

Abstract
In this study, silk fibroin and hyaluronic acid (HA) were enzymatically crosslinked to form biocompatible composite hydrogels with tunable mechanical properties similar to that of native tissues. The formation of di-tyrosine crosslinks between silk fibroin proteins via horseradish peroxidase has resulted in a highly elastic hydrogel but exhibits time-dependent stiffening related to silk self-assembly and crystallization. Utilizing the same method of crosslinking, tyramine-substituted HA forms hydrophilic and bioactive hydrogels that tend to have limited mechanics and degrade rapidly. To address the limitations of these singular component scaffolds, HA was covalently crosslinked with silk, forming a composite hydrogel that exhibited both mechanical integrity and hydrophilicity. The composite hydrogels were assessed using unconfined compression and infrared spectroscopy to reveal of the physical properties over time in relation to polymer concentration. In addition, the hydrogels were characterized by enzymatic degradation and for cytotoxicity. Results showed that increasing HA concentration, decreased gelation time, increased degradation rate, and reduced changes that were observed over time in mechanics, water retention, and crystallization. These hydrogel composites provide a biologically relevant system with controllable temporal stiffening and elasticity, thus offering enhanced tunable scaffolds for short or long term applications in tissue engineering.
Keywords: Polymer composites, hydrogel blends, enzymatic crosslinking, biomaterials, temporal stiffening
II. Introduction
Hydrogels are crosslinked hydrophilic polymer networks with high water content that are often utilized as scaffolds for a variety of biomedical applications including regenerative medicine, drug delivery, and tissue engineering [1, 2]. Due to their hydrophilic nature, hydrogels are permeable to oxygen and nutrients and possess mechanics similar to that of native extracellular matrix, creating a receptive environment for cell proliferation [3]. Silk fibroin, a fibrous protein derived from Bombyx mori cocoons, can form natural hydrogels with good biocompatibility, mechanical strength, and relatively slow degradation over a few months [4–7]. Silk-based hydrogels can be prepared through many methods including enzymatic crosslinking, sonicating, altering pH, vortexing, applying electric fields, and adding polyols or surfactants [5, 8–11]. Covalently crosslinking the phenolic groups of tyrosine residues, ~5% of amino acids in silk fibroin using horseradish peroxidase (HRP) and hydrogen peroxide (H2O2) results in a cytocompatible, mechanically tunable, elastomeric protein hydrogel [5]. Due to the resilience, lack of harsh crosslinking conditions, and mechanical properties similar to native tissues, enzymatically crosslinked silk hydrogels provided a useful system for tissue engineering applications [5]. However, these silk hydrogels lack cell specific epitopes and depending on the degree of crosslinking and environmental conditions, can undergo significant changes in mechanics over time due to gradual crystallization [5, 12, 13]. Modulating silk hydrogels through crosslinking of other polymers, peptides, and proteins can provide a route to address these limitations. For instance, the crosslinking of silk and cardiac extracellular matrix showed enhanced cell growth in vitro and cellular infiltration in vivo, but it did not significantly impact hydrogel stiffening over time compared to control silk hydrogels [13]. Therefore, there is a need to further understand and control temporal silk hydrogel stiffening, which can be accomplished through the incorporation of additional polymers, such as hyaluronic acid.
Hyaluronic acid (HA) is a naturally occurring, non-sulfated glucosaminoglycan, that consists of repeating disaccharide units of D-glucuronic acid and N-acetyl-D-glucosamine, linked by ß-1-3 and ß-1-4 glycosidic bonds [14]. HA is ubiquitous throughout the body, but is highly localized in the extracellular matrix of connective tissue, synovial joint fluid, vitreous humor, and brain tissue [14, 15]. HA is hydrophilic and has a net negative charge, providing tissues with hydration and structural support [16]. In addition, HA plays an important role in many biological functions such as embryonic development, inflammation, angiogenesis, cell-matrix interactions, and wound healing [15]. Due to its biological and structural importance, as well as its ease of modification, HA is an attractive polymer for biomedical applications [17]. Since the turnover of HA is rapid, about one-third of the total HA body content is degraded and reformed daily, covalent crosslinking is necessary to increase mechanical stability for tissue engineering purposes [14, 18, 19]. This can be accomplished either directly or by first modifying the hydroxyl or carboxyl groups of HA with functional moieties, which can then be crosslinked [15]. Tyramine-substituted HA has been previously synthesized to provide a biocompatible hydrogel that can be enzymatically crosslinked with HRP and H2O2, under physiological conditions [20–23]. But these hydrogels still have limited mechanical properties with moduli ranging under 10 kPa [24] and degrade by more than half of their initial weight by 1 month in vivo [25]. Thus, HA hydrogels can benefit from a composite system that can offer enhanced mechanical properties and resistance to degradation.
A composite scaffold consisting of silk and HA will retain the favorable properties of each polymer, while eliminating the limitations as seen in the singular component scaffolds. Silk fibroin can provide mechanical integrity and prolonged degradation for long-term applications, whereas HA can enhance hydrogel water retention and based on previous literature, can provide biological cues [15, 17]. Silk and HA have previously been combined in the form of hydrogels, films, microparticles, patches, and sponges to provide biologically active scaffolds for cartilage, neuronal, and cardiac tissue engineering [26–31]. One method of combining these two polymers into a bulk hydrogel was entrapping hyaluronic acid in a crystalline silk network through the application of ultrasonic frequencies [32]. This physically crosslinked hydrogel facilitated the attachment and growth of human mesenchymal stem cells (hMSCs) while providing mechanical integrity [32]. However, the reliance on physically crosslinked, β-sheet networks, which increased upon the incorporation of HA, resulted in brittle behavior, limiting applications and presenting challenges in material handling [32–34]. Covalently crosslinking silk and hyaluronic acid using 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) chemistry with hexamethylene diamine (HDMA) has been proposed to form hydrogels for skin and soft tissue treatments but the effect of HA on hydrogel properties has not be documented [35]. Although this method can be carried out under physiological conditions, the hydrogels are often washed prior to injection to extract the not reacted crosslinking agents [36] which are cytotoxic [37, 38] and can lead to loss of cell and tissue functions [21]. On the other hand, the enzymatic crosslinking method, using HRP and H2O2, has shown minimal cytotoxicity with the in situ gelation of HA hydrogels [24, 25]. Therefore, enzymatically crosslinking silk and HA can provide an alternative, cytocompatible method of forming composite hydrogels, while providing a format to understand how polymer concentration affects hydrogel properties.
In the present study we sought to combine the virtues of silk and HA into a composite hydrogel system, through enzymatic crosslinking, to provide a biologically friendly process and to overcome limitations that are associated with either polymer alone in hydrogel form. The effects of incorporating a hydrophilic bioactive polymer were explored through altering HA concentration and characterizing gelation, mechanical, and viscoelastic properties over time as well as determining in vitro degradation and cytotoxicity. These composite hydrogels offer a biocompatible, stable, tunable system that can be useful for biomaterials and tissue engineering applications.
III. Materials and Methods
Preparation of Silk-HA Hydrogels
Aqueous silk solutions were prepared using previously established methods [39]. Briefly, sericin protein was removed from B. mori silkworm cocoons by placing 5 g of cut cocoons in 2 L of a boiling 0.02 M sodium carbonate solution (Sigma-Aldrich, St. Louis, MO) for 60 minutes. After rinsing in deionized (DI) water three times, the degummed fibers were dried overnight and solubilized in 9.3 M lithium bromide (Sigma-Aldrich, St. Louis, MO) for 4 hours at 60°C. The resulting silk solution was then dialyzed against DI water using standard grade regenerated cellulose dialysis tubing (3.5 kD MWCO, Spectrum Labs Inc, Rancho Dominguez, CA). After 6 changes over 3 days, insoluble silk particulates were removed by centrifugation (two times at 9000 RPM, 5°C, 20 minutes). Silk concentration was determined by weighing a dried sample of a known volume.
