

Abstract
Bacterial cells have developed sophisticated systems to deal with the toxicity of metal ions. Escherichia coli CusCFBA is a complex efflux system, responsible for transferring Cu(I) and Ag(I) ions; this system, located in the periplasm, involves four proteins, CusA, CusB, CusC, and CusF. CusA, CusB, and CusC are connected to one another in an oligomerization ratio of 3:6:3 CusA/CusB/CusC to form the CusCBA periplasm membrane transporter. CusB is an adaptor protein that connects the two membrane proteins CusA (inner membrane) and CusC (outer membrane). CusF is a metallochaperone that transfers Cu(I) and Ag(I) to the CusCBA transporter from the periplasm. The crystal structures of CusB, CusC, CusF, and the CusBA complex have been resolved, shedding some light on the efflux mechanism underlying this intriguing system. However, since CusB is an adaptor protein, its role in operating this system is significant, and should be understood in detail. Here, we utilize EPR spectroscopy to target the conformational changes that take place in the full CusB protein upon binding Cu(I). We reveal that CusB is a dimer in solution, and that the orientation of one molecule with respect to the other molecule changes upon Cu(I) coordination, resulting in a more compact CusB structure. These structural and topological changes upon Cu(I) binding probably play the role of a switch for opening the channel and transferring metal ions from CusB to CusC and out of the cell.
Introduction
Bacteria cells have developed various mechanisms to overcome the effects of toxic environments (1, 2, 3, 4, 5). One such mechanism is the efflux system, which plays a role in exporting toxic compounds from the cytoplasm and periplasm environments (6, 7, 8). A detailed understanding of the cellular efflux mechanisms is essential for developing antibiotics that can overcome the inherent resistance of today’s bacteria. Herein, we seek to shed light on the Escherichia coli mechanism underlying the efflux of silver and copper, both of which are well-known bactericides that humans have been exploiting for centuries (9, 10, 11).
In E. coli, the CusCBA tetra-complex comprising CusCBA and its metallochaperone, CusF, is responsible for mediating Cu(I) and Ag(I) export across the inner and outer membranes of the periplasm via proton motive force. CusCFBA is composed of an inner membrane proton-substrate carrier (CusA) and an outer membrane pore (CusC) (12). These two components are connected by a linker protein, CusB, in an oligomerization ratio of 3:6:3 CusA/CusB/CusC (13). The CusF metallochaperone carries Cu(I) and Ag(I) from the periplasm to the CusCBA complex (8, 14) (see Fig. 1, inset). Owing to the large size and entanglement of the system, the crystal structure of the entire complex has not been solved. However, the crystal structures of the individual components CusF, CusC, CusA, and CusB have been determined, in addition to the that of the CusBA complex (12, 15, 16, 17, 18). Unfortunately, these structures are missing some important elements, including the first 88 aa of the CusB protein, or the CusB N-terminal domain (CusBNT). This domain plays a significant role in enabling the CusF chaperone to transfer Cu(I) from the periplasm into the CusCBA complex (14).
Figure 1.

A ribbon view of the CusB structure (PDB: 3H94), showing the protein’s various domains. The residue probes used here are denoted in blue. The upper part illustrates the site-directed spin-labeling method. The lower inset provides a schematic view of the E. coli periplasm efflux system, CusCFBA. To see this figure in color, go online.
According to its crystal structure, CusB is folded into an elongated structure (∼120 Å long and ∼40 Å wide) comprising four domains (see Fig. 1) (15, 17). The protein’s first three domains (domains 1–3: membrane proximal, β-barrel, and lipoyl) are mostly β-strands. The fourth domain (domain 4: α-helical) consists of only α-helices and is folded into a three-helix-bundle structure. Each promoter of CusA interacts specifically with two elongated molecules of CusB through the latter protein’s membrane-proximal (domain 1) and β-barrel (domain 2) domains. It has been suggested that the α-helical (domain 4) domain of CusB interacts with CusC (12).
Two different conformations of CusB have been identified using crystallography (17), which suggests that this protein is flexible. One conformation is more open, whereas the other is a more compact structure. Gel filtration chromatography suggests that CusB undergoes conformational changes upon Cu(I) coordination (19). However, the crystal structures of CusBA and CusB do not reveal any change in the CusB structure upon Cu(I)/Ag(I) binding. Nonetheless, CusA has been shown to undergo conformational changes upon metal binding (15, 16). Moreover, computational modeling suggests that CusF undergoes conformational changes upon interacting with the N-terminal domain of CusB (20).
In this study, we used electron paramagnetic resonance (EPR) spectroscopy in attempting to identify conformational changes in CusB upon Cu(I) coordination in solution, i.e., in the shift from the apo state to the holo state. The strength of EPR lies in its high sensitivity to target minor conformational changes that take place upon protein binding with ligands, DNA, small molecules, and metal ions (21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31). We show that CusB exists as a dimer in solution, and that upon Cu(I) binding, a shift from a more open structure to a more compact one occurs. We suggest that this shift might trigger the opening of the CusCBA channel.
