

Abstract
We report the design and synthesis of a series of BACE1 inhibitors incorporating mono-and bicyclic 6-substituted 2-oxopiperazines as novel P1′ and P2′ ligands and isophthalamide derivative as P2-P3 ligands. Among mono-substituted 2-oxopiperazinones, inhibitor 5a with N-benzyl-2-oxopiperazinone and isophthalamide showed potent BACE1 inhibitory activity (Ki = 2 nM). Inhibitor 5g, with N-benzyl-2-oxopiperazinone and substituted indole-derived P2-ligand showed a reduction in potency. The X-ray crystal structure of 5g-bound BACE1 was determined and used to design a set of disubstituted 2-oxopiperazinones and bicyclic derivatives that were subsequently investigated. Inhibitor 6j with an oxazolidinone derivative showed a BACE1 inhibitory activity of 23 nM and cellular EC50 of 80 nM.
Keywords: BACE-1, β-secretase, Alzheimer’s disease, Memapsin 2, Protease inhibitors, design, piperazinones, inhibitor, secretase
Graphical abstract
A series of BACE1 inhibitors incorporating mono- and bicyclic 2-oxopiperazines is described. Inhibitor 5a showed an enzyme Ki of 2 nM and a cellular EC50 value of 3.5 nM. An X-ray structural analysis of a realted compound 5g-bound BACE1 provided insight into the ligand-binding site interactions.
Alzheimer’s disease (AD) continues to be a major healthcare challenge in medicine.1,2 Currently, there is no effective treatment that will prevent or slow down the progression of AD.3,4 In recent years, a significant effort is being devoted toward the development of β-secretase inhibitors as potential drugs for AD treatment.5,6 There are at least three β-secretase (also known as memapsin 2 or BACE1) inhibitors that have entered advanced clinical trials.7 The pathological hallmark of AD has been the accumulation of β-amyloid (Aβ) peptides in the brain. This results in the accumulation of amyloid plaques and neurofibrillary tangles which leads to neuronal cell death.8,9,10
One of the major goals of today’s AD drug discovery efforts is to reduce Aβ-production through the inhibition of β-secretase. Aβ is produced by the sequential cleavage of the amyloid precursor protein (APP) by two key aspartic acid proteases, first by the β-secretase (β-site APP cleaving enzyme) and then by a γ-secretase. There are two BACE isoforms that are designated as BACE1 (memapsin 2) and BACE2 (memapsin 1). BACE1 (memapsin 2) is involved in the amyloidogenic pathway and is responsible for cleaving APP leading to the formation of 40/42 residue peptides, the major constituent of the brain amyloid plaques.11–13 Thus, development of potent and selective BACE1 inhibitors has become a major focus for AD drug discovery in many laboratories due to the central role of BACE1 in APP processing. Also, it is encouraging that BACE1 gene deletion in mice showed only a mild phenotypic response. It is important to note that recent studies revealed that besides APP, BACE has other important substrates.14,15
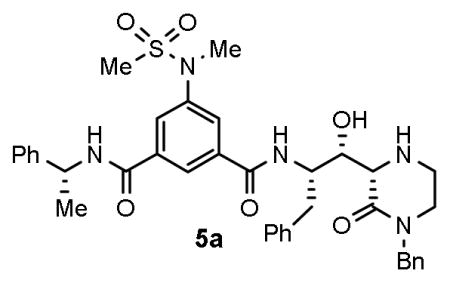
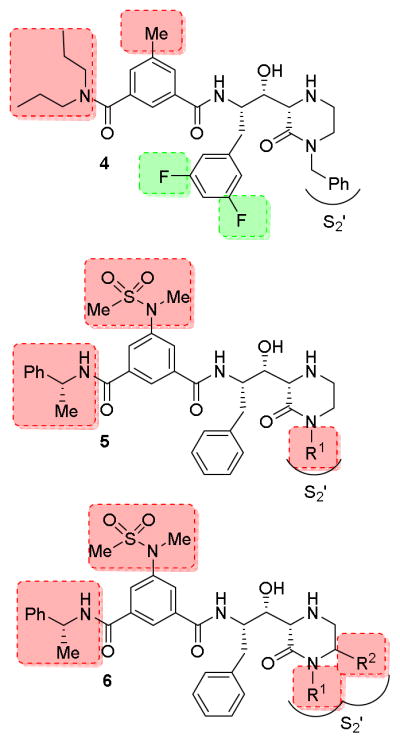
Over the years, many potent and selective BACE1 inhibitors have been developed based upon hydroxyethylene and hydroxyethylamine transition-state isosteres.5,13 We and others have reported BACE1 inhibitors incorporating substituted isophthalamides as P2-P3 ligands in combination with hydroxyethylene and hydroxyethylamine isosteres.16–19 As shown in Figure 1, BACE1 inhibitors 1 and 2 showed excellent BACE1 potency with low nM Ki values and they also showed reduction of cellular Aβ production. Our X-ray structural studies of inhibitors 1- and 2-bound BACE1 provided insight into the ligand-binding site interactions. In particular, the P2-isophthalamide derivative occupies the S2-site and makes extensive hydrogen bonding interactions with Asn 233, Ser 325, and Arg 235.19,20,21 The phenylmethyl moiety makes non-polar interactions with the extended hydrophobic binding site. We have subsequently designed a number of other potent and selective BACE1 inhibitors, including inhibitor 3 with a substituted isophthalamide ligand.13,19,21 Substituted isophthalamide derivatives without the 5-methylsulfonamides have also been utilized in the design of potent BACE1 inhibitors with hydroxyethylamine isosteres.5,13 Cumming and co-workers designed a series of BACE1 inhibitors incorporating conformationally constrained piperazine and piperazinone derivatives as the P1′ and P2′ ligands while keeping the basic hydroxyethylamine scaffold as the transition-state mimic.22,23,24 As shown in Figure 2, they have investigated dipropyl isophthalamide as the P2-ligand in combination with mono-substituted piperazinones.