Lyophilized tyramine-substituted HA (Corgel® powder 2.8%, MW 0.9–1 MDa, Lifecore Biomedical, Chaska, MN) was dissolved in Ultrapure™ distilled water (ThermoFisher Scientific, Waltham, MA) overnight at 4°C. Aqueous silk and HA were combined through gentle pipetting to yield a final silk concentration of 20 mg/mL and final HA concentrations of 0, 0.2, 1.05, 2.22, and 5 mg/mL. Samples are referred to as the weight percentage of HA (Table 1). HA only hydrogels consisted of 5 mg/mL of HA. Crosslinking was initiated by adding of 10 U/mL of horseradish peroxidase (HRP, type VI, Sigma-Aldrich, St. Louis, MO) followed by 0.01% H2O2 (final molarity of 3.27 mM, Sigma-Aldrich, St. Louis, MO), similar to previous reports [5, 13, 40]. Composite hydrogels with HA concentrations above 5 mg/mL were excluded from experiments due to an increase in viscosity and phase separation resulting in difficulties with handing and reproducibility. After mixing, the solutions were allowed to gel for 3–4 hours at 37°C prior to analysis, unless otherwise noted.
Table 1. Sample Conditions.
Samples are denoted by the mass percent of tyramine-substituted HA as compared to the total polymer concentration. Each condition was crosslinked with 10 U/mL of HRP and 0.01% H2O2 (final molarity of 3.27 mM).
| Sample | Silk Concentration (mg/mL) | HA Concentration (mg/mL) |
|---|---|---|
| 0% | 20 | 0.0 |
| 1% | 20 | 0.2 |
| 5% | 20 | 1.05 |
| 10% | 20 | 2.22 |
| 20% | 20 | 5.0 |
| HA only | 0 | 5.0 |
Gelation Kinetics
Gelation time was evaluated by a vial inversion test, where gelation time was designated as the time at which the solution no longer flowed after tilting the vials. Briefly, H2O2 was added into a silk-HA solution containing HRP in a 7 mL scintillation vial and mixed with a pipette for 10 seconds, yielding a total of 750 μL of hydrogel solution. Samples were kept at 37°C where gelation was monitored (n=4). Crosslinking kinetics were assessed through fluorescence spectroscopy using a SpectraMax M2 multi-mode microplate reader (Molecular Devices, Sunnyvale, CA). A 200 L aliquot of silk-HA solution was mixed in a black 96-well plate for 10 seconds with a pipette. Crosslinking was monitored at 37°C by measuring the intensity of intrinsic fluorescence emission at 415 nm after excitation at 315 nm of the crosslinked phenolic groups until plateau. Results are reported as a fraction of the maximum intensity after subtracting a blank measurement taken prior to adding H2O2 (n=7).
Rheology
Rheological measurements were performed at 37°C on a TA Instruments ARES-LS2 rheometer (TA Instruments, New Castle, DE) using a 25 mm stainless steel upper cone and peltier bottom plate. Samples were mixed as previously mentioned, without the addition of H2O2. 415.8 μL of silk-HA solution with HRP was loaded onto the plate and the cone was lowered to 46.8 m. During a 10 second precycle at a steady shear rate of 100/sec, 4.2 μL of 1% H2O2 was added into the gap. To prevent evaporation during analysis, mineral oil was placed around the system. A dynamic time sweep was performed at 1 Hz with a 1% applied strain until samples attained a steady modulus. Parameters were chosen in the linear viscoelastic region as determined in preliminary experiments (Supplement Figure 1). Following gelation, dynamic frequency sweeps (0.1 to 100 rad/s at 1% strain) and strain sweeps (0.1% to 500% or to failure, whichever came first, at 1 Hz) were conducted (n=3).
Unconfined Compression
Unconfined compression was performed on a TA Instruments RSA3 Dynamic Mechanical Analyzer (TA Instruments, New Castle, DE) between stainless steel parallel plates. Preformed hydrogels (4 mm diameter, ~2-3 mm height) were placed under a preload of ~0.5 g to ensure full contact. Two load-unload cycles to 30% strain at a rate of 0.667% per second were performed to eliminate artifacts. Stress response and elastic recovery were monitored during the third load-unload cycle at the same strain rate. Hydrogels were tested after incubation in 1× PBS at 37°C for 1 day and 1, 2, 3, and 4 weeks. Tangent modulus was calculated for each sample between 5 and 10% strain (n=5).
Swelling
Percentages of water in the hydrogels were calculated using Equation 1. Briefly, wet mass was determined by blotting preformed hydrogels with a WypAll paper towel (Kimberly-Clark Co., Neenah, WI) to remove residual surface water and weighing. After drying in a 60°C oven overnight, samples were weighed again to obtain their dry mass. The water content was determined initially and after incubation in 1× PBS at 37°C for 1 day, and 1, 2, 3, and 4 weeks (n=5).
Fourier Transform Infrared Spectroscopy (FTIR)
Secondary structure was analyzed by a JASCO FTIR 6200 spectrometer (JASCO, Tokyo, Japan) with a MIRacle™ attenuated total reflection with germanium crystal. Preformed hydrogels (4 mm diameter, ~2-3 mm height) were washed in deuterated water (Sigma-Aldrich, St. Louis, MO) three times for 30 minutes each prior to analysis to remove the interference of water in the amide I region. Data was obtained by averaging 32 scans with a resolution of 4 cm−1 within the wavenumber range of 600 and 4000 cm−1. Samples were allowed to incubate in 1× PBS at 37°C for 1 day prior to initial analysis and then at weeks 1, 2, 3, and 4 (n=5). Data was quantified by dividing the average peak absorbance at 1620–1625 and 1640–1650 cm−1 representing the ratio of ß-sheet to random coil secondary structures [41].
In Vitro Degradation
Preformed hydrogels (~ 22 mm diameter, 2 mm height) were first incubated at 37°C in 1× PBS for 3–4 hours. The gels were then placed in 2 mL of an enzyme cocktail consisting of lyophilized hyaluronidase (type I-S from bovine testes, Sigma-Aldrich, St. Louis, MO), and protease (type XIV from Streptomyces griseus, Sigma-Aldrich, St. Louis, MO) dissolved at 1 U/mL and 0.001 U/mL in 1× PBS, respectively. The enzyme solution was changed every 2 days. At days 0, 1, 2, 4, 6, and 8 a subset of gels were removed from the enzyme solution and washed in Ultrapure™ water over night at room temperature, lyophilized and weighed. Results are shown as the mass fraction of the initial weight (n=4).
In Vitro Cell Response
Human mesenchymal stem cells (hMSCs) were isolated from human bone marrow aspirates (Lonza, Gaithersburg, MD), as previously reported [42]. Cells were cultured in growth media consisting of Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, 1% Penicillin-Streptomycin (Life Technologies, Carlsbad, CA), and 1 ng/mL of basic fibroblast growth factor (bFGF) (Invitrogen, Carlsbad, CA). At passage 3, cells were plated onto preformed hydrogels (15 mm diameter, 1.5 mm height) containing silk only, silk-HA (10% HA), and HA only as well as tissue culture plastic (TCP) controls at a density of 10,000 cells/cm2. Media was changed 3–4 hours after initial plating and then every 2–3 days. Brightfield images were collected initially after seeding and at day 7 with a BZ-X700 Fluorescence Microscope (Keyence Corp., Itasca, IL). Cell proliferation was quantified using Quant-iT PicoGreen dsDNA Assay Kit as per manufacturer’s instructions (Invitrogen, Carlsbad, CA). In brief, cells were first lysed with 500 μL of 1× TE buffer solution (ThermoFisher Scientific, Waltham, MA). DNA content was determined in duplicates by measuring fluorescence intensity (ex/em: 480/520 nm) using a microplate reader after samples were diluted 5× with TE buffer and combined with PicoGreen reagent. Cell proliferation is shown as the fold change in DNA concentration at day 3, 5, and 7 compared to the initial DNA concentration (n=6).
Statistics
Data are expressed as means ± standard deviations. One- or Two-way ANOVA (analysis of variance) with Tukey’s post hoc multiple comparison tests were performed using GraphPad Prism (GraphPad Software, La Jolla, CA) to determine statistical significance (*p≤0.05, **p≤0.01, ***p≤0.001).