Materials and Methods
CusB cloning, expression, and purification
The CusB gene was isolated from E. coli genomic DNA by polymerase chain reaction using primers containing specific CusB sequences and flanking regions that correspond to the expression vector sequences of pYTB12 (5′ primer-GTTGTACAGAATGCTGGTCATATGAAAAAAATCGCGCTTATTATCG and 3′ primer-GTCACCCGGGCTCGAGGAATTTCAATGCGCATGGGTAGC). This amplicon was cloned into the pYTB12 vector using the free-ligation polymerase chain reaction technique (32). This construct, which encodes for the fusion protein composed of CusB, an intein, and a chitin-binding domain, was transformed into the E. coli strain BL21 (DE3). The CusB construct was expressed in BL21 cells, which were grown to an optical density of 0.6–0.8 at 600 nm and were induced with 1 mM isopropyl-β-d-thiogalactopyranoside (Calbiochem, San Diego, CA) for 20 h at 18°C. Cells were harvested by centrifugation, and the pellets were subjected to three freeze-thaw cycles. The pellet was resuspended in lysis buffer (25 mM Na2HPO4, 150 mM NaCl, and 200 μM phenylmethylsulfonyl fluoride (pH 7.5)). The cells were sonicated by 12 bursts of 30 s each with a 30 s cooling period between bursts (65% amplitude). After sonication, the cells were centrifuged, and the soluble fraction of the lysate was passed through a chitin bead column (New England Biolabs, Ipswich, MA), allowing the CusB fusion to bind to the resin via its chitin-binding domain. The resin was then washed with 30 column volumes of lysis buffer. To induce intein-mediated cleavage, beads were incubated in 50 mM dithiothreitol (DTT), 25 mM NaH2PO4, and 150 mM NaCl at pH 8.9 for 40 h at room temperature. CusB was collected in elution fractions and analyzed using silver-stained sodiumdodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) (10% glycine) (33). Mutant formation was carried out using an identical protocol.
CusB spin labeling
CusB was labeled by an initial incubation with 10 mM DTT overnight. Removal of DTT was carried out using a Microsep Advance Centrifugal Device (ref. no. MCO003C41, Pall, Port Washington, NY) with samples of up to 5 mL with a molecular weight cutoff (3 kDa cutoff) in lysis buffer. Samples were centrifuged four times at 3220 × g and 8°C for 20 min. S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl) methyl methanesulfonothioate (MTSSL; Toronto Research Chemicals, Toronto, Ontario, Canada) dissolved in dimethyl sulfoxide (New England Biolabs, Ipswich, MA). Next, 5 μL of MTSSL solution was added to 1 mL of protein solution whose concentration was 0.01–0.03 mM (a 10-fold molar excess of MTSSL). The protein solution was wrapped in tin foil and vortexed overnight at 4°C. The free spin label was removed using a Microsep Advance Centrifugal Device (Pall) with samples up to 5 mL, with a molecular weight cutoff (3 kDa) in lysis buffer. Samples were centrifuged 15 times at 3220 × g and 8°C for 20 min to remove free spin labels, and after each centrifuge, the samples were diluted with new buffer. Centrifugation was followed by a single dialysis (using 3.5 kDa Pierce cassettes (Thermo Fisher Scientific, Waltham, MA)) against lysis buffer at 4°C, overnight, to ensure that no free spin labels were left. A sample of the running buffer was taken from the last cycle of the Microsep Advance Centrifugal Device, and no free spin EPR signal was observed. Concentration was determined by a Lowry assay (34). The final concentration of CusB protein was 0.01–0.02 mM. Fig. S1 presents a comparison of the continuous-wave (CW)-EPR integrated area between the free spin label and CusB mutants, which indicates 100% spin labeling.
Addition of the metal ion
Cu(I) (tetrakis (acetonitrile) copper(I) hexafluorophosphate (Sigma-Aldrich, St. Louis, MO)) was added to the protein solution under nitrogen gas to preserve inert anaerobic conditions. No Cu(II) EPR signal was observed at any time. In this study, we chose a ratio of 3:1 [Cu(I)]/[CusB] based on the results obtained from CW-EPR (see Fig. S2).
Glutaraldehyde cross-linking
Treatment with glutaraldehyde (a cross-linker that crosses lysine residues) was conducted by mixing 20 μg (10 μL) of interacting protein in 20 mM (35 μL) of sodium phosphate and 0.15 M of NaCl solution at pH 8.5 (phosphate-buffered saline ×10), which was then reacted with various concentrations of glutaraldehyde solution and incubated for 10 min at 37°C. The reaction was terminated by the addition of an equivalent concentration of 1 M Tris-HCl at pH 8, as was gluteraldehyde, followed by SDS-PAGE (10% glycine) analysis.
EPR
A constant-time four-pulse double electron-electron resonance (DEER) experiment with pulse sequence π/2(νobs)-τ1-π(νobs)-t′−π(νpump)-(τ1 + τ2 − t′)-π(νobs)-τ2(νobs)-τ2-echo was performed at (80 ± 0.5 K) on a Q-band Elexsys E580 (equipped with a 2-mm probe head; bandwidth, 220 MHz). A two-step phase cycle was employed on the first pulse. The echo was measured as a function of t′, whereas τ2 was kept constant to eliminate relaxation effects. The pump pulse was set to the maximum of the EPR spectrum. The observer pulse was set to be 60 MHz higher than the pump pulse. The observer π/2 and π pulses, as well as the π pump pulse, had a duration of 40 ns; the dwell time was 20 ns. The observer frequency was 33.78 GHz. The power of the 40 ns π-pulse was 20.0 mW. The parameter τ1 was set to 200 ns, and τ2 was set to 1200 ns. The repetition time was set to 5 ms, and 30 shots per point were applied. The samples were measured in 1.6 mm capillary quartz tubes (Wilmad-Labglass, Vineland, NJ). The data were analyzed using the DeerAnalysis 2015 program and Tikhonov regularization (35, 36). The regularization parameter in the L curve was optimized by examining the fit of the time-domain data. The DEER raw data and the homogeneous background function are presented in the Supporting Material (see Figs. S3 and S4).