Figure 1.

BACE-1 inhibitors with isophthalamide P2-P3 ligands.
Figure 2.
Piperazinone and piperazine derivatives as BACE-1 inhibitors.
In our continuing efforts toward BACE1 inhibitor design, we are interested in the incorporation of classical transition state isosteres embedded in many FDA approved HIV-1 protease inhibitors.25–27 In the present studies, we have investigated various di- and tri-substituted piperazinone-derived hydroxyethylamine isosteres in combination with P2-P3 isophthalamide ligands shown in inhibitors 1 and 2. Herein, we report the design, synthesis, and evaluation of a series of BACE1 inhibitors with substituted piperazinones with donor and acceptor groups to interact with the S1′-S2′ subsites of the BACE1 active site. We opted to utilize the isophthalamide P2-P3 ligand that we have successfully incorporated in previous inhibitors. Asymmetric synthesis of these substituted piperazinones and aldol reaction with phenylalaninal provided convenient access to various piperazinone-derived hydroxylethylamine isosteres.
Initially, we focused our investigation on a small set of P2′ ligands. In particular, we explored the outcome of diverse alkyl substituents on the nitrogen of the oxopiperazine ring (Figure 2, structure class 5). We then focused on the incorporation of substituents at the C6-position of the oxopiperazine ring to modulate inhibitor binding at the S1′-subsite. Furthermore, we planned to explore reduction of the oxopiperazine carbonyl group and incorporation of a bicyclic ring involving R1 and R2 groups (Figure 2, structure class 6). The synthesis of inhibitors 5a–f is shown in Scheme 1. We carried out an aldol reaction between (S)-N,N-dibenzylphenylalaninal 7a and commercially available substituted 4-Boc-2-oxopiperazines 8a–f.28,29 As a representative example, for compound 9a, oxopiperazine 8a28 in THF was treated with 1.5 equivalents of LHMDS at −78 °C and the reaction mixture was stirred for 2 h. N,N-dibenzylphenylalaninal 7a was then added and the resulting mixture was warmed to 23 °C for 3 h. The reaction was quenched with aqueous NaHCO3 solution, then a standard work up and chromatography over silica gel provided 9a in 46% yield as a pure isomer.16 Other oxopiperazines 8b–f were converted to aldol products 9b–f in similar yields (40–46%). The stereochemical assignment of aldol products was based upon literature precedence.16,29,30 The removal of the N,N-dibenzyl groups by catalytic hydrogenation over Pearlman’s catalyst afforded the corresponding amines 10a–f in very good yields (85–90%). Amines 10a–f were coupled with known substituted isophthalic acid derivative 1119 in CH2Cl2 in the presence of EDCI, HOBt, and diisopropylethylamine (DIPEA), which afforded inhibitors 5a–f in good yields. For the synthesis of inhibitor 5g, substituted indole carboxylic acid 12 was prepared as described previously.31 Coupling of amine 10a with acid 12 provided the corresponding coupling product. Removal of the Boc-group by exposure to TFA in CH2Cl2 afforded inhibitor 5g in 60% yield for the two steps.
Scheme 1.

Reagents and conditions: (a) LHMDS, THF, −78 °C to 25 °C, 46%; (b) H2, Pd(OH)2, EtOH, AcOH, 23 °C, 90%; (c) 11, or 12, EDCI, HOBt, DIPEA, CH2Cl2; 23 °C, 60%; (d) TFA, CH2Cl2, 23 °C, 80%.