IV. Results
Hydrogel Gelation
Polymerization of the precursor hydrogel solutions with HRP and H2O2 generated phenolic radicals that could covalently bond with one another (Supplement Figure 2). In the silk-HA system, this reaction covalently linked the two polymers (Figure 1a), forming a hybrid, composite hydrogel with polymer crosslinking efficiency above 94% (Supplement Figure 3). The types of crosslinks that are possible within the composite system include dityrosine (silk-silk bonds), dityramine (HA-HA bonds), and tyramine-tyrosine (silk-HA bonds) (Supplement Figure 4a). All three crosslinks were identified in all composite hydrogels except that of 1% HA, indicating that the hydrogels contained both inter and intra-polymer bonds (Supplement Figure 4b). In the 1% HA samples, where the concentration of tyramine was relatively low initially, dityramine and tyramine-tyrosine peaks had similar areas to that of negative controls, and therefore they could not be reliably analyzed.
Figure 1. Hydrogel Gelation.
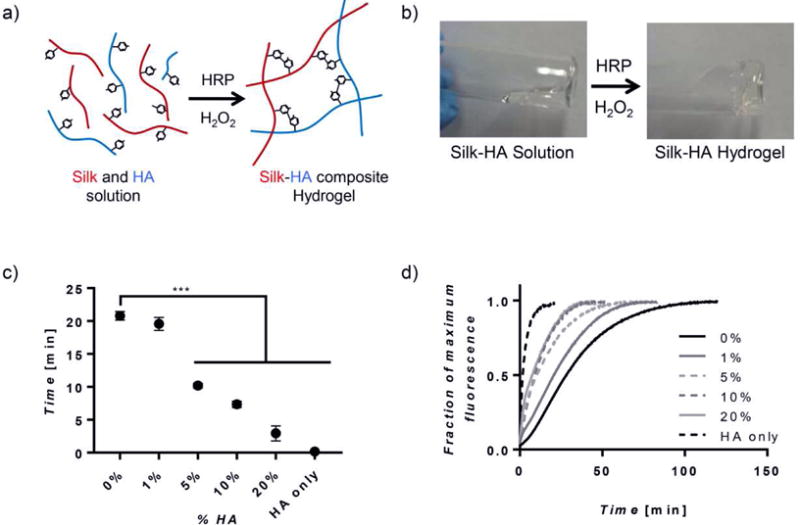
(a) A schematic representing the single step covalent crosslinking between tyrosine residues on silk and tyramine side chains on HA creating a composite hydrogel. (b) Images showing gelation of silk-HA hydrogels during a vial inversion test. (c) Gelation times, as determined by the vial inversion test, show hydrogels consisting of more than 1% HA decreased gelation time (n=4, ***p≤0.001). (d) In addition, the increasing of HA concentration affected crosslinking kinetics by reducing the lag period seen most prominently in 0% hydrogels and decreasing time at which crosslinking was complete (n=7).
The sol-gel transition of silk-HA hybrid hydrogels ranged from a few to approximately 20 minutes, as determined by a vial inversion test. When increasing the HA concentration greater than 1% gelation time decreased significantly, compared to silk only hydrogels (Figure 1c). As expected, HA only hydrogels gelled within seconds after crosslinking was induced [22]. To further assess gelation properties, crosslinking kinetics were explored by quantifying the rate of formation of phenolic crosslinking, which intrinsically emit fluorescence at 415 nm after excitation at 315 nm (Supplement Figure 5a) [5]. Both the shape of the curve and the time to plateau were dependent on HA content (Figure 1d). Silk only hydrogels experienced a delay in crosslinking, showing a sigmoidal shaped curve, whereas composite and HA only hydrogels showed hyperbolic shaped curves with no delay. HA only and silk only hydrogels completed crosslinking in approximately 15 and 110 minutes, respectively. Composite hydrogels completed crosslinking between these two extremes in which increasing HA concentration, lead to decreased crosslinking times (Supplement Figure 5b).
Rheology
Shear mechanical properties and the dependence on polymer concentration were determined through rheology. For pure silk and silk-HA hydrogels, mechanical stability occurred between 132 and 174 minutes, with no significant dependence on HA concentration (Figure 2a). However, HA only hydrogels reached their maximum storage modulus at ~15 minutes, much faster than that of silk only and silk-HA hybrids, revealing that silk component in the hybrid hydrogels is the limiting factor in mechanical stability during gelation. The silk and silk-HA hydrogels withstood approximately 100% strain before failing, whereas the pure HA hydrogels starting fracturing at ~30% strain (Figure 2b). Pure silk and pure HA hydrogels had storage moduli of 2.27 ± 0.09 and 0.55 ± 0.03 kPa, respectively. All composite hydrogels exhibited higher final storage moduli than that of the pure hydrogels, but those containing 20% HA showed the highest at 3.85 ± 0.08 kPa (Figure 2c).
Figure 2. Rheological Properties.
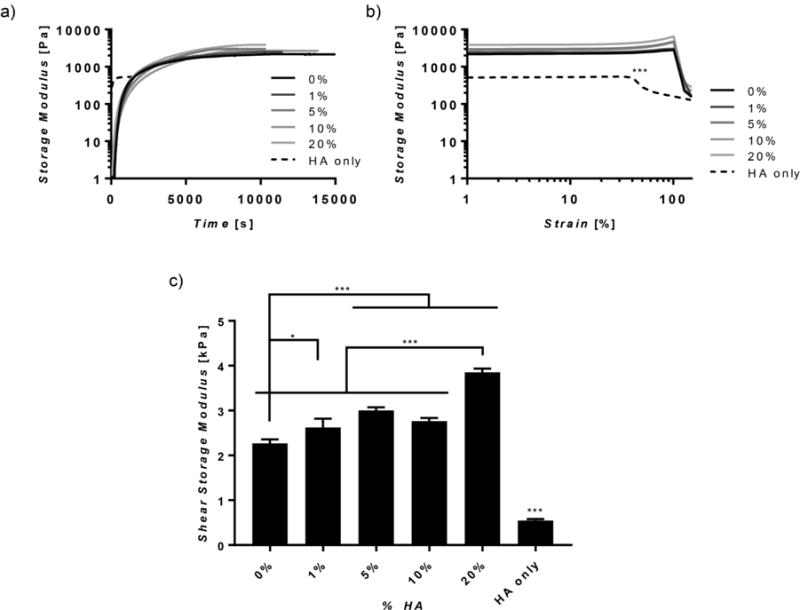
(a) Representative time sweeps, (b) strain sweeps, and (c) final shear storage moduli are shown. HA only hydrogels reached a final modulus much quicker (a), had a lower maximum strain (b), and had a much lower final storage modulus (c) as compared to composite and silk only hydrogels. (n=3, *p≤0.05, ***p≤0.001).
Unconfined Compression
Mechanical analysis was executed to determine the unconfined compressive properties of the hydrogels over a period of 1 month (Figure 3). Preformed hydrogels were soaked in PBS at 37°C and a subset of the samples were analyzed for compressive properties at weeks 0, 1, 2, 3, and 4. Initially, all hydrogels had moduli ranging between ~3 and 8 kPa and fully recovered as seen in the hysteresis curves (Figure 3). The times at which the elastic moduli of the hydrogels increased were significantly delayed with the addition of higher HA content. For instance, hydrogels containing 0 and 1% HA experienced a small increase in modulus at week 2 with a much larger increase at week 3. On the other hand, hydrogels with 5, 10, and 20% HA did not begin to stiffen until week 3 and HA only hydrogels remained consistent at ~3 kPa (Figure 3a). At week 4, all hybrid hydrogels except that of 1% HA were less stiff than silk only hydrogels. Particularly, increasing the amount of HA reduced the magnitude of stiffening. For instance, at week 4, hydrogels with 0, 1, 5, 10, and 20% HA stiffened approximately 240, 200, 100, 40, and 5 times their initial modulus (Supplement Table 1). A higher degree of resilience was also retained with higher HA concentrations, as seen through less hysteresis during a single load-unload cycle at week 4 (Figure 3b). Additionally, hydrogels containing 10% HA or less did not fully recover (Figure 3b).
Figure 3. Unconfined Compression.