CW- EPR data of the various mutants are presented in the Figs. S5 and S6.
Circular dichroism
Circular dichroism (CD) measurements were conducted using a Chirascan spectrometer (Applied Photophysics, Leatherhead, United Kingdom). Measurements were performed in a 1-cm-optical-pathlength cell, and the spectra were recorded from 270 to 190 nm with a step size and bandwidth of 0.5 nm. The CD signal was averaged for 10 s every 2 nm, with three scans per sample.
Results and Discussion
SDS-PAGE analysis
The full-length CusB protein (408 residues) was expressed and purified. Fig. 2 A shows a native gel analysis at three different protein concentrations, which confirmed the formation of CusB oligomerization in solution. Cross-linking experiments with glutaraldehyde (Fig. 2 B; Fig. S7) showed a band at ∼40 KDa in the absence of a cross-linker, corresponding to the CusB monomer. However, when the glutaraldehyde concentration increases, a transition from a mixed monomer-dimer solution to a solely dimer solution (a band close to 80 KDa) occurs. Higher oligomerization states were not detected even at high cross-linker concentrations. Native gel for apo-CusB and holo-CusB (bound to Cu(I)) in the presence of reference proteins is presented in Fig. S8. Both the native gel and the cross-linking experiments suggest that in solution, the full CusB protein exists as a dimer in the apo and holo states. This proposition is compatible with the crystal structures of CusB and CusBA, which indicate that two molecules of CusB interact closely with each other via hydrogen bonds. Specifically, CusB domains 1–3 (the membrane proximal and β-barrel domains) are involved in the interaction between the two CusB molecules; residues E118, Y119, R186, E252, and R292 of one CusB molecule participate in hydrogen bonds with T139, D142, T206, N312, and N113, respectively, of the other CusB molecule (15). Hence, higher oligomerization (trimers and hexamers) can probably occur only in the presence of the CusA and CusC proteins (13).
Figure 2.
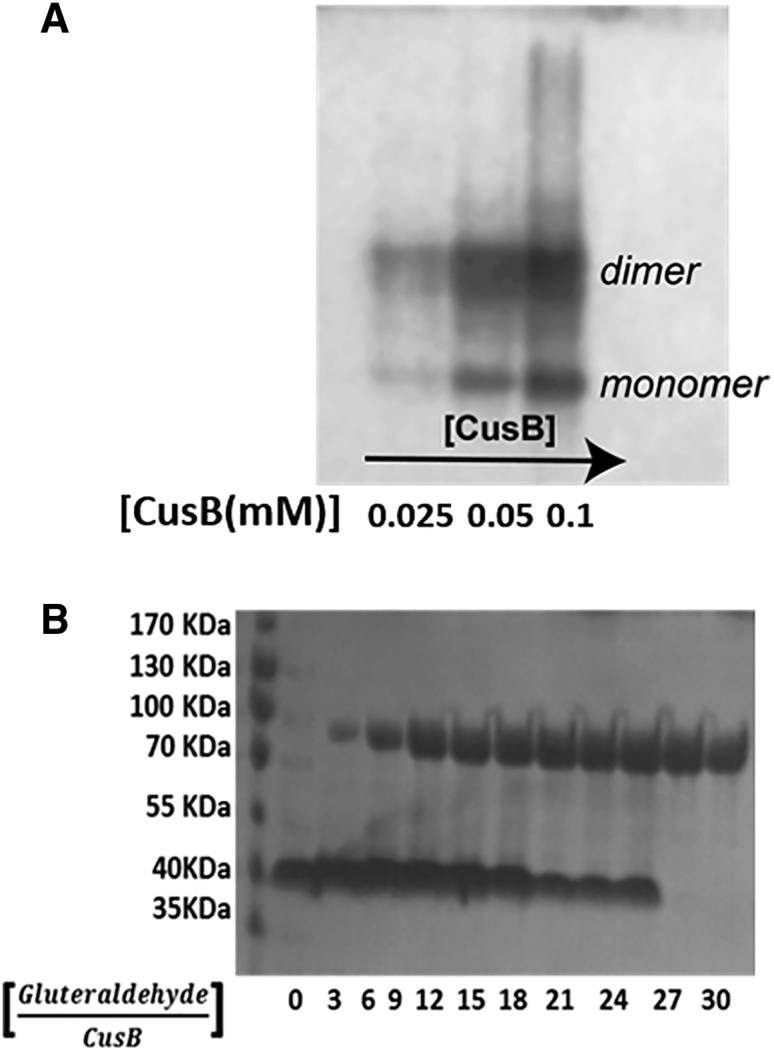
(Upper) Native glycine (10%) gel of the full-length wt-CusB (the various lanes represent different concentrations of CusB). (Lower) Chemical cross-linking with glutaraldehyde SDS-PAGE glycine (10%) gel at various glutaraldehyde concentrations, with [CusB] = 0.05 mM.
CD experiments
To compare the thermodynamic stability levels of apo-CusB and a Cu(I)-bound CusB (holo-CusB), we ran CD experiments at various temperatures. Fig. 3, A and B, present the CD spectra for apo- and holo-CusB, and Fig. 3 C shows the change at 218 nm as a function of temperature. Table 1 presents the secondary structure distribution at various temperatures for both apo- and holo-CusB. Although both states of the protein are quite stable even at 90°C (the prevalence of random-coil structures is ∼15% greater at 90°C than at 20°C), holo-CusB is slightly more thermodynamically stable than at the apo state.
Figure 3.

