WN, Elichová M, Kubová J, Burda H. 2004. Microclimatic stability in burrows of an Afrotropical solitary bathyergid rodent, the silvery mole-rat (Heliophobius argenteocinereus). J Zool 263:409–416. doi: 10.1017/S095283690400545X. [DOI] [Google Scholar]
- 46.Horwitz MA. 1983. Formation of a novel phagosome by the Legionnaires' disease bacterium (Legionella pneumophila) in human monocytes. J Exp Med 158:1319–1331. doi: 10.1084/jem.158.4.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bozue JA, Johnson W. 1996. Interaction of Legionella pneumophila with Acanthamoeba castellanii: uptake by coiling phagocytosis and inhibition of phagosome-lysosome fusion. Infect Immun 64:668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olofsson J, Axelsson-Olsson D, Brudin L, Olsen B, Ellstrom P. 2013. Campylobacter jejuni actively invades the amoeba Acanthamoeba polyphaga and survives within non digestive vacuoles. PLoS One 8:e78873. doi: 10.1371/journal.pone.0078873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huynh KK, Grinstein S. 2007. Regulation of vacuolar pH and its modulation by some microbial species. Microbiol Mol Biol Rev 71:452–462. doi: 10.1128/MMBR.00003-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shannon JG, Bosio CF, Hinnebusch BJ. 2015. Dermal neutrophil, macrophage and dendritic cell responses to Yersinia pestis transmitted by fleas. PLoS Pathog 11:e1004734. doi: 10.1371/journal.ppat.1004734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gonzalez RJ, Miller VL. 2016. A deadly path: bacterial spread during bubonic plague. Trends Microbiol 24:239–241. doi: 10.1016/j.tim.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beard CB, Butler JF, Hall DW. 1990. Prevalence and biology of endosymbionts of fleas (Siphonaptera: Pulicidae) from dogs and cats in Alachua County, Florida. J Med Entomol 27:1050–1061. doi: 10.1093/jmedent/27.6.1050. [DOI] [PubMed] [Google Scholar]
- 53.Sinclair JL, McClellan JF, Coleman DC. 1981. Nitrogen mineralization by Acanthamoeba polyphaga in grazed Pseudomonas paucimobilis populations. Appl Environ Microbiol 42:667–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gendlina I, Held KG, Bartra SS, Gallis BM, Doneanu CE, Goodlett DR, Plano GV, Collins CM. 2007. Identification and type III-dependent secretion of the Yersinia pestis insecticidal-like proteins. Mol Microbiol 64:1214–1227. doi: 10.1111/j.1365-2958.2007.05729.x. [DOI] [PubMed] [Google Scholar]
- 55.Higuchi K, Smith JL. 1961. Studies on the nutrition and physiology of Pasteurella pestis. VI. A differential plating medium for the estimation of the mutation rate to avirulence. J Bacteriol 81:605–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Surgalla MJ, Beesley ED. 1969. Congo red-agar plating medium for detecting pigmentation in Pasteurella pestis. Appl Microbiol 18:834–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Debnath A, Tunac JB, Silva-Olivares A, Galindo-Gomez S, Shibayama M, McKerrow JH. 2014. In vitro efficacy of corifungin against Acanthamoeba castellanii trophozoites and cysts. Antimicrob Agents Chemother 58:1523–1528. doi: 10.1128/AAC.02254-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Biddick CJ, Rogers LH, Brown TJ. 1984. Viability of pathogenic and nonpathogenic free-living amoebae in long-term storage at a range of temperatures. Appl Environ Microbiol 48:859–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ryckman RE. 1971. Plague vector studies part II. The role of climatic factors in determining seasonal fluctuations of flea species associated with the California ground squirrel. J Med Entomol 8:541–549. [DOI] [PubMed] [Google Scholar]
- 60.Martínez-Chavarría LC. 2016. Yersinia pestis-host immune cells interactions at early events during bubonic plague infection. Curr Trop Med Rep 3:51–59. doi: 10.1007/s40475-016-0071-5. [DOI] [Google Scholar]
- 61.Vadyvaloo V, Jarrett C, Sturdevant DE, Sebbane F, Hinnebusch BJ. 2010. Transit through the flea vector induces a pretransmission innate immunity resistance phenotype in Yersinia pestis. PLoS Pathog 6:e1000783. doi: 10.1371/journal.ppat.1000783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ivanov MI, Noel BL, Rampersaud R, Mena P, Benach JL, Bliska JB. 2008. Vaccination of mice with a Yop translocon complex elicits antibodies that are protective against infection with F1− Yersinia pestis. Infect Immun 76:5181–5190. doi: 10.1128/IAI.00189-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goguen JD, Yother J, Straley SC. 1984. Genetic analysis of the low calcium response in Yersinia pestis mu d1(Ap lac) insertion mutants. J Bacteriol 160:842–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deng W, Burland V, Plunkett G III, Boutin A, Mayhew GF, Liss P, Perna NT, Rose DJ, Mau B, Zhou S, Schwartz DC, Fetherston JD, Lindler LE, Brubaker RR, Plano GV, Straley SC, McDonough KA, Nilles ML, Matson JS, Blattner FR, Perry RD. 2002. Genome sequence of Yersinia pestis KIM. J Bacteriol 184:4601–4611. doi: 10.1128/JB.184.16.4601-4611.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fetherston JD, Perry RD. 1994. The pigmentation locus of Yersinia pestis KIM6+ is flanked by an insertion-sequence and includes the structural genes for pesticin sensitivity and Hmwp2. Mol Microbiol 13:697–708. doi: 10.1111/j.1365-2958.1994.tb00463.x. [DOI] [PubMed] [Google Scholar]
- 66.Parkhill J, Wren BW, Thomson NR, Titball RW, Holden MT, Prentice MB, Sebaihia M, James KD, Churcher C, Mungall KL, Baker S, Basham D, Bentley SD, Brooks K, Cerdeno-Tarraga AM, Chillingworth T, Cronin A, Davies RM, Davis P, Dougan G, Feltwell T, Hamlin N, Holroyd S, Jagels K, Karlyshev AV, Leather S, Moule S, Oyston PC, Quail M, Rutherford K, Simmonds M, Skelton J, Stevens K, Whitehead S, Barrell BG. 2001. Genome sequence of Yersinia pestis, the causative agent of plague. Nature 413:523–527. doi: 10.1038/35097083. [DOI] [PubMed] [Google Scholar]
- 67.Lucier TS, Brubaker RR. 1992. Determination of genome size, macrorestriction pattern polymorphism, and nonpigmentation-specific deletion in Yersinia pestis by pulsed-field gel electrophoresis. J Bacteriol 174:2078–2086. doi: 10.1128/jb.174.7.2078-2086.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.