Besides N-substituted oxopiperazines, we also planned to explore N-alkyl-6-substituted 2-oxopiperazines. Previous literature reports32,33 for the synthesis of these substituted oxopiperazine, involved low-yielding coupling reactions or harsh reaction conditions. We therefore designed an improved route using readily available amino acids as shown in Scheme 2. (S)-N,N-dibenzylphenylalaninal 7a and (S)-N,N-dibenzylalaninal 7b were prepared from the corresponding alcohol as described previously.29 Reductive amination34 of these aldehydes with glycine ethyl ester hydrochloride with Na(OAc)3BH in a mixture of acetic acid and 1,2-dichloroethane at 23 °C for 10 h provided the corresponding amines. These were protected as Boc-derivatives 13a and 13b by treatment with Boc2O and triethylamine in MeOH. Catalytic hydrogenation of 13a,b over Pearlman’s catalyst in ethanol in the presence of AcOH at 23 °C afforded the corresponding amines. Treatment of the resulting aminoester with NaH and benzylbromide in THF at 23 °C furnished N-benzyl 6-alkyl-2-oxopiperazines 14a,b in good yields. The synthesis of enantiomeric 2-oxopiperazines ent-14a,b were carried out using analogous procedures utilizing (R)-N,N-dibenzylphenylalaninal ent-7a or (S)-N,N-dibenzylalaninal ent-7b. For the synthesis of 2-oxo-piperazine 15, amino ester derivative ent-13a was hydrogenated over Pearlman’s catalyst and the resulting amino ester was treated with NaH and methyl iodide in THF at 23 °C to provide 15 in 68% yield over two-steps.
Scheme 2.
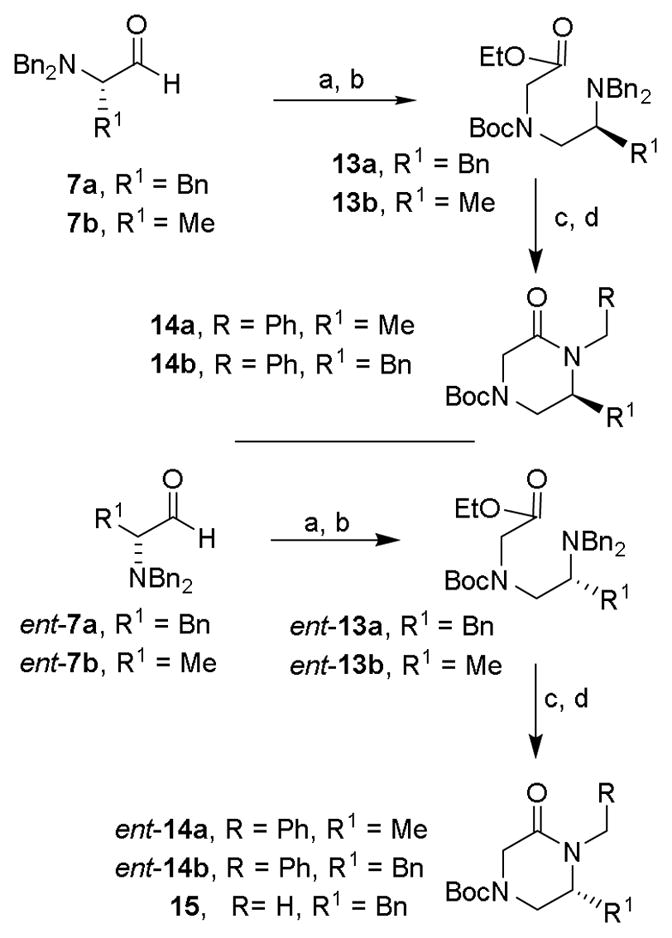
Reagents and conditions: (a) glycine ethyl ester hydrochloride salt, NaBH(OAc)3, AcOH, 1,2-DCE, 23 °C, 85%; (b) Boc2O, TEA, MeOH, 50 °C, 78%; (c) H2, Pd(OH)2, EtOH, AcOH, 23 °C; (d) BnBr or MeI, NaH, dry THF, 68% (over 2 steps).
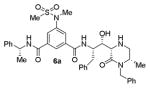
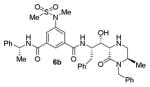
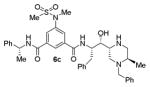
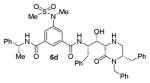
The synthesis of the 6-substituted-2-oxopiperazine-derived inhibitors is shown in Scheme 3. Aldol reaction of substituted-2-oxopiperazines 14a,b with (R)-N,N-dibenzylphenylalaninal 7 provided the corresponding aldol products in moderate yields (38–45%). The absolute and relative stereochemistry of the hydroxyl group and cyclic piperazinone ring were unambiguously set via chirality transfer from the α-amino acid center in a non-chelation controlled aldol reaction as described previously.16,29,30 Catalytic hydrogenation of these aldol products provided amines 16a,b. Coupling of amines 16a,b with isophthalamide derivative 11 in the presence of EDCI, HOBt in CH2Cl2 afforded the corresponding coupling products in good yields (55–60%). Exposure of these coupling products to TFA in CH2Cl2 at 23 °C afforded inhibitors 6a,b. Inhibitors 6c–i were prepared by aldol reaction of aldehyde 7 and 6-substituted-2-oxopiperazines ent-14a,b and 15 followed by the conversion of the resulting aminoalcohols 17a–c to inhibitors 6c–f following a similar reaction sequence as described above. For the synthesis of inhibitor 6g, known17,22 3-(dipropylcarbamoyl)-5-methylbenzoic acid 18 was coupled with amine 17a and the resulting product was treated with TFA in CH2Cl2 to provide 6g in 67% yield for the two steps.