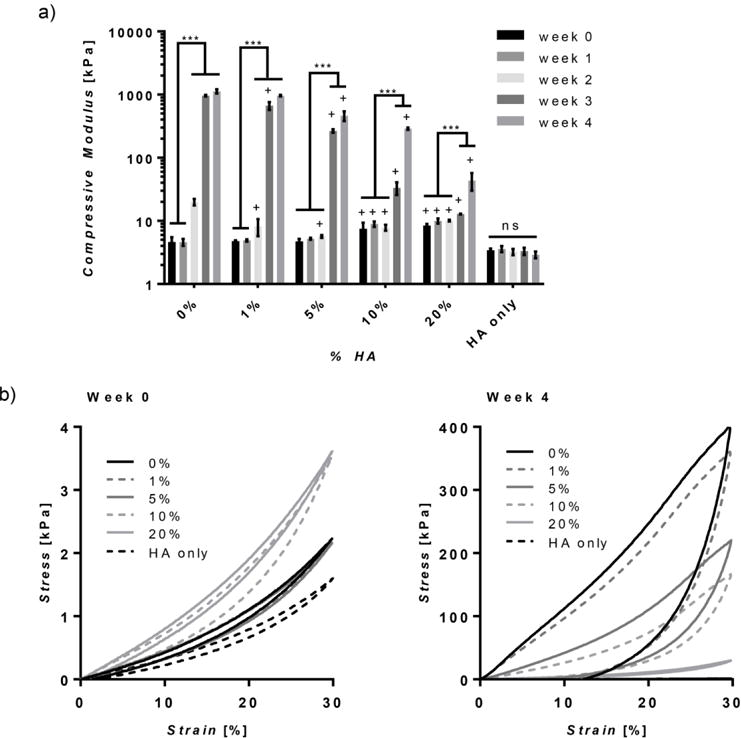
(a) The compressive moduli of the hydrogels over time, expressed on a log scale, are dependent on HA concentration where increasing concentration reduces the amount the modulus increases after 1 month. (b) Average stress-strain curves show that stress and hysteresis increase for all samples after 4 weeks, except for HA only hydrogels, with the extent of hysteresis reduced by increasing HA concentration. (n=5, +p≤0.05 compared to 0%, *p≤0.05, ***p≤0.001. Statistical analysis was performed after log transformation).
Fourier Transform Infrared Spectroscopy (FTIR)
Secondary structure was assessed through FTIR to determine the conformational changes over time. Over 1 month, there was a peak shift in the FTIR spectra from ~1640 cm-1 to ~1620 cm−1 in silk only and low HA concentration hydrogels, suggesting a change in silk secondary structure from random coil to crystalline β-sheet (Figure 4a) [41]. This shift was quantified by calculating the ratio of the average peak absorbance at 1620–1625 and 1640–1650 cm−1 (Figure 4b). At week 3, the ratio of 0% and 1% HA hydrogels increased to over 1, signifying there was a larger contribution from ß-sheet than random coil secondary structures. The 5% hydrogels followed this trend at week 4. Additionally, at week 4, the 5%, 10% and 20% hydrogels had significantly lower ratios as compared to silk only hydrogels.
Figure 4. FTIR Absorbance Spectra.
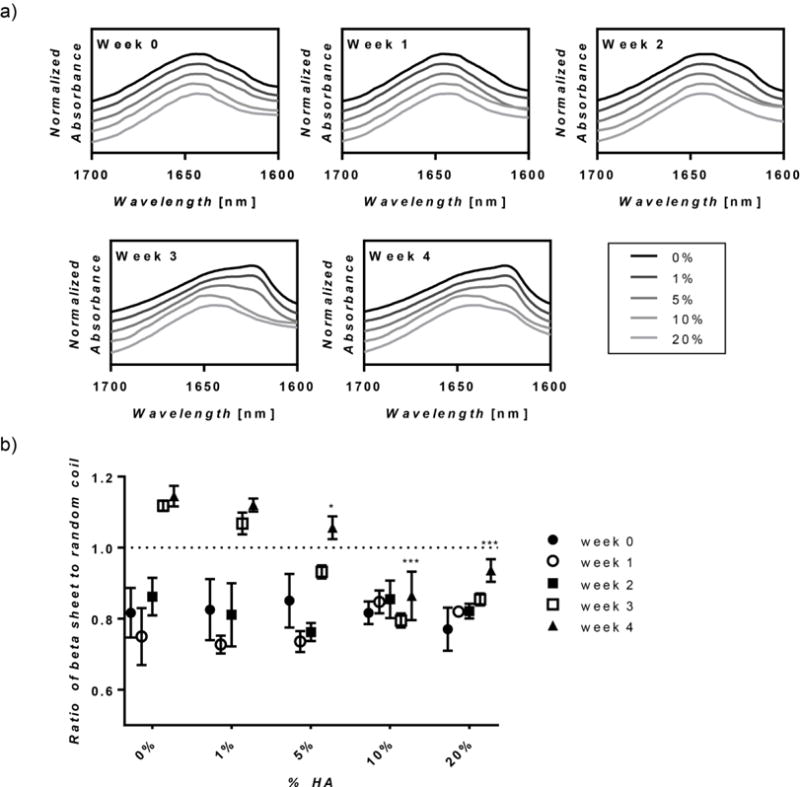
(a) The average FTIR absorbance spectrum in the amide I region is shown for hydrogels over time. Hydrogels with lower HA concentration exhibit a peak shift from ~1640cm−1 to ~1620cm−1 at 3 weeks. Additionally, the peak at ~1620 cm−1, which is representative of ß-sheet formation, becomes larger and wider as HA concentration decreases. (b) The ratio of ß-sheet to random coil conformation was calculated by dividing the average of peak absorbance at 1620–1625 and 1640–1650cm−1 showing that increasing HA concentration reduced the ratio over time. (n=5, *p≤0.05 and ***p≤0.001 compared to week 0).
Swelling
The water retention capabilities of the hydrogels were determined by calculating percentage of water in the hydrogels over 1 month (Equation 1). Figure 5 shows that initially, all composite hydrogels contained ~97% water, similar to that of silk only hydrogels. Starting at week 1, the composite hydrogels contained higher percentages of water as compared to silk only hydrogels. HA concentration was directly correlated to water retention, where after 1 month the 0, 10, and 20% HA hydrogels contained 83%, 88%, and 94% percent water, respectively. This shows that increased HA concentrations help maintain water content over time.
Figure 5. Percent Water.
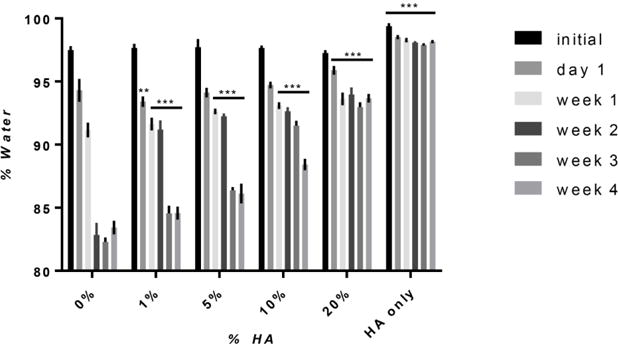
The percent water, based on mass, was calculated for hydrogels over time. Initially, all hydrogels, with the exception of HA only hydrogels, contained ~97% water. At weeks1, 2, 3, and 4, all composite hydrogels had higher percentages of water as compared to 0% hydrogels, showing that the addition of HA reduces the amount of water loss over time. (n=5, **p≤0.01, ***p≤0.001 compared to 0% HA).
In Vitro Degradation
The degradation profiles of hydrogels were determined in vitro after soaking hydrogels in an enzyme solution consisting of protease and hyaluronidase (Figure 6). The composite hydrogels exhibited degradation rates ranging between that of their singular component counterparts. HA only hydrogels completely degraded at day 6 whereas silk only hydrogels were 70% their original weight at day 8. In the composite hydrogels, HA concentration was correlated to degradation rate as increasing HA concentration above 5% increased degradation at day 8. More specifically, hydrogels with 10 and 20% HA were approximately 50% and 15% their initial weight, respectively.
Figure 6. In vitro Degradation.