(A and B) CD spectra at various temperatures from 20 to 90°C for (A) apo-CusB and (B) holo-CusB. (C) Temperature effects at 218 nm for holo-CusB are denoted by black squares and those for apo-CusB are denoted by red squares. [CusB]= 0.025 mM. To see this figure in color, go online.
Table 1.
CD Spectral Analysis Carried Out Using CDNN Software at Various Temperatures for apo-CusB and holo-CusB
| α-Helix (%) | β-Sheet (%) | Random Coil (%) | |
|---|---|---|---|
| 20°C (apo-CusB) | 14.9 ± 0.1 | 54.6 ± 0.4 | 30.4 ± 0.2 |
| 20°C (holo-CusB) | 14.4 ± 0.1 | 54.8 ± 0.4 | 30.8 ± 0.2 |
| 60°C (apo-CusB) | 12.6 ± 0.1 | 54.6 ± 0.4 | 32.8 ± 0.25 |
| 60°C (holo-CusB) | 12.7 ± 0.1 | 56.1 ± 0.4 | 31.2 ± 0.2 |
| 90°C (apo-CusB) | 9.2 ± 0.05 | 48.1 ± 0.3 | 42.7 ± 0.3 |
| 90°C (holo-CusB) | 9.3 ± 0.05 | 49.1 ± 0.3 | 41.6 ± 0.3 |
CDNN software is described in (59).
Pulsed-EPR spectroscopy for CusB domains 2–4
We used pulsed-EPR spectroscopy to identify the conformational differences between apo-CusB and holo-CusB in solution. The most common EPR experiment used to determine nanoscale structural information is the DEER experiment, also known as PELDOR (37, 38, 39, 40, 41, 42, 43, 44, 45). In DEER, two microwave channels are applied to target the dipolar interaction between two paramagnetic centers. Here, we used the site-directed spin-labeling method to introduce paramagnetic centers into our system (46, 47, 48, 49). In site-directed spin labeling, a nitroxide radical, usually the methanesulfonothioate spin label (MTSSL, see inset in Fig. 1), is attached to a cysteine residue in the biomolecule. Since CusB lacks cysteine residues, we selected a set of probe residues in the protein, and for each DEER experiment, we mutated one of those probes to cysteine and spin-labeled the mutants using MTSSL (the selected residues are marked in Fig. 1). Therefore, the inter-monomer distance distribution was evaluated at different regions of the protein. Native gel pictures of non-spin-labeled and spin-labeled mutants confirmed that the dimerization of CusB was not affected by the spin-labeling procedure (see Fig. S9).
Fig. 4 shows the DEER time-domain signals for apo-CusB and holo-CusB and the corresponding distance-distribution functions for the various CusB mutants. The DEER data were acquired up to ∼1.0–1.2 μs. For this time domain, we are restricted to distances of up to 4.0 nm. We also observed that although several mutants underwent large changes in the DEER distance-distribution functions upon Cu(I) binding, in other mutants, the changes were minor. The most substantial changes were observed for the A248C, A236C, and S109C mutants, whereas for N312C and G173C, only slight changes in the DEER distance-distribution functions were observed. Moderate changes in the DEER signals were observed for A303C and A188C.
Figure 4.

(A and B) Q-band DEER signals for various spin-labeled CusB mutants in the (A) apo (black line) and (B) holo (gray line) states at a ratio of 3:1 Cu(I)/CusB. (C) The distance-distribution functions corresponding to the DEER signals in (A) and (B) are shown. The dashed red line corresponds to the distance distribution obtained from MMM modeling on the PDB: 3NE5 crystal structure. (D) Positions of the selected spin labels (Fig. S10 shows a magnification of these views). [CusB] = 0.02 mM. To see this figure in color, go online.
To compare the DEER data with the crystal structure of CusB, we performed multiscale modeling of macromolecular systems (MMM software, 2015 version) (50). MMM is a computational approach (51) for deriving the rotamer library based on a coarse-grained representation of the conformational space of the spin label (52). This method describes spin labels by a set of alternative conformations or rotamers, which can be attached without serious clashes with atoms of other residues or cofactors. The rotamer library is derived from molecular dynamics simulation with a total length of 100 ns at a temperature of 175 K, which is an estimate of the glass transition of a protein sample. The program uses third-party software, SCWRL4, developed by the group of Dunbrack (53). The Protein Data Bank structure of the CusB-CusA complex (PDB: 3NE5) was the input to the MMM program. This structure contains one CusA molecule and two CusB molecules. The dashed-red-line distribution in Fig. 4 C denotes the distance distribution obtained from MMM. Fig. 4 D shows the conformation of the attached spin labels obtained from the MMM simulations. For all mutants besides the CusB_A303C mutant, the DEER distance-distribution function obtained for the apo state is consistent with the distance distribution derived for the spin labels attached to the crystal-structure conformation of the CusB dimer. The DEER distances do not preclude the formation of a symmetrical trimer in solution; however, when coupled with cross-linking and native-gel experiments (Fig. 2), the combined data strongly confirm the formation of a CusB dimer in solution. Moreover, based on the crystal structure (15), a symmetrical trimer is not physically possible. For CusB_A303C, the crystal structure suggests a larger mean distance of 4.0 ± 0.8 nm, whereas the DEER shows a distribution of 2.8 ± 0.7 nm. The DEER time domain traces up to 1.0 μs, and thus, distances of up to 4.0 nm are reliable. However, the distribution width may be different owing to the restriction in the resolution. This difference indicates that the most probable distance on the crystal-structure conformation at this site is larger than that obtained by DEER.