Scheme 3.

Reagents and conditions: (a) LHMDS, dry THF, −78 °C to 23 °C, then add aldehyde 7, 38%; (b) H2, Pd(OH)2, EtOH, AcOH 23 °C; (c) 11 or 18, EDCI, HOBt, DIPEA, CH2Cl2; 23 °C; (d) TFA, CH2Cl2, 23 °C, (31% over 3 steps); (e) BH3•SMe, dry THF, reflux, 78%.
Synthesis of bicyclic piperazinone-derived compound 6h is shown in Scheme 4. Cbz-protected (R)-prolinal 19 was employed as the starting material. Reductive amination of 19 with glycine ethyl ester provided amine 20. N-Boc protection with Boc2O afforded Boc-derivative 21. Catalytic hydrogenation of 21 over Pearlman’s catalyst provided the corresponding amine which was concommitantly cyclized to bicyclic derivative 22. Aldol reaction of 22 with aldehyde 7a provided aldol product 23 which was converted to final compound 6h as described above. Compound 6i was similarly obtained starting from Cbz-protected (S)-prolinal.
Scheme 4.
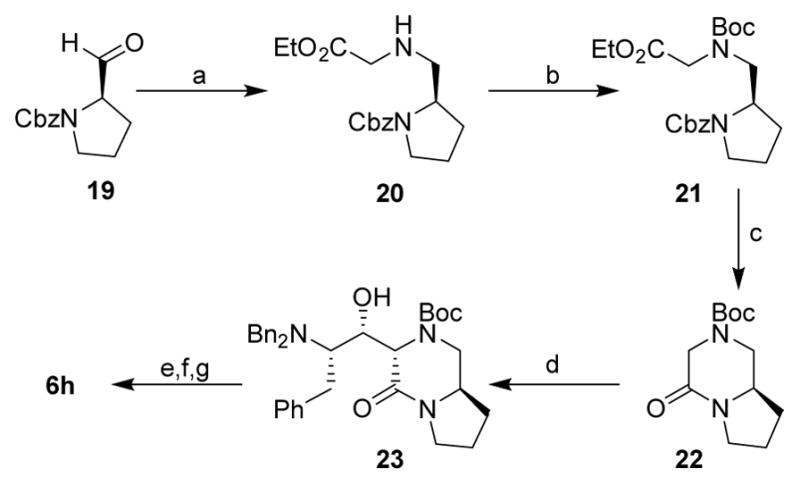
Reagents and conditions: (a) glycine ethyl ester hydrochloride salt, NaBH(OAC)3, AcOH, 1,2-DCE, 25 °C, 80%; (b) Boc2O, TEA, MeOH, 50 °C, 80%; (c) H2, Pd(OH)2, EtOH, AcOH, 25 °C, quant.; (d) 7a, LHMDS, dry THF, −78 °C to 25 °C, 26%; (e) H2, Pd(OH)2, EtOH, AcOH, 25 °C; (f) 11, EDCI, HOBt, DIPEA, CH2Cl2; 25 °C; (g) TFA, CH2Cl2, 25 °C, (30% over 3 steps).
Synthesis of oxazolidinone-derived BACE1 inhibitor 6j is shown in Scheme 5. Aminoalcohol derivative 24 was prepared from aldol reaction of a suitable oxopiperazine and aldehyde 7a as described above. The lactam carbonyl was reduced by treatment with borane-dimethyl sulfide complex in THF at reflux to provide amine derivative 25. Catalytic hydrogenation of 25 over Pearlman’s catalyst provided the corresponding amine which was coupled with isophthalic acid derivative 11 to provide the corresponding amide coupling product. Treatment of this product with TBAF in THF removed the TBS-group and provided aminoalcohol 26. Exposure of 26 to 1,1-carbonyldiimidazole in dioxane at reflux for 36 h afforded the corresponding oxazolidinone product. Treatment of the resulting oxazolidinone with TFA in CH2Cl2 at 23 °C for 1.5 h afforded inhibitor 6j in excellent yield.
Scheme 5.