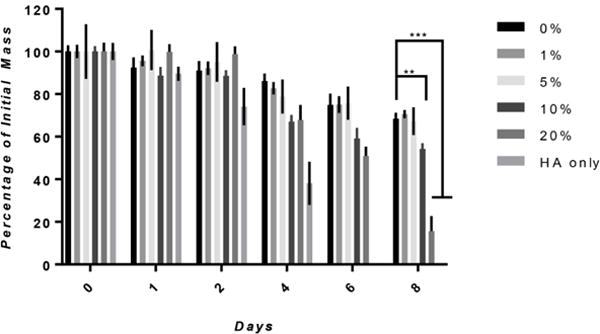
Degradation was determined by calculating the fraction of initial mass after soaking in an enzymatic cocktail consisting of 1 U/mL and 0.001 U/mL of hyalruonidase and protease. A direct relation between degradation rate and HA concentration was seen when increasing HA concentration above 5% significantly increased the degradation rate at day 8 (n=4, **p≤0.01, ***p≤0.001).
hMSC Responses
hMSCs from bone marrow aspirates were plated onto preformed hydrogels containing silk only, silk-HA, HA only, or TCP to determine in vitro cell response. Brightfield images showed cell attachment and morphology at day 0 and 7, where cells on TCP, silk, and silk-HA (10% HA) hydrogels became confluent (Figure 7a). Cellular proliferation at day 3, 5, and 7 are shown as the fold change of initial DNA content, quantified with PicoGreen (Figure 7b). Both silk and silk-HA hydrogels supported cellular growth with spread morphologies and proliferation, similar to that of TCP controls, revealing the cytocompatibility of the hydrogels. On the other hand, HA only hydrogels inhibited hMSC growth with cell morphology remaining spherical over 1 week.
Figure 7. 2D hMSC Response.
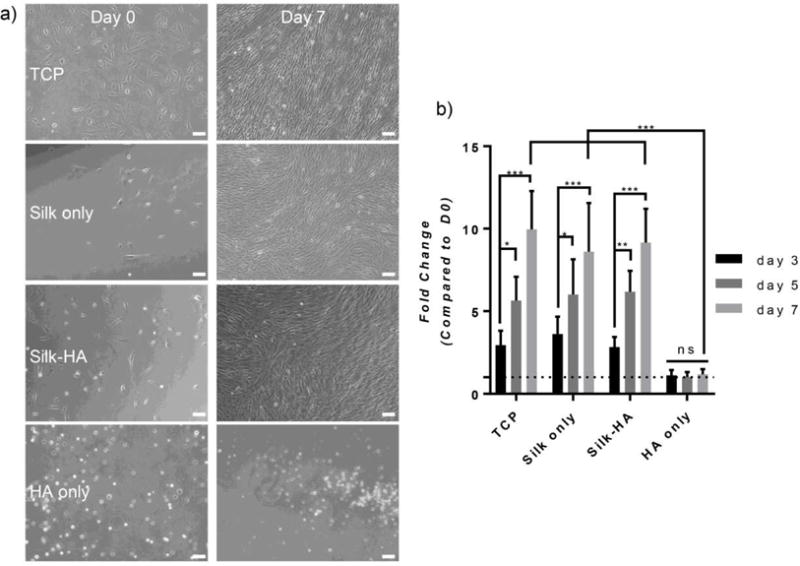
(a) Bright-field images of 2D hMSCs are shown at day 0 and 7. Hydrogels containing silk fibroin enhanced cell spreading whereas HA only hydrogels inhibited spreading (scale bar=100 μm). (b) The fold change of hMSC DNA content as compared to day 0 was calculated for days 3, 5, and 7. Silk only and silk-HA (10% HA) hydrogels promoted cellular growth similar to that of TCP whereas HA only hydrogels showed inhibited growth (n=6, * p≤0.05, **p≤0.01, ***p≤0.001).
V. Discussion
In the present study, enzymatically crosslinked silk-based hydrogels were further explored by investigating the effects of covalently binding HA to the silk. The oxidative crosslinking of tyrosine groups found in silk using HRP and H2O2 leads to highly elastic, cytocompatible hydrogels with tunable mechanical properties [5]. Although these hydrogels provide limited biological cues attributed to the absence of cell-specific signaling epitopes in the silk fibroin primary sequence, they are nonetheless advantageous due to the reaction simplicity, mechanical integrity, relatively slow degradation, and control of properties, when considered in the context of other natural polymeric systems such as HA [21], alginate [43, 44], dextran [45], and gelatin [46]. These other degradable polymers often require the addition of functional groups to facilitate crosslinking, have mechanical properties limited to a few kPa and rapidly degrade in vivo. As observed in silk-based enzymatically crosslinked hydrogels, depending on the degree of crosslinking and external environmental stimuli, the tendency of silk to self-assemble into ß-sheet secondary structures results in stiffening of the initial crosslinked gels over time [13, 47, 48]. Therefore, silk-HA composite hydrogels were formed in hopes to develop a hybrid scaffold with unique properties that cannot be achieved with single polymer materials. The hybrid hydrogel can thus possess the positive attributes from each polymer where silk can provide mechanical integrity and resistance to degradation while HA can offer enhanced hydration and potentially provide biofunctionality such as facilitating cell differentiation [49] and angiogenesis [50]. In this study, we sought to explore the physical material properties of silk-HA composite hydrogels, with the hypothesis that HA would maintain hydration and provide physical control of silk self-assembly.
The most notable effect of HA concentration in the composite hydrogels was the control of mechanical changes over time. Initially, all hydrogels were highly elastic and had compressive moduli between 3 and 8 kPa, similar to that of soft tissues [51]. After 1 month, silk only hydrogels became inelastic and stiffened to approximately 1 MPa, driven by a thermodynamically favorable shift in secondary structure from random coil to ß-sheet as determined by a peak shift in the FTIR spectra from ~1640 cm−1 to ~1620 cm−1. In addition, the increase in crystallinity is associated with concentrated hydrophobic regions [52] and decreased hydrogel water retention, as determined through swelling studies. With the composite hydrogels, the increasing of HA concentration delayed and reduced this shift in conformation, leading to more stable mechanics, elasticity, and water retention over time. For instance, HA concentrations of at least 5% did not show significant stiffening until week 3, whereas silk only and hydrogels with lower HA concentrations started to stiffen at week 2 with larger increases seen at week 3 This is reflective on the ratio of ß-sheet to random coil where silk only and lower HA hydrogels exhibited a ratio over 1 by week 3. By 1 month, there was an inverse relationship between HA concentration and compressive modulus where 1, 5 and 10% HA hydrogels had moduli of approximately 960, 460, and 290 kPa, with ß-sheet to random coil ratios of 1.12, 1.06, and 0.86, and water percentages of 85%, 86%, and 88%, respectively. Hydrogels containing 20% HA exhibited a comparatively small amount of stiffening to approximately 40 kPa with 94% water, revealing that high HA concentration impedes the extensive changes in mechanical properties, crystallinity, and water retention as seen in neat silk hydrogels.
The interaction between silk and HA was previously explored with physically crosslinked matrices, where increasing HA increased silk ß-sheet content, resulting in stiffer scaffolds [32, 34]. Unlike in our system, HA was physically blended with silk, which fostered phase separation and silk self-assembly, resulting in the encapsulation of HA within the matrix with increased crystallinity. However, an intermixed hydrogel is formed when HA is covalently crosslinked within the silk matrix. The large hydrophilic polymer thus helps to retain water molecules within the network and forces the separation of the polymer backbones, retarding changes in secondary structure and stabilizing mechanical properties over time.
Overall, the results show that by changing HA concentration we can select and predict the rate and extent of matrix stiffening, without further external stimuli, which can negatively impact cell function or damage tissue in vivo. Temporal hydrogel stiffening has been exploited with a number of hydrogel systems to study tissue development [13, 53], stem cell differentiation [54, 55], disease models [56], and tumor growth suppression [47]. In particular, methacrylated HA hydrogels initially crosslinked through Michael type reactions could be stiffened to ~30 kPa, about a 10-fold increase from the initial modulus, upon stimulation from UV [54] or visible light [56] after addition of a photoinitiator. Thiolated HA hydrogels exhibited a 3-fold increase in stiffness after about 3 days, through crosslinking of poly(ethylene glycol) diacrylate [53]. In comparison, the silk-HA hydrogels studied here exhibited stiffening that covers a larger range of increased moduli from an order of 10 kPa to just under 1 MPa, by varying initial HA concentration. This range of mechanics, which is generally unattainable by natural polymers, covers most intermediate stiffness ranges of biological tissues [57], thus extending the potential for these composite hydrogels to serve as biomimetic scaffolds to study tissue fibrosis [13] or the stiffer of soft tissues such as muscle and cartilage [57, 58].