To understand better how Cu(I) binding affects the CusB structure, we used the elastic network model (ENM) implemented in the MMM software. The inputs to the ENM are the PDB structures of CusB and the distance-distribution functions obtained by DEER. Based on the DEER constraints, the program generates a new PDB file (51).
ENM
We used the ENM software to model the structure of the CusB dimer in the apo and holo states, using the seven distance-distribution constraints (Table 2) obtained from the DEER data in the solution. The CusAB crystal structure (PDB: 3NE5) was used as a basis for this model. The 3NE5 structure contains one CusA molecule and two CusB molecules. There may be small deviations from the reported ENM structures owing to the restricted resolution of the time-domain DEER traces. Fig. 5 A shows the structure of the CusB dimer (molecule 1 is red and molecule 2 is blue) constructed using the seven DEER constraints in the apo state overlaid onto the PDB: 3NE5 crystal structure (gray). Although some minor differences between the two structures are observed, in general, we can assume that the two structures are similar (taking into account the experimental error, as well as the fact that the DEER data were acquired in solution, not from a crystal structure). This similarity suggests that the DEER results obtained for apo-CusB are in good agreement with the crystal structure. Fig. 5 B shows the structure obtained using the DEER constraints in the holo state (blue and red) overlaid on the structure achieved using the DEER constraints in the apo state (gray). Fig. 5 C shows the structure of apo-CusB and holo-CusB using the seven DEER constraints in the presence of CusA. A clear structural difference is observed between the apo and holo CusB states.
Table 2.
Distance Distribution Constraints Used in ENM
| Apo State (nm) | Holo State (nm) | |
|---|---|---|
| CusB_N312C | 2.4 ± 0.4 | 2.5 ± 0.4 |
| CusB_A303C | 2.8 ± 0.7 | 1.8 ± 0.3 |
| CusB_A248C | 2.1 ± 0.2 | 3.5 ± 0.3 |
| CusB_A236C | 2.1 ± 0.3 | 3.7 ± 0.5 |
| CusB_A188C | 2.3 ± 0.8 | 3.1 ± 0.7 |
| CusB_G173C | 2.8 ± 0.3 | 2.0 ± 0.4 |
| CusB_S109C | 2.1 ± 0.3 | 3.9 ± 0.5 |
Figure 5.

(A) A ribbon view of the CusB structure (red, molecule 1; blue, molecule 2) obtained using the ENM implemented in the MMM program, using the seven DEER constraints in the apo state (solid colors) overlaid onto the CusBA crystal structure (PDB: 3NE5; gray). (B) CusB structure using the seven DEER constraints in the holo state (solid colors) overlaid onto the structure obtained in the apo state (gray structure). (C) CusB structure using the seven DEER constraints in the apo state (gray) and holo state in the presence of CusA (solid colors). To see this figure in color, go online.
The ENM indicates that upon Cu(I) binding, the structure of the CusB dimer becomes much more compact. Specifically, the β-barrel domains (domains 2 and 3) of the two CusB molecules spread apart, leading to the α-helical domain (domain 4) of each molecule getting closer to the β-barrel domains (domains 2 and 3). In other words, the dimer undergoes variations that affect the coordination of one CusB molecule with respect to the second CusB molecule, resulting in a denser structure. We suggest that these structural changes might lead to the opening of the whole CusCBA channel, which might lead to the export of Cu(I) from the cell.
DEER measurements on residues in the N-terminal domain of CusB
DEER measurements were also performed on two residues in the N-terminal domain of the full CusB protein that are missing from the crystal structure (thus, the MMM simulations cannot be used for these sites). Fig. 6 shows the DEER signal and the corresponding distance distribution functions for M61C and A28C in the apo and holo states of the full CusB. For M61C, we observed a decrease in the mean distance upon Cu(I) coordination from 2.4 ± 0.4 to 1.5 ± 0.5 nm. In contrast, for A28C we observed a large increase in the mean distance upon Cu(I) coordination from 1.5 ± 0.3 to 2.9 ± 0.3 nm. The N-terminal domain of CusB is suggested to be disordered where it faces the periplasm liquid to be able to interact with CusF. This suggests that the two N-terminal domains (at least the first 30 aa) of the two molecules of the dimer spread apart upon Cu(I) coordination, whereas the M61 residues, which are connected to the main body of CusB, get a bit closer to each other. This observation is in line with our findings from a previous investigation that focused exclusively on CusBNT (54). In that study, we showed that in solution, the two N-terminal domains (the first 60 aa) are close to each other, but that they spread apart in the presence of a CusF metallochaperone or in the presence of Cu(I).
Figure 6.

Q-band DEER signals for spin-labeled CusB at the M61C and A28C positions in (A) the apo state (black line) and (B) the holo state (gray line). (C) Corresponding distance-distribution functions for various DEER signals for apo-CusB (black line) and holo-CusB (gray line). [CusB] = 0.02 mM.