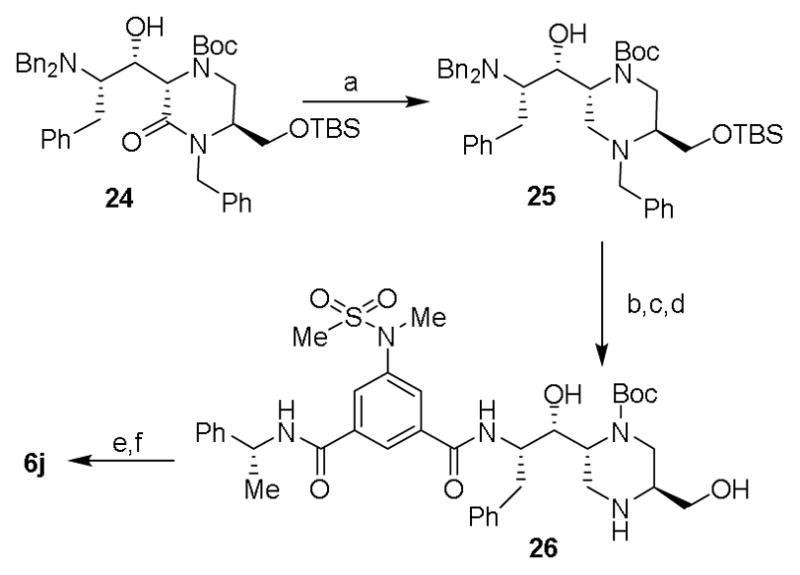
Reagents and conditions: (a) BH3·SMe, dry THF, reflux, 84%; (b) 11, EDCI, HOBt, DIPEA, CH2Cl2; 23 °C, 58%; (c) TBAF, THF, 0 °C to 23 °C, 2 h, 82%; (d) H2, Pd(OH)2, EtOH, AcOH, 23 °C, quant.; (e) CDI, dioxane, reflux, 36 h, 55%; (f) TFA, CH2Cl2, 23 °C, 1.5 h, quant.
The structures and BACE1 inhibitory activity of various inhibitors containing N-alkyl piperazinones are shown in Table 1. The BACE-1 inhibitory activity of 5a–g was determined against recombinant enzyme using our previously reported assay protocols.35–37 As can be seen, the size and electronic effects on the N-alkyl derivatives are important for potency. Compound 5a with N-benzyl substituent is significantly more potent than compound 5b with an N-isobutyl derivative (entries 1 and 2). A small linear chain proproyl derivative 5c is less potent than 5a and 5b. We incorporated an electron rich OMe group at the para, meta, and ortho position of the phenyl ring on inhibitor 5a. The resulting inhibitors, 5d, 5e, and 5f, showed significant reductions in potency. Incorporation of a substituted indole carboxamide as the P2 ligand in place of the isophthalamide provided inhibitor 5g. However, this inhibitor showed a reduction in potency compared to isophthalamide derivative 5a. Inhibitor 5g displayed enhanced BACE2 selectivity with a Ki =7.14 μM for BACE2.
Table 1.
Enzyme inhibitory activity for inhibitors 5a–g
| Entry | Inhibitor | Ki (nM) |
|---|---|---|
| 1. |
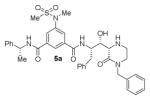
|
2.0 |
| 2. |
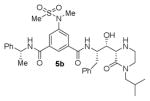
|
12.9 |
| 3. |
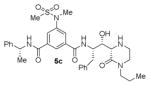
|
20.5 |
| 4. |
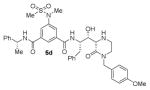
|
81.4 |
| 5. |
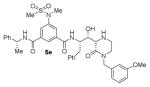
|
129 |
| 6. |
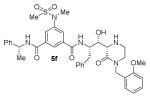
|
144 |
| 7. |
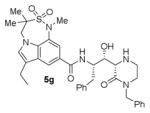
|
67.1 |
To obtain molecular insight into the inhibitor-BACE1 interactions, we determined the X-ray structure of 5g bound to BACE1 to 2.2 Å resolution (RFree = 0.192, Rwork = 0.16).38 As shown in Figure 3, the transition-state hydroxyl group forms a tight hydrogen bond with one of the catalytic aspartates Asp32. The 2-oxopiperazinone moiety fits nicely in the S1′ and S2′ subsites. The C2-carbonyl oxygen appears to form a strong hydrogen bond with the Thr72-backbone amide hydrogen. The P2-indole ligand makes a number of interactions in the active site including the formation of a pair of hydrogen bonds between one of the oxygen atoms of the sulfonamide group and the backbone amide hydrogens of Thr232 and Asn233. The benzyl group fits into a hydrophobic pocket formed by Leu 30, Tyr 71 and Trp 115 shown in Figure 4.
Figure 3.