Due to the simplicity of the reaction, it may be possible to achieve similar results by crosslinking silk to other high molecular weight, unbranched, hydrophilic polymers such as polyethylene glycol, extending the utility of the system. Additionally, other silk-based hydrogels using a similar method of preparation, showed mechanical properties and extent of stiffening controllable by altering silk molecular weight and concentration [13, 59], as well as the ratios of the cross-linking components [48], which if applied to the silk-HA hybrid system, can further the versatility and tunability of the hydrogels.
HA concentration not only affected the mechanical properties but also altered gelation kinetics, where increasing HA concentration above 1% steadily decreased the sol-gel transition time from 20 minutes to just over 1 minute. A similar trend was seen with crosslinking kinetics where HA concentration was inversely correlated to the completion of crosslinking. These results may be in part due to an increase in potential crosslinks from an increase in HA concentration. The ability to tune the sol-gel transition within this time frame shows the potential for in situ gelation. Since there are no known human peroxidases that facilitate phenolic crosslinking, in order to let HRP crosslinked hydrogels form in situ, sufficient time prior to gelation after mixing is required [60].
Although HA concentration affected the gelation kinetics, in terms of sol-gel transition and crosslinking kinetics, it had no significant effect in mechanical stability as determined through rheological time sweeps. Additionally, HA only hydrogels became mechanically stable after crosslinking was induced at ~15 minutes, a similar time as to when crosslinking was completed. However, the silk only hydrogel completed crosslinking at ~110 minutes but did not reach the final modulus until 170 minutes. This variation in time may be due to the intramolecular interactions between silk protein domains, driving the shear mechanical properties after covalent crosslinking is completed. As previously reported, the time to reach stable shear mechanics can be decreased by increasing silk concentration and decreasing molecular weight, thus potentially increasing the rate of intramolecular silk interactions [5]. Therefore, in the present study, since silk concentration and molecular weight were held constant, it was not surprising that HA concentration had no effect on the time to reach stable shear mechanics, confirming that the silk component in the composite system drove initial mechanical stability. However, the addition of HA, led to increased moduli as compared to silk only hydrogels, due to the increase in overall polymer concentration and potential crosslinking sites.
To test the potential and viability of the hydrogels for in situ applications, hydrogels were evaluated for cytotoxicity by culturing hMSCs directly on the gels without preconditioning or extracting crosslinking agents. HA only hydrogels evaluated did not support hMSC growth, as expected, due to its low mechanical strength and anti-adhesive properties [61–63]. HA-based hydrogels are often combined with small peptides or other polymers to enhance cell adhesion and proliferation through bioactive or mechanical cues [54, 61, 64]. In the present study, the combination of silk and HA led to composite hydrogels that supported hMSC growth, similar to that of silk hydrogels and TCP controls. Additional experiments show that composite hydrogels can also successfully encapsulate hMSCs with good cell survival and increased metabolic activity (Supplement Figure 7). These results show that the composite hydrogels developed are cytocompatible and support cell growth, which demonstrate the potential use of these hydrogels as a biomaterial.
HA hydrogels, like many naturally-based polymers, often degrade quickly due to the presence of hyaluronidases in the body [50, 63]. To overcome this rapid degradation, HA modifications and cross-linking have been attempted with varying degrees of success. For instance, thiolated HA hydrogels degraded completely in vivo in 2 weeks [65] whereas tyramine-HA hydrogels degraded about half their initial weight after 1 month [25]. On the other hand, silk-based hydrogels have exhibited slow degradation, maintaining their original size and shape up to 15 weeks [6]. Due to these differences, by covalently crosslinking silk and HA, the degradation rates of the composite hydrogels can be tuned for either long or short-term applications. Through in vitro studies,we showed that the composite silk-HA hydrogels degraded more slowly than HA only hydrogels upon exposure to protease XIV and hyaluronidase. As anticipated, increasing the HA concentration in the composite hydrogels led to faster degradation rates due to the rapid degradation of HA, disrupting the overall polymer network. These data show we have formed a highly interconnected hybrid hydrogel that has tunable degradation rates dependent on the concentration of HA. With the control of biodegradation, there needs to be a balance between scaffold degradation and mechanical integrity. This balance is often application specific and depends on the function of the scaffold. Particularly, in tissue regeneration applications, scaffold degradation should occur at the same rate as new ECM deposition to avoid scaffold failure [66]. This balance is often facilitated by cell-mediated degradation and can be accomplished through enzyme-sensitive scaffolds. In addition, for drug delivery applications, the rate of degradation will depend on the rate of therapeutic delivery [3]. The balance between biodegradation and scaffold integrity can be further controlled by adding less-degradable hydrophilic polymers, such as polyethylene glycol [67], or increasing the crosslink density by substituting additional phenolic groups on the side chains of the polymers.
VI. Conclusions
Silk-HA hybrid hydrogels with controllable gelation and degradation rates and predictive temporal stiffening were successfully developed. These binary hydrogels are advantageous over other natural polymer-based systems that tend to rapidly degrade and possess narrow mechanical ranges limited to softer tissue types. By changing the concentration of a single polymer in the binary system, we were able to show a wide range of tunable properties resulting in a versatile scaffold that can be specifically designed for many tissue engineering applications.
| Equation 1 |
Supplementary Material
Acknowledgments
We thank the NIH [grant numbers R01 EB02126, P41 EB002520]; AFOSR [FA9550-14-1-0015] for support of this work. The authors would like to thank Whitney L. Stoppel, Morgan Hawker, Siran Wang, Aswin Sundarakrishnan, and Matthew Applegate for valuable experimental advice and intellectual discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
VIII. References
- 1.Caló E, Khutoryanskiy VV. Biomedical applications of hydrogels: a review of patents and commercial products. European Polymer Journal. 2015;65:252–267. [Google Scholar]
- 2.Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Hydrogels in biology and medicine: from molecular principles to bionanotechnology. Advanced Materials. 2006;18(11):1345–1360. [Google Scholar]
- 3.Hoffman AS. Hydrogels for biomedical applications. Advanced drug delivery reviews. 2012;64:18–23. doi: 10.1016/s0169-409x(01)00239-3. [DOI] [PubMed] [Google Scholar]
- 4.Bae KH, Wang LS, Kurisawa M. Injectable biodegradable hydrogels: progress and challenges. Journal of Materials Chemistry B. 2013;1(40):5371–5388. doi: 10.1039/c3tb20940g. [DOI] [PubMed] [Google Scholar]