Conclusions
The CusCBA complex can transfer Cu(I) ions from the periplasm through the N-terminal domain of CusB. It has also been demonstrated that the chaperone, CusF, can directly transfer its bound Cu(I) to the CusBNT domain (54, 55, 56, 57). Since the N-terminal domain of CusB consists of 18.3% negatively charged amino acids and 40% polar amino acids, it has the potential to interact well with the electropositive CusF (58). However, it seems likely that CusB must adopt a specific conformation to enable the metal ion to be transferred from CusF through CusBNT and out of the cell. Here, we have succeeded in showing, for the first time, to our knowledge, that the full CusB protein is a dimer in solution, and that it undergoes major conformational changes upon Cu(I) coordination, especially in the orientation of the two CusB molecules with respect to each other. We utilized EPR spectroscopy to target structural changes in the apo and holo states of CusB in four regions of the proteins: the N-terminal domain, the β-barrel domains (domain 2 and 3), and the α-helical domain (domain 4). We observed that upon Cu(I) coordination, the two β-barrel domains (domains 2 and 3) of the CusB dimer spread apart, and that the α-helical domain (domain 4) of each CusB molecule approaches the β-barrel domains (domains 2 and 3). Therefore, the most significant changes are observed in those areas surrounding the A248, S109, A236, and A188 residues. These conformational changes result in a holo-CusB structure that is more compact than the apo-CusB structure. The formed compact structure expands the channel pore between CusB and CusC and probably allows the transition of Cu(I) ions to CusC. The EPR results are consistent with the gel filtration chromatography experiments suggesting that CusB undergoes conformational changes upon Cu(I) coordination, such that it becomes more compact (19).
We also showed that the two N-terminal domains of the CusB dimer undergo structural changes upon Cu(I) binding, namely, they spread apart. This observation is consistent with previous data that we obtained from experiments focusing exclusively on CusBNT (54).
The flexibility of CusB enables it to receive Cu(I) from the CusF chaperone in the periplasm. On the basis of the data described herein, together with previous findings, we propose that the CusCFBA complex functions according to the following efflux mechanism. 1) Initially, Cu(I) ions are accumulated by CusBNT from CusF in the periplasm. 2) Then, conformational changes occur in the β-barrel domains (domains 2 and 3) of CusB. 3) Those changes induce the α-helical domain (domain 4) of CusB to approach the β-barrel domains (domains 2 and 3), which leads to 4) formation of a compact CusB structure that enables the CusCBA pore channel to open. These structural changes drive the Cu(I) ions to the CusC protein and out of the cell. Additional structural studies on CusBA and CusCB complexes in solution are required to fully map the Cu(I) efflux mechanism through this intriguing system.
Author Contributions
A.M. designed and performed all research and assisted in writing the article. A.A. assisted with the EPR measurements and the expression of proteins. Y.M. calibrated the expression and purification protocol of CusB. S.R. designed the research, analyzed the data, and wrote the article.
Acknowledgments
This study was supported by a Marie Curie Career Integration Grant 303636, and by the Israel Science Foundation (grant 176/16). The Elexsys E580 Bruker EPR spectrometer was partially supported by the Israel Science Foundation (grant 564/12).
Editor: Julie Biteen.
Footnotes
Supporting Materials and Methods and ten figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(17)30550-7.
Supporting Material
References
- 1.Beswick P.H., Hall G.H., Lott K.A. Copper toxicity: evidence for the conversion of cupric to cuprous copper in vivo under anaerobic conditions. Chem. Biol. Interact. 1976;14:347–356. doi: 10.1016/0009-2797(76)90113-7. [DOI] [PubMed] [Google Scholar]
- 2.Boal A.K., Rosenzweig A.C. Structural biology of copper trafficking. Chem. Rev. 2009;109:4760–4779. doi: 10.1021/cr900104z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burkhead J.L., Reynolds K.A., Pilon M. Copper homeostasis. New Phytol. 2009;182:799–816. doi: 10.1111/j.1469-8137.2009.02846.x. [DOI] [PubMed] [Google Scholar]
- 4.Prohaska J.R. Role of copper transporters in copper homeostasis. Am. J. Clin. Nutr. 2008;88:826S–829S. doi: 10.1093/ajcn/88.3.826S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rensing C., Grass G. Escherichia coli mechanisms of copper homeostasis in a changing environment. FEMS Microbiol. Rev. 2003;27:197–213. doi: 10.1016/S0168-6445(03)00049-4. [DOI] [PubMed] [Google Scholar]
- 6.Delmar J.A., Su C.-C., Yu E.W. Structural mechanisms of heavy-metal extrusion by the Cus efflux system. Biometals. 2013;26:593–607. doi: 10.1007/s10534-013-9628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Franke S., Grass G., Nies D.H. Molecular analysis of the copper-transporting efflux system CusCFBA of Escherichia coli. J. Bacteriol. 2003;185:3804–3812. doi: 10.1128/JB.185.13.3804-3812.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su C.-C., Long F., Yu E.W. The Cus efflux system removes toxic ions via a methionine shuttle. Protein Sci. 2011;20:6–18. doi: 10.1002/pro.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fung M.C., Bowen D.L. Silver products for medical indications: risk-benefit assessment. J. Toxicol. Clin. Toxicol. 1996;34:119–126. doi: 10.3109/15563659609020246. [DOI] [PubMed] [Google Scholar]