Stereoview of the X-ray structure of inhibitor 5g (green carbon chain) with BACE1 (PDB code: 5VON). Possible hydrogen bonds between the inhibitor and BACE1 are shown in black dotted lines.
Figure 4.

Stereoview (Wall-eye) of the X-ray structure of BACE1 showing the two hydrophobic pockets that bind the benzyl and N-benzyl groups of inhibitor 5g (PDB code: 5VON).
The N-benzyl group also fits into a hydrophobic pocket formed from residues Ser35, Val69, Pro 70, Tyr 71, Ile126 and Tyr198 (Figure 4) and provides critical data on the molecular interactions of N-benzylpiperazinone in the S2′ site of the active site. It appears that the unsubstituted N-benzyl group is quite optimum for this hydrophobic pocket. Further increases in steric bulk by incorporating methoxy group likely change the conformation of the oxapiperazinone’s carbonyl group and destabilizes the hydrogen bond interaction with the Thr 72 flap residue’s backbone -NH group (Figure 3). This may explain significant loss of potency for the methoxybenzyl derivatives compound to benzyl derivative 5a. Since inhibitor 5a with a P2-isophthalamide derivative and P2′-N-benzylpiperazinone showed the best potency, we decided to further modify the piperazinone scaffold with a combination of a various N-alkyl and 6-alkyl substituted 2-piperazinones. The structures and potency of a set of disubstituted 2-piperazinone-derived inhibitors 6a–j are shown in Table 2.
Table 2.
Enzyme inhibitory activity for inhibitors 6a–j
| Entry | Inhibitor | Ki (nM) |
|---|---|---|
| 1. |
|
52900 |
| 2. |
|
180 |
| 3. |
|
300 |
| 4. |
|
32600 |
| 5. |
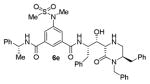
|
22200 |
| 6. |
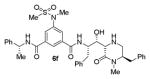
|
12 |
| 7. |
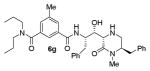
|
320 |
| 8. |
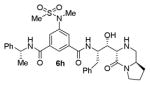
|
26 |
| 9. |
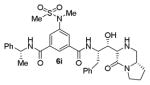
|
5690 |
| 10. |
|
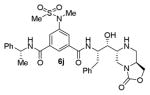
23 |
As can be seen, the set of 6-substituted-piperazinone- and piperazine-containing compounds show well-defined structure-activity relationships (SARs). First of all, the stereochemistry at the 6-position was found to be critical to inhibitory activity, as demonstrated by the dramatic difference in potency between compounds 6a and 6b (Ki = 52.9 μM vs Ki = 180 nM respectively). The removal of the lactam carbonyl oxygen in 6c resulted in a reduction of potency. Beyond the preference for the (R)-configuration at 6-position, the preference for a small alkyl group is also highlighted (6b vs 6e, Ki = 175.5 nM vs Ki = 222 μM, respectively). Inhibitor 6f with a combination of N-Me and 6-(R)-benzyl piperazinone displayed best inhibitory activity. Incorporation of N,N-dipropyl isophthalamide derivative in inhibitor 6g resulted in a significant loss of potency (Ki = 320 nM vs Ki = 12 nM) compared to N-methyl sulfonamide-derivatives as the P2-ligand. We then designed a set of bicyclic P1′ and P2′ ligands in inhibitors 6h–6j. Inhibitor 6h with C-6(R) stereochemistry is significantly more potent than inhibitor 6i with 6-(S)-stereochemistry. The oxazolidinone derivative 6j displayed comparable potency to inhibitor 6h.
We measured the potency of a subset of inhibitors in different assays. Regarding the first series of inhibitors 5a–g, we sought to determine the cell activity of the representative compound 5a, bearing the P2-isophthalamide derivative and the P2′ N-benzylpiperazinone. Inhibitor 5a displayed an EC50 value of 3.5 nM in a cell-based assay. Also, we assessed the cellular outcome for compound 5g, in order to appraise the contribution of the substituted indole carboxamide as the P2 ligand. Compound 5g showed an EC50 value of 18.5 nM. For the prototype of the series 5a we also evaluated the selectivity over β-site APP-cleaving enzyme 2 (BACE2), for its close similarity to BACE1, and cathepsin D (CatD), for its ubiquitous presence in nearly all the cells. Compound 5a showed marginal selectivity against BACE2 (Ki = 134 nM) and good selectivity against CatD (Ki = 1.5 μM). We also evaluated the BACE2 inhibitory activity of compound 5g, which showed improved selectivity (BACE2 Ki = 7.14 μM). Finally, we sought to compare the cell-based activity of two bicyclic inhibitors, namely the bicyclic piperazinone inhibitor 6g and the oxazolidinone derivative 6i, displaying comparable enzyme inhibitory potency. Inhibitor 6g showed an EC50 of 184 nM, while inhibitor 6j is slightly more potent in the same settings (EC50 = 80 nM).