- 5.Partlow BP, Hanna CW, Rnjak‐Kovacina J, Moreau JE, Applegate MB, Burke KA, Marelli B, Mitropoulos AN, Omenetto FG, Kaplan DL. Highly tunable elastomeric silk biomaterials. Advanced functional materials. 2014;24(29):4615–4624. doi: 10.1002/adfm.201400526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leng X, Liu B, Su B, Liang M, Shi L, Li S, Qu S, Fu X, Liu Y, Yao M. In situ ultrasound imaging of silk hydrogel degradation and neovascularization. Journal of tissue engineering and regenerative medicine. 2015 doi: 10.1002/term.1981. [DOI] [PubMed] [Google Scholar]
- 7.Cao Y, Wang B. Biodegradation of silk biomaterials. International journal of molecular sciences. 2009;10(4):1514–1524. doi: 10.3390/ijms10041514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Kluge JA, Leisk GG, Kaplan DL. Sonication-induced gelation of silk fibroin for cell encapsulation. Biomaterials. 2008;29(8):1054–1064. doi: 10.1016/j.biomaterials.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yucel T, Cebe P, Kaplan DL. Vortex-induced injectable silk fibroin hydrogels. Biophysical journal. 2009;97(7):2044–2050. doi: 10.1016/j.bpj.2009.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim UJ, Park J, Li C, Jin HJ, Valluzzi R, Kaplan DL. Structure and properties of silk hydrogels. Biomacromolecules. 2004;5(3):786–792. doi: 10.1021/bm0345460. [DOI] [PubMed] [Google Scholar]
- 11.Kang GD, Nahm JH, Park JS, Moon JY, Cho CS, Yeo JH. Effects of poloxamer on the gelation of silk fibroin. Macromolecular rapid communications. 2000;21(11):788–791. [Google Scholar]
- 12.Partlow BP, Bagheri M, Harden JL, Kaplan DL. Tyrosine templating in the self-assembly and crystallization of silk fibroin. Biomacromolecules. 2016 doi: 10.1021/acs.biomac.6b01086. [DOI] [PubMed] [Google Scholar]
- 13.Stoppel WL, Gao AE, Greaney AM, Partlow BP, Bretherton RC, Kaplan DL, Black LD. Elastic silk‐cardiac extracellular matrix hydrogels exhibit time‐dependent stiffening that modulates cardiac fibroblast response. Journal of Biomedical Materials Research Part A. 2016 doi: 10.1002/jbm.a.35850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garg HG, Hales CA. Chemistry and biology of hyaluronan. Elsevier; 2004. [Google Scholar]
- 15.Borzacchiello A, Russo L, Malle BM, Schwach-Abdellaoui K, Ambrosio L. Hyaluronic acid based hydrogels for regenerative medicine applications. BioMed research international. 2015;2015 doi: 10.1155/2015/871218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta V, Werdenberg JA, Blevins TL, Grande-Allen KJ. Synthesis of glycosaminoglycans in differently loaded regions of collagen gels seeded with valvular interstitial cells. Tissue engineering. 2007;13(1):41–49. doi: 10.1089/ten.2006.0091. [DOI] [PubMed] [Google Scholar]
- 17.Kogan G, Šoltés L, Stern R, Gemeiner P. Hyaluronic acid: a natural biopolymer with a broad range of biomedical and industrial applications. Biotechnology letters. 2007;29(1):17–25. doi: 10.1007/s10529-006-9219-z. [DOI] [PubMed] [Google Scholar]
- 18.Chung CW, Kang JY, Yoon IS, Hwang HD, Balakrishnan P, Cho HJ, Chung KD, Kang DH, Kim DD. Interpenetrating polymer network (IPN) scaffolds of sodium hyaluronate and sodium alginate for chondrocyte culture. Colloids and Surfaces B: Biointerfaces. 2011;88(2):711–716. doi: 10.1016/j.colsurfb.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Xu X, Jha AK, Harrington DA, Farach-Carson MC, Jia X. Hyaluronic acid-based hydrogels: from a natural polysaccharide to complex networks. Soft matter. 2012;8(12):3280–3294. doi: 10.1039/C2SM06463D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chin L, Calabro A, Walker E, Derwin KA. Mechanical properties of tyramine substituted‚Äêhyaluronan enriched fascia extracellular matrix. Journal of Biomedical Materials Research Part A. 2012;100(3):786–793. doi: 10.1002/jbm.a.34025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Darr A, Calabro A. Synthesis and characterization of tyramine-based hyaluronan hydrogels. Journal of Materials Science: Materials in Medicine. 2009;20(1):33–44. doi: 10.1007/s10856-008-3540-0. [DOI] [PubMed] [Google Scholar]
- 22.Lee F, Chung JE, Kurisawa M. An injectable hyaluronic acid–tyramine hydrogel system for protein delivery. Journal of Controlled Release. 2009;134(3):186–193. doi: 10.1016/j.jconrel.2008.11.028. [DOI] [PubMed] [Google Scholar]
- 23.Xu K, Lee F, Gao SJ, Chung JE, Yano H, Kurisawa M. Injectable hyaluronic acid-tyramine hydrogels incorporating interferon-Œ±2a for liver cancer therapy. Journal of Controlled Release. 2013;166(3):203–210. doi: 10.1016/j.jconrel.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 24.Abu-Hakmeh A, Kung A, Mintz BR, Kamal S, Cooper JA, Lu XL, Wan LQ. Sequential gelation of tyramine-substituted hyaluronic acid hydrogels enhances mechanical integrity and cell viability. Medical & biological engineering & computing. 2016:1–10. doi: 10.1007/s11517-016-1474-0. [DOI] [PubMed] [Google Scholar]
- 25.Kurisawa M, Chung JE, Yang YY, Gao SJ, Uyama H. Injectable biodegradable hydrogels composed of hyaluronic acid–tyramine conjugates for drug delivery and tissue engineering. Chemical communications. 2005;34:4312–4314. doi: 10.1039/b506989k. [DOI] [PubMed] [Google Scholar]
- 26.Chi NH, Yang MC, Chung TW, Chen JY, Chou NK, Wang SS. Cardiac repair achieved by bone marrow mesenchymal stem cells/silk fibroin/hyaluronic acid patches in a rat of myocardial infarction model. Biomaterials. 2012;33(22):5541–5551. doi: 10.1016/j.biomaterials.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 27.Foss C, Merzari E, Migliaresi C, Motta A. Silk fibroin/hyaluronic acid 3D matrices for cartilage tissue engineering. Biomacromolecules. 2012;14(1):38–47. doi: 10.1021/bm301174x. [DOI] [PubMed] [Google Scholar]
- 28.Park SH, Cho H, Gil ES, Mandal BB, Min BH, Kaplan DL. Silk-fibrin/hyaluronic acid composite gels for nucleus pulposus tissue regeneration. Tissue Engineering Part A( 2011;17(23–24):2999–3009. doi: 10.1089/ten.tea.2010.0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ren YJ, Zhou ZY, Liu BF, Xu QY, Cui FZ. Preparation and characterization of fibroin/hyaluronic acid composite scaffold. International journal of biological macromolecules. 2009;44(4):372–378. doi: 10.1016/j.ijbiomac.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Serban MA, Kluge JA, Laha MM, Kaplan DL. Modular elastic patches: Mechanical and biological effects. Biomacromolecules. 2010;11(9):2230–2237. doi: 10.1021/bm1007772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang LS, Yan SQ, Li MZ. Silk Fibroin/Hyaluronic Acid Blend Film with Good Water Stability and Cytocompatibility, Applied Mechanics and Materials. Trans Tech Publ. 2013:209–214. [Google Scholar]
- 32.Hu X, Lu Q, Sun L, Cebe P, Wang X, Zhang X, Kaplan DL. Biomaterials from ultrasonication-induced silk fibroin− hyaluronic acid hydrogels. Biomacromolecules. 2010;11(11):3178–3188. doi: 10.1021/bm1010504. [DOI] [PubMed] [Google Scholar]
- 33.Matsumoto A, Chen J, Collette AL, Kim UJ, Altman GH, Cebe P, Kaplan DL. Mechanisms of silk fibroin sol-gel transitions. The Journal of Physical Chemistry B. 2006;110(43):21630–21638. doi: 10.1021/jp056350v. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Fuentes M, Giger E, Meinel L, Merkle HP. The effect of hyaluronic acid on silk fibroin conformation. Biomaterials. 2008;29(6):633–642. doi: 10.1016/j.biomaterials.2007.10.024. [DOI] [PubMed] [Google Scholar]
- 35.Pavlovic E, Serban MA, Yu X, Manesis NJ. Cross-linked silk-hyaluronic acid compositions. Google Patents. 2013 [Google Scholar]
- 36.Yeom J, Bhang SH, Kim BS, Seo MS, Hwang EJ, Cho IH, Park JK, Hahn SK. Effect of cross-linking reagents for hyaluronic acid hydrogel dermal fillers on tissue augmentation and regeneration. Bioconjugate chemistry. 2010;21(2):240–247. doi: 10.1021/bc9002647. [DOI] [PubMed] [Google Scholar]