- 10.Lok C.-N., Ho C.-M., Che C.M. Silver nanoparticles: partial oxidation and antibacterial activities. J. Biol. Inorg. Chem. 2007;12:527–534. doi: 10.1007/s00775-007-0208-z. [DOI] [PubMed] [Google Scholar]
- 11.Silver S., Phung T., Silver G. Silver as biocides in burn and wound dressings and bacterial resistance to silver compounds. J. Ind. Microbiol. Biotechnol. 2006;33:627–634. doi: 10.1007/s10295-006-0139-7. [DOI] [PubMed] [Google Scholar]
- 12.Lei H.-T., Bolla J.R., Yu E.W. Crystal structures of CusC review conformational changes accompanying folding and transmembrane channel formation. J. Mol. Biol. 2014;426:403–411. doi: 10.1016/j.jmb.2013.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janganan T.K., Bavro V.N., Walmsley A.R. Evidence for the assembly of a bacterial tripartite multidrug pump with a stoichiometry of 3:6:3. J. Biol. Chem. 2011;286:26900–26912. doi: 10.1074/jbc.M111.246595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chacón K.N., Mealman T.D., Blackburn N.J. Tracking metal ions through a Cu/Ag efflux pump assigns the functional roles of the periplasmic proteins. Proc. Natl. Acad. Sci. USA. 2014;111:15373–15378. doi: 10.1073/pnas.1411475111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su C.-C., Long F., Yu E.W. Crystal structure of the CusBA heavy-metal efflux complex of Escherichia coli. Nature. 2011;470:558–562. doi: 10.1038/nature09743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su C.-C., Long F., Yu E.W. Charged amino acids (R83, E567, D617, E625, R669, and K678) of CusA are required for metal ion transport in the Cus efflux system. J. Mol. Biol. 2012;422:429–441. doi: 10.1016/j.jmb.2012.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su C.-C., Yang F., Yu E.W. Crystal structure of the membrane fusion protein CusB from Escherichia coli. J. Mol. Biol. 2009;393:342–355. doi: 10.1016/j.jmb.2009.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loftin I.R., Franke S., McEvoy M.M. A novel copper-binding fold for the periplasmic copper resistance protein CusF. Biochemistry. 2005;44:10533–10540. doi: 10.1021/bi050827b. [DOI] [PubMed] [Google Scholar]
- 19.Bagai I., Liu W., McEvoy M.M. Substrate-linked conformational change in the periplasmic component of a Cu(I)/Ag(I) efflux system. J. Biol. Chem. 2007;282:35695–35702. doi: 10.1074/jbc.M703937200. [DOI] [PubMed] [Google Scholar]
- 20.Melek N., Chakravorty D.K., Merz J.K.M. Models for the metal transfer complex of the N-terminal region of CusB and CusF. Biochemistry. 2015;54:4226–4235. doi: 10.1021/acs.biochem.5b00195. [DOI] [PubMed] [Google Scholar]
- 21.Dockter C., Volkov A., Paulsen H. Refolding of the integral membrane protein light-harvesting complex II monitored by pulse EPR. Proc. Natl. Acad. Sci. USA. 2009;106:18485–18490. doi: 10.1073/pnas.0906462106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fajer P.G., Gyimesi M., Song L.K. Myosin cleft closure by double electron-electron resonance and dipolar EPR. J. Phys. Condens. Matter. 2007;19:285208. [Google Scholar]
- 23.Galiano L., Bonora M., Fanucci G.E. Interflap distances in HIV-1 protease determined by pulsed EPR measurements. J. Am. Chem. Soc. 2007;129:11004–11005. doi: 10.1021/ja073684k. [DOI] [PubMed] [Google Scholar]
- 24.Klare J.P. Site-directed spin labeling EPR spectroscopy in protein research. Biol. Chem. 2013;394:1281–1300. doi: 10.1515/hsz-2013-0155. [DOI] [PubMed] [Google Scholar]
- 25.Krstic I., Hansel R., Prisner T.F. Long-range distance measurements on nucleic acids in cells by pulsed EPR spectroscopy. Angew Chem Int. Ed. Engl. 2011;50:5070–5074. doi: 10.1002/anie.201100886. [DOI] [PubMed] [Google Scholar]
- 26.Martorana A., Bellapadrona G., Goldfarb D. Probing protein conformation in cells by EPR distance measurements using Gd3+ spin labeling. J. Am. Chem. Soc. 2014;136:13458–13465. doi: 10.1021/ja5079392. [DOI] [PubMed] [Google Scholar]
- 27.Schiemann O., Piton N., Engels J.W. Spin labeling of oligonucleotides with the nitroxide TPA and use of PELDOR, a pulse EPR method, to measure intramolecular distances. Nat. Protoc. 2007;2:904–923. doi: 10.1038/nprot.2007.97. [DOI] [PubMed] [Google Scholar]
- 28.Cafiso D.S. Identifying and quantitating conformational exchange in membrane proteins using site-directed spin labeling. Acc. Chem. Res. 2014;47:3102–3109. doi: 10.1021/ar500228s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freed D.M., Lukasik S.M., Cafiso D.S. Monomeric TonB and the Ton box are required for the formation of a high-affinity transporter-TonB complex. Biochemistry. 2013;52:2638–2648. doi: 10.1021/bi3016108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim M., Xu Q., Cafiso D.S. Solutes alter the conformation of the ligand binding loops in outer membrane transporters. Biochemistry. 2008;47:670–679. doi: 10.1021/bi7016415. [DOI] [PubMed] [Google Scholar]
- 31.Levy A.R., Yarmiayev V., Ruthstein S. Probing the structural flexibility of the human copper metallochaperone Atox1 dimer and its interaction with the CTR1 c-terminal domain. J. Phys. Chem. B. 2014;118:5832–5842. doi: 10.1021/jp412589b. [DOI] [PubMed] [Google Scholar]
- 32.Saiki R.K., Gelfand D.H., Erlich H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1988;239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- 33.Cleveland D.W., Fischer S.G., Laemmli U.K. Peptide mapping by limited proteolysis in sodium dodecyl sulfate and analysis by gel electrophoresis. J. Biol. Chem. 1977;252:1102–1106. [PubMed] [Google Scholar]