In summary, we have designed and synthesized a series of BACE1 inhibitors containing substituted 2-oxopiperazines as the P1′ and P2′ ligands and substituted isophthalamide as the P2 ligand. We have developed methodologies for the synthesis of 6-alkyl and N-alkyl piperazinones, as well as bicyclic piperazinones. Among the N-substituted piperazinones examined, N-benzyl derivative 5a showed the best result. The choice of substitution on the isophthalamide is critical to potency. The combination of N-alkyl and 6-alkyl is important and the stereochemistry at the 6-alkyl position is also critical. Inhibitors 6f with N-methyl and 6-benzyl derivatives provided best BACE1 activity. Also, bicyclic piperazinone 6g and oxazolidinone derivative 6i showed good BACE1 inhibitory activity as well as inhibition of cellular production of Aβ in neuroblastoma cells. We determined the X-ray structure of inhibitor 5g-bound BACE1 to 2.2Å resolution. The structure provided important molecular insight into the ligand-binding site interactions in the S1′ and S2′ subsites of β-secretase. Further design and improvement of inhibitor properties are in progress.
Supplementary Material
Acknowledgments
Financial support by the National Institutes of Health and the Purdue University Office of the Executive Vice President for Research are gratefully acknowledged. We would also like to thank Dr. Kalapala Venkateswara Rao for helpful discussions. BACE1 crystallization, NMR and Mass Spectrometry were all performed using shared resources that are partially supported by the Purdue Center for Cancer Research through NIH grant (P30CA023168). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility.
Footnotes
Supplementary data associated with this article can be found in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. 2016 Available at: www.alz.org/alzheimers_disease_facts_figures.asp.
- 2.Rutten BP, Steinbusch HW. Mol Neurodegener. 2013;8:33. doi: 10.1186/1750-1326-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lansdall CJ. Bioscience Horizons. 2014;7:1–11. [Google Scholar]
- 4.Prince M, Bryce R, Albanese E. Alzheimer’s and Dementia. 2013;9:63–75. doi: 10.1016/j.jalz.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh AK, Osswald HL. Chem Soc Rev. 2014;43:6765–6813. doi: 10.1039/c3cs60460h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vassar R, Kovacs DM, Yan R, Wong PC. J Neurosci. 2009;29:12787–12794. doi: 10.1523/JNEUROSCI.3657-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vassar R. Alzheimer’s Res Ther. 2014;6:89. doi: 10.1186/s13195-014-0089-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanzi RE. Cold Spring Harb Perspect Med. 2012:2. doi: 10.1101/cshperspect.a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selkoe DJ, Schenk D. Annu Rev Pharacol. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- 10.Hardy J, Selkoe DJ. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 11.Evin G, Barakat A, Masters CL. Internat J Biochem & Cell Biology. 2010;42:1923–1926. doi: 10.1016/j.biocel.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 12.Citron M. Trends in Pharmacological Sciences. 2004;25:92–97. doi: 10.1016/j.tips.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh AK, Brindisi M, Tang J. J Neurochem. 2012;120(Suppl 1):71–83. doi: 10.1111/j.1471-4159.2011.07476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R. Nature Neuroscience. 2001;4:231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- 15.Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. Nature Neuroscience. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- 16.Cumming JN, Le TX, Babu S, Carroll C, Chen X, Favreau L, Gaspari P, Guo T, Hobbs DW, Huang Y, Iserloh U, Kennedy ME, Kuvelkar R, Li G, Lowrie J, McHugh NA, Ozgur L, Pan J, Parker EM, Saionz K, Stamford AW, Strickland C, Tadesse D, Voigt J, Wang L, Wu Y, Zhang L, Zhang Q. Bioorg Med Chem Lett. 2008;18:3236–3241. doi: 10.1016/j.bmcl.2008.04.050. [DOI] [PubMed] [Google Scholar]
- 17.Cumming JN, Babu S, Huang Y, Carroll C, Chen X, Favreau L, Greenlee W, Guo T, Kennedy ME, Kuvelkar R, Le T, Li G, McHugh NA, Orth P, Ozgur L, Parker EM, Saionz K, Stamford AW, Strickland C, Tadesse D, Voigt J, Zhang L, Zhang Q. Bioorg Med Chem Lett. 2010;20:2837–2842. doi: 10.1016/j.bmcl.2010.03.050. [DOI] [PubMed] [Google Scholar]