- 37.Kennedy GL., Jr Toxicity of Hexamethylenediamine (HMDA) Drug and chemical toxicology. 2005;28(1):15–33. doi: 10.1081/dct-39681. [DOI] [PubMed] [Google Scholar]
- 38.Moshnikova A, Afanasyev V, Proussakova O, Chernyshov S, Gogvadze V, Beletsky I. Cytotoxic activity of 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide is underlain by DNA interchain cross-linking. Cellular and Molecular Life Sciences CMLS. 2006;63(2):229–234. doi: 10.1007/s00018-005-5383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rockwood DN, Preda RC, Yücel T, Wang X, Lovett ML, Kaplan DL. Materials fabrication from Bombyx mori silk fibroin. Nature protocols. 2011;6(10):1612–1631. doi: 10.1038/nprot.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Applegate MB, Coburn J, Partlow BP, Moreau JE, Mondia JP, Marelli B, Kaplan DL, Omenetto FG. Laser-based three-dimensional multiscale micropatterning of biocompatible hydrogels for customized tissue engineering scaffolds. Proceedings of the National Academy of Sciences. 2015;112(39):12052–12057. doi: 10.1073/pnas.1509405112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu X, Kaplan D, Cebe P. Determining beta-sheet crystallinity in fibrous proteins by thermal analysis and infrared spectroscopy. Macromolecules. 2006;39(18):6161–6170. [Google Scholar]
- 42.Altman G, Horan R, Martin I, Farhadi J, Stark P, Volloch V, Vunjak-Novakovic G, Richmond J, Kaplan DL. Cell differentiation by mechanical stress. Faseb Journal. 2002;16(2):270–272. doi: 10.1096/fj.01-0656fje. [DOI] [PubMed] [Google Scholar]
- 43.Ganesh N, Hanna C, Nair SV, Nair LS. Enzymatically cross-linked alginic–hyaluronic acid composite hydrogels as cell delivery vehicles. International journal of biological macromolecules. 2013;55:289–294. doi: 10.1016/j.ijbiomac.2012.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakai S, Kawakami K. Development of porous alginate-based scaffolds covalently cross-linked through a peroxidase-catalyzed reaction. Journal of Biomaterials Science, Polymer Edition. 2011;22(18):2407–2416. doi: 10.1163/092050610X540468. [DOI] [PubMed] [Google Scholar]
- 45.Jin R, Teixeira LM, Dijkstra P, Van Blitterswijk C, Karperien M, Feijen J. Enzymatically-crosslinked injectable hydrogels based on biomimetic dextran–hyaluronic acid conjugates for cartilage tissue engineering. Biomaterials. 2010;31(11):3103–3113. doi: 10.1016/j.biomaterials.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 46.Amini AA, Nair LS. Enzymatically cross-linked injectable gelatin gel as osteoblast delivery vehicle. Journal of Bioactive and Compatible Polymers. 2012;27(4):342–355. [Google Scholar]
- 47.Yan L-P, Silva-Correia J, Ribeiro VP, Miranda-Gonçalves V, Correia C, da Silva Morais A, Sousa RA, Reis RM, Oliveira AL, Oliveira JM. Tumor Growth Suppression Induced by Biomimetic Silk Fibroin Hydrogels. Scientific Reports. 2016;6 doi: 10.1038/srep31037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao S, Chen Y, Partlow BP, Golding AS, Tseng P, Coburn J, Applegate MB, Moreau JE, Omenetto FG, Kaplan DL. Bio-functionalized silk hydrogel microfluidic systems. Biomaterials. 2016;93:60–70. doi: 10.1016/j.biomaterials.2016.03.041. [DOI] [PubMed] [Google Scholar]
- 49.Chung C, Burdick JA. Influence of three-dimensional hyaluronic acid microenvironments on mesenchymal stem cell chondrogenesis. Tissue Engineering Part A. 2008;15(2):243–254. doi: 10.1089/ten.tea.2008.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cui F, Tian W, Hou S, Xu Q, Lee IS. Hyaluronic acid hydrogel immobilized with RGD peptides for brain tissue engineering. Journal of Materials Science: Materials in Medicine. 2006;17(12):1393–1401. doi: 10.1007/s10856-006-0615-7. [DOI] [PubMed] [Google Scholar]
- 51.Sartori S, Chiono V, Tonda-Turo C, Mattu C, Gianluca C. Biomimetic polyurethanes in nano and regenerative medicine. Journal of Materials Chemistry B. 2014;2(32):5128–5144. doi: 10.1039/c4tb00525b. [DOI] [PubMed] [Google Scholar]
- 52.Floren M, Migliaresi C, Motta A. Processing Techniques and Applications of Silk Hydrogels in Bioengineering. Journal of Functional Biomaterials. 2016;7(3):26. doi: 10.3390/jfb7030026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Young JL, Engler AJ. Hydrogels with time-dependent material properties enhance cardiomyocyte differentiation in vitro. Biomaterials. 2011;32(4):1002–1009. doi: 10.1016/j.biomaterials.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guvendiren M, Burdick JA. Stiffening hydrogels to probe short-and long-term cellular responses to dynamic mechanics. Nature communications. 2012;3:792. doi: 10.1038/ncomms1792. [DOI] [PubMed] [Google Scholar]
- 55.Yang C, DelRio FW, Ma H, Killaars AR, Basta LP, Kyburz KA, Anseth KS. Spatially patterned matrix elasticity directs stem cell fate. Proceedings of the National Academy of Sciences. 2016;113(31):E4439–E4445. doi: 10.1073/pnas.1609731113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caliari SR, Perepelyuk M, Cosgrove BD, Tsai SJ, Lee GY, Mauck RL, Wells RG, Burdick JA. Stiffening hydrogels for investigating the dynamics of hepatic stellate cell mechanotransduction during myofibroblast activation. Scientific reports. 2016;6 doi: 10.1038/srep21387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levental I, Georges PC, Janmey PA. Soft biological materials and their impact on cell function. Soft Matter. 2007;3(3):299–306. doi: 10.1039/b610522j. [DOI] [PubMed] [Google Scholar]
- 58.Pal S. Design of artificial human joints & organs. Springer. 2014 [Google Scholar]
- 59.Golding A, Guay JA, Herrera-Rincon C, Levin M, Kaplan DL. A Tunable Silk Hydrogel Device for Studying Limb Regeneration in Adult Xenopus Laevis. PloS one. 2016;11(6):e0155618. doi: 10.1371/journal.pone.0155618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teixeira LSM, Feijen J, van Blitterswijk CA, Dijkstra PJ, Karperien M. Enzyme-catalyzed crosslinkable hydrogels: emerging strategies for tissue engineering. Biomaterials. 2012;33(5):1281–1290. doi: 10.1016/j.biomaterials.2011.10.067. [DOI] [PubMed] [Google Scholar]
- 61.Tsou YH, Khoneisser J, Huang PC, Xu X. Hydrogel as a bioactive material to regulate stem cell fate. Bioactive Materials. 2016 doi: 10.1016/j.bioactmat.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Necas J, Bartosikova L, Brauner P, Kolar J. Hyaluronic acid hyaluronan: a review. Veterinarni medicina. 2008;53(8):397–411. [Google Scholar]
- 63.Burdick JA, Prestwich GD. Hyaluronic acid hydrogels for biomedical applications. Advanced materials. 2011;23(12) doi: 10.1002/adma.201003963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khetan S, Katz JS, Burdick JA. Sequential crosslinking to control cellular spreading in 3-dimensional hydrogels. Soft Matter. 2009;5(8):1601–1606. [Google Scholar]
- 65.Hahn SK, Park JK, Tomimatsu T, Shimoboji T. Synthesis and degradation test of hyaluronic acid hydrogels. International journal of biological macromolecules. 2007;40(4):374–380. doi: 10.1016/j.ijbiomac.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 66.Nicodemus GD, Bryant SJ. Cell encapsulation in biodegradable hydrogels for tissue engineering applications. Tissue Engineering Part B: Reviews. 2008;14(2):149–165. doi: 10.1089/ten.teb.2007.0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lam J, Truong NF, Segura T. Design of cell–matrix interactions in hyaluronic acid hydrogel scaffolds. Acta biomaterialia. 2014;10(4):1571–1580. doi: 10.1016/j.actbio.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van den Hoogen BM, van Weeren PR, Lopes-Cardozo M, van Golde LM, Barneveld A, van de Lest CH. A microtiter plate assay for the determination of uronic acids. Analytical biochemistry. 1998;257(2):107–111. doi: 10.1006/abio.1997.2538. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.