- 34.Peterson G.L. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- 35.Jeschke G. Deeranalysis 2006: Distance measurements on nanoscopic length scales by pulse ESR. In: Hemminga M.A., Berliner L.J., editors. ESR Spectroscopy in Membrane Biophysics. Springer; 2007. pp. 287–288. [Google Scholar]
- 36.Jeschke G. DEER distance measurements on proteins. Annu. Rev. Phys. Chem. 2012;63:419–446. doi: 10.1146/annurev-physchem-032511-143716. [DOI] [PubMed] [Google Scholar]
- 37.Abé C., Klose D., Steinhoff H.J. Orientation selective DEER measurements on vinculin tail at X-band frequencies reveal spin label orientations. J. Magn. Reson. 2012;216:53–61. doi: 10.1016/j.jmr.2011.12.024. [DOI] [PubMed] [Google Scholar]
- 38.Aitha M., Moritz L., Crowder M.W. Conformational dynamics of metallo-β-lactamase CcrA during catalysis investigated by using DEER spectroscopy. J. Biol. Inorg. Chem. 2015;20:585–594. doi: 10.1007/s00775-015-1244-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joseph B., Korkhov V.M., Bordignon E. Conformational cycle of the vitamin B12 ABC importer in liposomes detected by double electron-electron resonance (DEER) J. Biol. Chem. 2014;289:3176–3185. doi: 10.1074/jbc.M113.512178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larsen R.G., Singel D.J. Double electron-electron resonance spin-echo modulation: spectroscopic measurements of electron spin pair separations in orientationally disordered solids. J. Chem. Phys. 1993;98:5134–5146. [Google Scholar]
- 41.Milov A.D., Maryasov A.G., Tsvetkov Y.D. Pulsed electron double resonance (PELDOR) and its applications in free-radicals research. Appl. Magn. Reson. 1998;15:107–143. [Google Scholar]
- 42.Pannier M., Veit S., Spiess H.W. Dead-time free measurement of dipole-dipole interactions between electron spins. J. Magn. Reson. 2000;142:331–340. doi: 10.1006/jmre.1999.1944. [DOI] [PubMed] [Google Scholar]
- 43.Sahu I.D., McCarrick R.M., Lorigan G.A. DEER EPR measurements for membrane protein structures via bifunctional spin labels and lipodisq nanoparticles. Biochemistry. 2013;52:6627–6632. doi: 10.1021/bi4009984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sicoli G., Mathis G., Gambarelli S. Double electron-electron resonance (DEER): a convenient method to probe DNA conformational changes. Angew. Chem. Int. Ed. Engl. 2008;47:735–737. doi: 10.1002/anie.200704133. [DOI] [PubMed] [Google Scholar]
- 45.Glaenzer J., Peter M.F., Hagelueken G. PELDOR spectroscopy reveals two defined states of a sialic acid TRAP transporter SBP in solution. Biophys. J. 2017;112:109–120. doi: 10.1016/j.bpj.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Altenbach C., Kusnetzow A.K., Hubbell W.L. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc. Natl. Acad. Sci. USA. 2008;105:7439–7444. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Columbus L., Hubbell W.L. A new spin on protein dynamics. Trends Biochem. Sci. 2002;27:288–295. doi: 10.1016/s0968-0004(02)02095-9. [DOI] [PubMed] [Google Scholar]
- 48.Hubbell W.L., López C.J., Yang Z. Technological advances in site-directed spin labeling of proteins. Curr. Opin. Struct. Biol. 2013;23:725–733. doi: 10.1016/j.sbi.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.López C.J., Oga S., Hubbell W.L. Mapping molecular flexibility of proteins with site-directed spin labeling: a case study of myoglobin. Biochemistry. 2012;51:6568–6583. doi: 10.1021/bi3005686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boal A.K., Rosenzweig A.C. Crystal structures of cisplatin bound to a human copper chaperone. J. Am. Chem. Soc. 2009;131:14196–14197. doi: 10.1021/ja906363t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Polyhach Y., Bordignon E., Jeschke G. Rotamer libraries of spin labelled cysteines for protein studies. Phys. Chem. Chem. Phys. 2011;13:2356–2366. doi: 10.1039/c0cp01865a. [DOI] [PubMed] [Google Scholar]
- 52.Jeschke G. Conformational dynamics and distribution of nitroxide spin labels. Prog. Nucl. Magn. Reson. Spectrosc. 2013;72:42–60. doi: 10.1016/j.pnmrs.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 53.Krivov G.G., Shapovalov M.V., Dunbrack R.L.J., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meir A., Natan A., Ruthstein S. EPR spectroscopy identifies Met and Lys residues that are essential for the interaction between the CusB N-terminal domain and metallochaperone CusF. Metallomics. 2015;7:1163–1172. doi: 10.1039/c5mt00053j. [DOI] [PubMed] [Google Scholar]
- 55.Mealman T.D., Bagai I., McEvoy M.M. Interactions between CusF and CusB identified by NMR spectroscopy and chemical cross-linking coupled to mass spectrometry. Biochemistry. 2011;50:2559–2566. doi: 10.1021/bi102012j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mealman T.D., Zhou M., McEvoy M.M. N-terminal region of CusB is sufficient for metal binding and metal transfer with the metallochaperone CusF. Biochemistry. 2012;51:6767–6775. doi: 10.1021/bi300596a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chakravorty D.K., Wang B., Merz J.K.M. Insights into the cation-π interaction at the metal binding site of the copper metallochaperone CusF. J. Am. Chem. Soc. 2011;133:19330–19333. doi: 10.1021/ja208662z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Argüello J.M., Patel S.J., Quintana J. Bacterial Cu+-ATPases: models for molecular structure-function studies. Metallomics. 2016;8:906–914. doi: 10.1039/c6mt00089d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Böhm G., Muhr R., Jaenicke R. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 1992;5:191–195. doi: 10.1093/protein/5.3.191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.