- 18.Stachel SJ, Coburn CA, Steele TG, Jones KG, Loutzenhiser EF, Gregro AR, Rajapakse HA, Lai MT, Crouthamel MC, Xu M, Tugusheva K, Lineberger JE, Pietrak BL, Espeseth AS, Shi XP, Chen-Dodson E, Holloway MK, Munshi S, Simon AJ, Kuo L, Vacca JP. J Med Chem. 2004;47:6447–6450. doi: 10.1021/jm049379g. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh AK, Kumaragurubaran N, Hong L, Kulkarni S, Weerasena V, Turner R, Koelsch G, Bilcer G, Tang J. J Med Chem. 2007;50:2399–2407. doi: 10.1021/jm061338s. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh AK, Kumaragurubaran N, Hong L, Kulkarni S, Xu X, Miller HB, Reddy DS, Weerasena V, Turner R, Chang W, Koelsch G, Tang J. Bioorg Med Chem Lett. 2008;18:1031–1036. doi: 10.1016/j.bmcl.2007.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosh AK, Kumaragurubaran N, Hong L, Lei H, Hussain KA, Liu CF, Devasamudram T, Weerasena V, Turner R, Koelsch G, Bilcer G, Tang J. J Am Chem Soc. 2006;128:5310–5311. doi: 10.1021/ja058636j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iserloh U, Wu Y, Cumming JN, Pan J, Wang LY, Stamford AW, Kennedy ME, Kuvelkar R, Chen X, Parker EM, Strickland C, Voigt J. Bioorg Med Chem Lett. 2008;18:414–417. doi: 10.1016/j.bmcl.2007.10.116. [DOI] [PubMed] [Google Scholar]
- 23.Maillard MC, Hom RK, Benson TE, Moon JB, Mamo S, Bienkowski M, Tomasselli AG, Woods DD, Prince DB, Paddock DJ, Emmons TL, Tucker JA, Dappen MS, Brogley L, Thorsett ED, Jewett N, Sinha S, John V. J Med Chem. 2007;50:776–781. doi: 10.1021/jm061242y. [DOI] [PubMed] [Google Scholar]
- 24.Iserloh U, Cummins JN. In: Peptidomimetic BACE1 Inhibitors for Treatment of Alzheimer’s Disease: design and evolution in Aspartic Acid proteases as Therapeutic Targets. Ghosh AK, editor. Wiley-VCH; 2010. pp. 441–479. [Google Scholar]
- 25.Virgil S. First Generation HIV-1 Protease Inhibitors for the treatment of HIV/AIDS. In: Ghosh AK, editor. Aspartic Acid proteases as Therapeutic Targets. Wiley-VCH; 2010. pp. 139–168. [Google Scholar]
- 26.Ghosh AK, Chapsal B. Second-Generation Approved HIV Protease Inhibitors for the Treatment of HIV/AIDS. In: Ghosh AK, editor. Aspartic Acid proteases as Therapeutic Targets. Wiley-VCH; 2010. pp. 169–204. [Google Scholar]
- 27.Ghosh AK, Osswald HL, Prato G. J Med Chem. 2016;59:5172–5208. doi: 10.1021/acs.jmedchem.5b01697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernotas RC, Adams G. Tetrahedron Lett. 1996;37:7339–7342. [Google Scholar]
- 29.Reetz MT, Drewes MW, Schmitz A. Angew Chem Int Ed. 1987;26:1141–1143. [Google Scholar]
- 30.Reetz MT. Chem Rev. 1999;99:1121–1162. doi: 10.1021/cr980417b. [DOI] [PubMed] [Google Scholar]
- 31.Ghosh AK, Rao KV, Yadav ND, Anderson DD, Gavande N, Huang X, Terzyan S, Tang J. J Med Chem. 2012;55:9195–9207. doi: 10.1021/jm3008823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boger DL, Goldberg J, Satoh S, Ambroise Y, Cohen SB, Vogt PK. Helv Chim Acta. 2000;83:1825–1845. [Google Scholar]
- 33.Whitby LR, Lee AM, Kunz S, Oldstone MBA, Boger DL. 2009;19:3771–3774. doi: 10.1016/j.bmcl.2009.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abdel-Magid AF, Mehrman SJ. Org Process Res. 2006;10:971–1031. [Google Scholar]
- 35.BACE1 inhibition was measured using recombinant enzyme produced from E. coli expression as described in reference 36. A fluorogenic substrate Arg-Glu(EDANS)-Glu-Val-Asn-Leu-Asp-Ala-Glu-Phe-Lys (Dabcyl)-Arg was used with 0.47 μM of the enzyme in 0.1 M Na-acetate + 5% dimethylsulfoxide, pH 4.5 at 37 °C. The excitation wavelength was 350 nm and the emission wavelength was 490 nm.
- 36.Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J. Proc Natl Acad Sci, USA. 2000;97:1456–1460. doi: 10.1073/pnas.97.4.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ermolieff J, Loy JA, Koelsch G, Tang J. Biochem. 2000;39:12450–12456. doi: 10.1021/bi001494f. [DOI] [PubMed] [Google Scholar]
- 38.PDB code: 5VON. For details of X-ray studies, please see Supplementary information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.